Sevá-Pereira A, Magalhães AFN. Digestão e Absorção de Proteínas - Fisiologia e
Fisiopatologia. In: Castro L de P, Rocha PRS, Cunha-Melo JR. Tópicos em
Gastroenterologia 5. Editora Medsi, Rio de Janeiro, 1994. cap 11. p. 163-185.
Intestinos
11) Digestão e Absorção de Proteínas: Fisiologia e Fisiopatologia
*Adriana Sevá-Pereira
Professora Adjunta da Disciplina de Gastroenterologia do Departamento de Clínica
Médica da Faculdade de Ciências Médicas da UNICAMP.
Doutora em Medicina pela Unicamp.
Pesquisadora do CNPQ.
Antonio Frederico Novaes de Magalhães
Professor Titular da Disciplina de Gastroenterologia do Departamento de Clínica
Médica da Faculdade de Ciências Médicas da UNICAMP.
1. Introdução
Uma das maiores funções do trato gastrintestinal é transformar a energia dos alimentos
ingeridos em combustível a ser utilizado pelo organismo. Eles são classificados em
carboidratos, gorduras e proteínas, e se apresentam na natureza como polímeros (amido,
triglicerídeos, proteínas), que, ao serem ingeridos, necessitam ser transformados em
moléculas suficientemente pequenas, os monômeros, para que sejam absorvidos e
transformados em energia. O processo de quebra das moléculas, a digestão, ocorre na
luz ou na mucosa intestinal. A absorção é o transporte dos substratos da luz intestinal
através da barreira de células epiteliais para a circulação.
Este capítulo discute primeiramente os processos através dos quais as proteínas são
digeridas, em segundo lugar, os mecanismos através dos quais os produtos da digestão
são absorvidos, e, por último, os mecanismos fisiopatológicos das alterações de várias
doenças.
2. Proteínas: conceito e fontes (exógena e endógena)
2.1. Conceito
Cerca de três quartos dos sólidos corporais são proteínas, as quais incluem as proteínas
estruturais, as enzimas, os genes, as proteínas que transportam oxigênio, as proteínas
que efetuam a contração muscular e vários outros tipos que executam funções
específicas, tanto dentro quanto fora das células em todo o organismo 17.
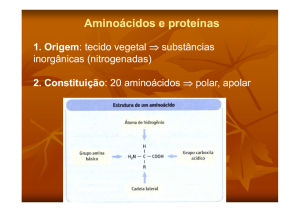
Os principais constituintes das proteínas são os aminoácidos, que têm em comum: um
grupo ácido (-COOH) e um radical nitrogenado ligado à molécula próxima ao radical
ácido, em geral representado pelo grupo amino (-NH2).
Os aminoácidos podem ser agrupados de acordo com suas estruturas químicas em nove
grupos 28:
1. Ácidos monoamino-monocarboxílicos alifáticos: glicina, alanina, serina e treonina.
2. Ácidos monoamino-monocarboxílicos alifáticos de cadeia ramificada: valina, leucina
e isoleucina.
3. Aminoácidos aromáticos: fenilalanina e tirosina.
4. Aminoácidos heterocíclicos: triptofano e histidina.
5. Ácidos monoamino-dicarboxílicos: ácido glutâmico e ácido aspártico.
6. Ácidos diamino-monocarboxílicos: lisina, ornitina e arginina.
7. Aminoácidos sulfúricos: cisteína, homocisteína, cistina, homocistina e metionina.
8. Iminoácidos: prolina e hidroxiprolina.
9. Beta-aminoácidos: beta-aminoácido isobutírico
Para a presente discussão, que envolve os mecanismos de transporte dos aminoácidos, é
feita uma divisão em quatro grupos que representam uma segregação natural e
genética28:
Grupo A: Ácidos monoamino-monocarboxílicos (cadeia simples e cadeia ramificada) e
metionina.
Grupo B: Ácidos diamino-monocarboxílicos.
Grupo C: Iminoácidos.
Grupo D: Ácidos monoamino-dicarboxílicos.
As proteínas são formadas de aminoácidos unidos em longas cadeias por meio das
ligações peptídicas. Nessa ligação, um íon hidroxila é removido de um aminoácido,
enquanto um íon hidrogênio é removido do próximo aminoácido. Assim, os
aminoácidos combinam-se, através de um processo de condensação, ao mesmo tempo
em que perdem uma molécula de água. Algumas proteínas são compostas de v rias
cadeias peptídicas, que se ligam umas às outras por outras ligações, quase sempre por
pontes de hidrogênio entre os radicais CO e NH dos peptídeos. Além disso, várias
cadeias peptídicas enovelam-se ou dobram-se, e os sucessivos novelos ou dobras são
presos em espiral ou em outras formas por pontes similares de hidrogênio e também por
pontes hidrofóbicas, ligações sulfidrílicas, fenólicas e salinas.
2.2. Proteínas exógenas
Os adultos necessitam de aproximadamente 0,5 a 1g de proteína, enquanto crianças em
fase de crescimento necessitam de 4g por quilo de peso corporal por dia, para manterem
balanço positivo de nitrogênio. Estima-se que a dieta do adulto forneça 70 a 100g de
proteína exógena, quantidade correspondente a 10 a 15% da ingestão calórica total. O
valor nutritivo da proteína, sinônimo de sua qualidade, é dado por três características:
quantidade e qualidade dos aminoácidos, digestibilidade da proteína e seu processo de
preparo.
2.2.1. Aminoácidos essenciais e não essenciais
A proteína da dieta deve suprir os aminoácidos essenciais, cujo esqueleto de carbono
não pode ser formado pelo homem. Eles incluem: leucina, isoleucina, lisina, triptofano,
fenilalanina, metionina, treonina e valina. Cistina e tirosina também são consideradas
essenciais pois são derivados respectivamente da metionina e da fenilalanina.
Os aminoácidos não essenciais são produzidos a partir do esqueleto de carbono de
produtos intermediários do metabolismo de carboidratos e gordura, enquanto os grupos
aminas vêm da transaminação.
Ambos são necessários para manter balanço positivo de nitrogênio e síntese protéica
eficaz.
2.2.2. Digestibilidade das proteínas exógenas
As proteínas ocorrem nos alimentos geralmente associadas a gorduras e carboidratos
complexos, os quais podem limitar a proteólise eficiente, levando a variação da
digestibilidade das proteínas. As técnicas modernas de processamento de alimentos,
especialmente as de cozimento e de estocagem, podem provocar mudanças químicas,
tais como criar novas ligações éster ou amidas com e entre pontes pépticas ou causar
condições resistentes à hidrólise enzimática, além de causar efeitos adversos nos valores
nutricionais das proteínas11.
2.3. Proteínas endógenas
Em adição à proteína exógena, as fontes endógenas de proteínas, secreções digestivas,
células descamadas e pequenas quantidades de proteínas plasmáticas, aumentam
significantemente o pool de proteínas utilizáveis, as quais são, então, digeridas e
absorvidas. Somente cerca de 10% da ingestão diária são eliminados nas fezes sob a
forma de bactérias, células descamadas e mucoproteínas.
A proteína endógena perfaz pelo menos metade da proteína utilizável pelo organismo.
Ela corresponde a 20 a 30g das secreções digestivas ricas em muco e enzimas, 30g das
descamações celulares, 1 a 2g de proteínas plasmáticas. Apesar de nem sempre ser
lembrada, é importante fonte protéica que entra no processo de digestão/absorção, sendo
reutilizada como fonte de nitrogênio.
Em condições normais os processos de digestão e absorção das proteínas exógenas (50100 g/dia) e endógenas (40-70 g/dia) são eficientíssimos, visto que a perda fecal é de
cerca de 1 a 2 g de nitrogênio por dia, equivalente a 6 a 12 g de proteína por dia 25.
3. Fisiologia da digestão/absorção das proteínas
Normalmente, a absorção pelo intestino delgado consiste, diariamente, de centenas de
gramas de carboidratos, 100 ou mais gramas de gordura, 50 a 100 gramas de
aminoácidos, 50 a 100 gramas de íons e 7 a 8 litros de água. Contudo, a capacidade
absortiva do intestino delgado é muito maior que isso, podendo absorver até vários
quilogramas de carboidratos por dia, 500 a 1000 gramas de gordura por dia, 500 a 700
gramas de aminoácidos por dia e 20 ou mais litros de água diariamente.
Quando o alimento tiver sido mastigado corretamente e não tiver sido ingerido em
quantidades excessivamente grandes, cerca de 98% de todas as proteínas são
transformadas, de uma única vez, quer em aminoácidos, quer em dipeptídios, que são as
formas pelas quais as proteínas passam para a circulação.
A taxa de liberação da proteína para o duodeno, e conseqüentemente sua velocidade de
digestão e absorção, varia muito. Cinqüenta por cento de uma refeição composta de
carne magra pode deixar o estômago na primeira hora e 83%, no final da terceira hora.
Uma vez no duodeno, a proteína é rapidamente digerida. O fluído aspirado do duodeno
humano subseqüente à alimentação contém 200-800 micro g/ml de tripsina e
quimotripsina. Isto é suficiente para digerir 50% da proteína em 10 minutos.
A digestão das proteínas envolve um processo de hidrólise em que as enzimas
proteolíticas fazem a água voltar às moléculas protéicas, separando-as em seus
aminoácidos constituintes. Todas as enzimas proteolíticas, incluindo aquelas do suco
gástrico, do suco pancreático e da borda em escova das células do epitélio intestinal, são
muito específicas para hidrolisar tipos individuais de ligações peptídicas. As ligações
entre certos pares de aminoácidos diferem em sua energia de ligação e em outras
características físicas das ligações entre outros pares. Portanto, é necessária uma enzima
específica para tipos específicos de ligação. Isto é responsável pela multiplicidade das
enzimas proteolíticas.
Os aminoácidos livres são absorvidos muito rapidamente, no máximo em cinco
minutos. Cerca de um décimo escapa da digestão no intestino delgado e penetra no
cólon, onde sua digestão é completada por microorganismos. A proteína contida nas
fezes, aproximadamente 10% da proteína ingerida não é, portanto, proteína da
alimentação, mas derivada de bactérias e de fragmentos celulares.
Para melhor entendimento, o processo de digestão/absorção das proteínas é dividido em
fases gástrica, pancreática e intestinal. Entretanto, após uma refeição, estas fases
ocorrem quase simultaneamente, influenciadas por fatores como o esvaziamento
gástrico e a velocidade na qual o alimento caminha através do intestino.
A figura 11.1 mostra esquematicamente o processo de digestão/absorção das proteínas,
o qual vai ser discutido detalhadamente a seguir.
3.1. Fase gástrica
A digestão das proteínas da dieta começa no estômago, onde a proteólise é iniciada por
dois tipos de pepsina, cuja função é ótima em meio ácido. Estas enzimas são ativadas
em pH ácido (entre 2 e 3), por autocatálise dos seus precursores zimogênios ou
pepsinogênios. As glândulas gástricas, através das células oxínticas ou parietais,
secretam grande quantidade de ácido clorídrico, em quantidade tal que o pH cai para
menos de 1. Porém, no momento em que se mistura com os conteúdos gástricos e com
as secreções procedentes das células não oxínticas das glândulas do estômago, o pH
gástrico passa para 2 a 3, que é uma faixa de acidez altamente favorável à atividade
péptica. Conseqüentemente, para que essas enzimas tenham alguma ação digestiva
sobre a proteína, é necessário que os sucos gástricos sejam ácidos.
Para a maioria dos autores 5,7,9,16, a digestão gástrica não tem muita importância e é
dispensável. Seus argumentos são: no máximo 10 a 15% das proteínas são
transformadas em aminoácidos antes de chegar ao duodeno, deixando para o intestino
delgado a maior parte da digestão protéica; indivíduos com acloridria podem
permanecer em equilíbrio nitrogenado enquanto estiverem ingerindo proteína de forma
usual.
A importância da fase gástrica da digestão protéica tem sido recentemente reavaliada
11,25. Alguns aminoácidos derivados da digestão péptica (fenilalanina e triptofano) são
potentes estimulantes da secreção de ácido e de gastrina e podem, conseqüentemente,
assumir o papel de moduladores da digestão péptica. Em segundo lugar, os produtos da
digestão péptica estimulam muito mais eficazmente do que as proteínas íntegras os
receptores do duodeno produtores dos hormônios entéricos, a secretina e a
colecistocinina, os quais modulam a secreção pancreática exócrina, respectivamente
hidrolítica e enzimática. A ação da pepsina resulta em uma mistura de polipeptídios
grandes, oligopeptídios menores e alguns aminoácidos livres. Estes produtos da
hidrólise influenciam muitas funções gástricas que estão sob controle hormonal, como a
secreção de ácido e de pepsinogênio, a velocidade de esvaziamento gástrico e o controle
do esfíncter pilórico 9,11.
Além disto, a pepsina é capaz de digerir praticamente todos os diferentes tipos de
proteínas da dieta. Uma das características importantes da digestão péptica é sua
capacidade de digerir o colágeno, um albuminóide que é pouco afetado pelas outras
enzimas digestivas. O colágeno é um grande constituinte do tecido conjuntivo
intercelular das carnes, e, para que as enzimas digestivas penetrem nas carnes para
hidrolisar as proteínas celulares, é necessário, primeiramente, que as fibras colágenas
sejam digeridas. Conseqüentemente, nas pessoas com deficiências de atividade péptica
no estômago, as carnes ingeridas não são bem penetradas pelas enzimas digestivas e,
por conseguinte, a digestão não é totalmente realizada.
Quando o estômago está funcionando normalmente, a intensidade da digestão gástrica
varia amplamente, dependendo dos fatores que afetem a secreção, do tamanho e da
divisão das proteínas alimentares ingeridas, misturadas no corpo e no antro, e da
velocidade de esvaziamento gástrico. Na pior das hipóteses, somente uma pequena
quantidade de proteína pode ser atacada no estômago; e na melhor das hipóteses, 10 a
15% podem ser cindidos em aminoácidos. Portanto, a proteína da dieta liberada para o
intestino é uma mistura de feixes de fibras musculares não digeridos, de proteínas
naturais em solução e de produtos da digestão péptica, variando de grandes
polipeptídios a alguns aminoácidos livres. Somente os últimos estão prontos para a
absorção; conseqüentemente, a maior parte da digestão protéica deve ocorrer no
intestino delgado.
3.2. Fase pancreática
A maior parte da digestão das proteínas ocorre principalmente dentro do intestino
delgado, pela ação das enzimas proteolíticas da secreção pancreática. Quando as
proteínas deixam o estômago, em geral encontram-se principalmente na forma de
proteoses, peptonas e grandes polipeptídios. Logo após penetrarem no intestino delgado,
os produtos da desintegração parcial são atacados pelas proteases pancreáticas, as
endopeptidases (tripsina, quimotripsina, elastase) e a exopeptidase
(carboxipolipeptidase).
As enzimas pancreáticas são secretadas, por estímulos hormonais, principalmente a
colecistocinina e a secretina, na forma inativa de zimogênios (tripsinogênio,
quimotripsinogênio, pró-elastase, pró-carboxipeptidase), que são ativados no duodeno,
quando o pH está neutro. A ativação do tripsinogênio requer uma enteropeptidase
intestinal, a enteroquínase, enzima da mucosa do intestino delgado, que rompe sua única
ligação peptídica. Ela inicia a ativação das pró-enzimas pancreáticas pela tripsina, que
provoca eventos proteolíticos em cascata, resultando nas enzimas totalmente ativas:
tripsina, quimotripsina, elastase e carboxipeptidase A e B9. A ativação dos outros
precursores requer a tripsina.
As endopeptidases (tripsina, quimotripsina, elastase) quebram as ligações peptídicas
internas das proteínas e peptídios. Todas têm um aminoácido serina no seu local ativo, e
são coletivamente chamadas de proteases da serina. A tripsina quebra a ponte peptídica
no local carboxila dos aminoácidos básicos (arginina, lisina), enquanto a quimotripsina
quebra a ponte onde o grupo carboxila é aromático (tirosina, fenilalanina, triptofano). A
elastase hidrolisa a elastina e quebra a ponte nos peptídios onde o grupo carboxila dos
aminoácidos é alifático (alanina, leucina, glicina, valina, isoleucina). Então, tanto a
tripsina quanto a quimotripsina podem fracionar as moléculas protéicas em pequenos
polipeptídios. A seguir, as carboxipolipeptidases A e B, as maiores exopeptidases
encontradas na secreção pancreática, retiram os aminoácidos individuais das
extremidades carboxilas dos polipeptídios. Elas são metalopeptidases que contêm zinco
de especificidade definida para remover aminoácidos simples dos terminais carboxílicos
das proteínas e dos oligopeptídios.
As especificidades das cinco proteases pancreáticas se complementam umas com as
outras 9. Como resultado, a maioria das proteínas é reduzida a uma mistura de
aminoácidos livres e oligopeptídios (2 a 6 ou 8 aminoácidos). A fase luminal da
digestão de proteínas leva principalmente ao aparecimento de oligopeptídios, mas
somente a pequenas quantidades de aminoácidos livres 5. Os produtos da digestão
pancreática das proteínas perfazem aproximadamente 40 a 60% do total luminal de
nitrogênio alfa-aminado10. Deste modo, a fase pancreática da digestão é mais
importante do que a fase gástrica, para a assimilação das proteínas 9.
O fluxo de suco pancreático começa 10-20 minutos após a ingestão de uma refeição, e a
concentração de enzimas pancreáticas permanece alta por todo o jejuno e o íleo durante
o período de digestão e absorção. Uma fração de enzimas pancreáticas está presente nas
fezes de homens, assim como nas dos cães e ratos normais, mas sua quantidade é
desconhecida.
Não há dúvida que a ausência de suco pancreático prejudica a digestão das proteínas. V
rios exemplos, na prática clínica, podem demonstrar que pacientes com fibrose cística
do pâncreas, com pancreatite crônica e que foram submetidos à pancreatectomia, em
que as enzimas pancreáticas estão quase completamente ausentes, têm baixa absorção
de proteína, que pode ser corrigida pela ingestão de enzimas pancreáticas.
3.3. Fase intestinal
A superfície absortiva da mucosa intestinal mostra muitas pregas denominadas válvulas
coniventes (ou pregas de Kerckring), que aumentam a área de absorção da mucosa cerca
de três vezes. Essas pregas estendem-se circularmente, por quase todo o intestino e
estão bastante desenvolvidas especialmente no duodeno e no jejuno, onde muitas vezes
fazem uma protrusão de até 8mm na luz intestinal. Localizadas em toda a superfície do
intestino delgado, desde aproximadamente o ponto em que o ducto biliar comum
esvazia-se no duodeno, até a válvula ileocecal, existem milhões de pequenas
vilosidades, que se projetam cerca de 1mm, a partir da superfície da mucosa. Essas
vilosidades situam-se tão próximas umas das outras, na porção superior do intestino
delgado, que na realidade tocam-se entre si, e elas aumentam em dez vezes a área
absortiva. As células epiteliais intestinais, também chamadas de enterócitos,
caracterizam-se pela borda em escova, contendo, cada célula, cerca de 600
microvilosidades com 1 milimícron de comprimento e 0,1 milimícron de diâmetro. Isto
aumenta a superfície de exposição às substâncias no intestino mais outras 20 vezes.
Assim, a combinação das pregas de Kerckring com as vilosidades e as microvilosidades
aumenta a área absortiva da mucosa em cerca de 600 vezes, atingindo a grande área
total de cerca de 250 metros quadrados para o intestino delgado, aproximadamente a
área de uma quadra de tênis.
O resultado da ação coletiva das proteases do estômago, do pâncreas e da membrana
celular do enterócito é a redução das proteínas da dieta a uma mistura de aminoácidos
livres, dipeptídios e tripetídeos, os quais se tornam próprios para serem transportados
pelos carregadores de membrana do enterócito.
Após a hidrólise das proteínas na luz intestinal pelas enzimas pancreáticas que as
quebram em aminoácidos e oligopeptídios, estes são digeridos pelas peptidases da
membrana celular das microvilosidades. Estas peptidases são amino-oligopeptidases
que hidrolisam as ligações peptídicas finais dos pequenos polipeptídios quando entram
em contato com o epitélio das vilosidades. Existem também várias dipeptidases e
tripeptidases diferentes que, ou na membrana ou no citoplasma hidrolisam os
respectivos di e tripeptídios.
Os polipeptídios, após serem liberados das proteínas por digestão pela tripsina dentro da
luz intestinal, difundem-se na borda em escova das células epiteliais intestinais. Os
polipeptídios são hidrolisados até seus aminoácidos constituintes dentro da borda em
escova ou imediatamente subjacentes a ela. Uma pequena fração dos aminoácidos
liberados retorna à luz por difusão; os remanescentes são carregados pelos sistemas
celulares responsáveis pelo transporte ativo de aminoácidos, acumulados dentro das
células e liberados no sangue portal.
Existem muitas enzimas capazes de catalisar a hidrólise de polipeptídios nas células
mucosas, mas a quantidade secretada para a luz intestinal é muito pequena. Sua
distribuição ao longo do tubo digestivo é semelhante à das oligossacaridases: a
concentração é baixa no estômago, duodeno proximal e cólon, e alta no jejuno e no íleo.
3.3.1. Digestão intestinal
Somente nos últimos 15 a 20 anos foi compreendida a importância da célula epitelial da
mucosa intestinal, o enterócito na digestão das proteínas. Acreditava-se que as proteínas
da dieta eram hidrolisadas rápida e completamente em seus aminoácidos pela ação das
proteases pancreáticas e gástricas. Então, a função principal do epitélio do intestino
delgado seria somente a da absorção dos aminoácidos e de seu transporte para o sangue
portal.
Atualmente sabe-se que as etapas finais da digestão dos peptídios ocorrem no intestino
delgado e estão associadas com os enterócitos. Eles são morfologicamente polarizados e
apresentam, no seu lado voltado para a luz intestinal, uma membrana diferenciada que
contém peptidases2 que participam da fase final da digestão protéica. Estas peptidases,
usualmente anfipáticas com o pólo interno hidrofóbico e o pólo externo hidrofílico, são
proteínas que integram a membrana, presas por sua porção hidrofóbica que fica
mergulhada entre as camadas carbônicas fosfolipídicas da membrana ou por ligações
covalentes ao glicosil-fosfatidilinositol 21. O local ativo está associado com a porção
hidrofílica da cadeia peptídica que se projeta na luz do intestino. O pólo externo é
constituído predominantemente por sete aminoácidos hidrofílicos, ou seja, histidina,
arginina, ácido aspártico, ácido glutâmico, serina e treonina. Estas enzimas são
sintetizadas no retículo endoplasmático rugoso, glicosiladas na membrana do aparelho
celular de Golgi e inseridas na membrana plasmática.
Outras enzimas da mucosa intestinal têm papel na hidrólise de peptídios. Foram
detectadas dipeptidases e aminotripeptidases no citoplasma dos enterócitos.
Pelo menos sete peptidases, que hidrolisam peptídios pequenos, foram identificadas no
enterócito 11,29. A glicil-L-leucina dipeptidase é uma das maiores enzimas
citoplasmáticas, com especificidade para dipeptídios contendo aminoácidos neutros. A
aminotripeptidase citoplasmática com especificidade para tripeptídios contendo prolina
na amina terminal. A dipeptidase da prolina (prolidase ou iminopeptidase)
citoplasmática com atividade sobre os peptídios contendo prolina ou sarcosina na
penúltima posição. A dipeptidil peptidase IV, presente na borda em escova, é capaz de
quebrar dois resíduos de aminoácidos da amina terminal de peptídios tem papel
importante na hidrólise de oligopeptídios contendo prolina. A prolil- carboxipeptidase,
ou carboxipeptidase P, ou dipeptidil-aminopeptidase IV, presente na borda em escova,
hidrolisa prolil- peptídios do terminal carboxílico. H também as endopeptidases
intrínsecas da borda em escova.
A aspartato aminopeptidase, a amino-oligopeptidase, a glicil-L-leucina dipeptidase da
membrana e a Asp-Lis peptidase zinco estável 29 hidrolisam os dipeptídios. Há,
também, a gama-glutamil transferase ou glutamil-aminopeptidase que hidrolisa
peptídios que contêm ácido glutâmico ou aspártico no N-terminal, a carboxipeptidase e
uma ou mais dipeptil aminopeptidases 29.
A peptidase mais abundante é, provavelmente, a aminopeptidase N, que remove
seqüencialmente os aminoácidos com terminal N dos oligopeptídios curtos. A
membrana celular contém, pelo menos, quatro peptidases com altas velocidades de
hidrólise dos peptídios no local da prolina 29. A dipeptidil-aminopeptidase IV e a
aminopeptidase P quebram na ligação da prolina terminal, enquanto a enzima
conversora da angiotensina (ou dipeptidil carboxipeptidase) e a carboxipeptidase P
hidrolisam prolina do terminal carboxílico dos peptídios 31.
As especificidades destas enzimas, acredita-se, devem ser complementares com as
proteases pancreáticas, as quais têm pouca ou nenhuma capacidade para hidrolisar
pontes de peptídios envolvendo a prolina.
A membrana celular na borda em escova contém muitas metaloendopeptidases neutras,
que quebram as pontes peptídicas interiores de proteínas e de polipeptídios
relativamente longos, mas são diferentes das endopeptidases pancreáticas discutidas
anteriormente 14. Estas enzimas podem iniciar a hidrólise de proteínas longas, tais
como alfa-caseína, fibrinogênio e histona, reduzindo-as a peptídios pequenos e
aminoácidos, mesmo sem a ação de proteases pancreáticas 14. Então, estas
endopeptidases intestinais devem ter importância nutricional nos pacientes com
insuficiência pancreática exócrina.
3.3.2. Absorção intestinal
O estômago constitui uma área do tubo gastrintestinal de absorção deficiente, porque
não contém o tipo de vilosidades da membrana absortiva, bem como as junções entre as
células epiteliais são muito estreitas. Apenas algumas substâncias altamente
lipossolúveis, como o álcool e a aspirina, podem ser absorvidas em pequenas
quantidades.
A absorção através da mucosa gastrintestinal dá-se pelo transporte ativo e por difusão.
O transporte ativo confere energia à substância, à medida que ela é transportada, com o
propósito de concentrá-la do outro lado da membrana ou de movê-la contra um
potencial elétrico. Por outro lado, o termo difusão significa simplesmente o transporte
de substâncias através da membrana em conseqüência do movimento molecular mais a
favor que contra um gradiente eletroquímico.
Como resultado da digestão intraluminal, é oferecida ao enterócito uma mistura de
oligopeptídios e aminoácidos livres. Durante muitos anos acreditou-se que a absorção
das proteínas se fizesse apenas sob a forma de aminoácidos. Atualmente admite-se que a
membrana celular clareia os produtos da digestão intraluminal através de dois
mecanismos: hidrólise na borda em escova dos oligopeptídios com conseqüente
transporte dos aminoácidos resultantes por diferentes sistemas transportadores de
aminoácidos; e translocação pela membrana de peptídios pequenos com subseqüente
hidrólise destes peptídios pelas peptidases citosólicas. O sistema de transporte de
peptídios opera para di e tripetídeos, enquanto grandes peptídios são quebrados
predominantemente na membrana da borda em escova com subseqüente translocação
dos aminoácidos livres por sistemas transportadores específicos mediados por
carregadores para aminoácidos neutros, básicos e ácidos 5,6,9.
3.3.2.1. Absorção dos aminoácidos
Os aminoácidos são absorvidos pelo intestino delgado tão rapidamente quanto são
liberados.
Pelo menos quatro sistemas diferentes transportam os aminoácidos. Um transporta os
aminoácidos neutros (aminoácidos monoamino monocarboxílicos), outro transporta os
aminoácidos básicos (aminoácidos dibásicos e cistina), um terceiro transporta os
aminoácidos ácidos (aminoácidos dicarboxílicos) e um quarto possui especificidade
para os aminoácidos L-prolina, hidroxiprolina, sarcolina, dimetil-glicina e betaína. Da
mesma forma, os mecanismos de transporte têm afinidade muito maior para o transporte
dos L-estereoisômeros dos aminoácidos, que para os D-estereoisômeros.
O transporte do aminoácido, da mesma forma que o transporte da glicose, só ocorre em
presença do transporte simultâneo de sódio. Além disso, os sistemas de transporte dos
aminoácidos, da mesma forma que aqueles para o transporte da glicose, encontram-se
na borda em escova da célula epitelial. Devido ao gradiente de sódio através da borda
em escova, a difusão do sódio para o interior puxa o carregador, bem como o
aminoácido a ele unido, para o interior, onde o aminoácido é aprisionado. Este processo
é descrito como uma “porta giratória”, que conduz o substrato da superfície da célula
para o compartimento citoplasmático, em mecanismo bastante específico 2. Por
conseguinte, as concentrações de aminoácidos aumentam no interior da célula e
difundem-se daí, através dos lados ou da base da célula, para os espaços intercelulares e
para a circulação porta.
1. Os aminoácidos neutros são levados por um sistema de transporte simples e
competem entre si pela absorção. Há alguma, mas não absoluta, especificidade óptica; a
D-metionina é transportada com cerca de um terço da velocidade da L-metionina, e altas
concentrações das formas D inibem a absorção das formas L. O ácido transportado deve
ter um grupo carboxila livre; ácidos esterificados ou reduzidos a um álcool não são
transportados. Deve haver um alfa-hidrogênio livre; nenhuma substituição é permitida.
A cadeia lateral deve ser neutra. Nas séries alifáticas, quase nada além de H (glicina) a
NH2-CO-CH2CH2CH2 (citrulina) é permitido, e nas séries aromáticas grupos
permitidos variam de fenil (fenilalanina) a imidazol (triptofano). A afinidade do
aminoácido (Kt) por seu carregador diminui com o aumento da polaridade da cadeia
lateral. O defeito neste sistema de transporte resulta na doença de Hartnup.
2. Os aminoácidos básicos são levados por um sistema separado numa velocidade de 5 a
10% da de transporte de ácidos neutros. Os aminoácidos transportados incluem a Larginina, a L-lisina, a DL-ornitina e a L-cistina. O defeito neste sistema de transporte de
aminoácidos resulta na cistinúria.
3. Um terceiro sistemas transporta L-prolina, hidroxiprolina, sarcolina, dimetil-glicina e
betaína. A prolina e a hidroxiprolina têm uma afinidade muito maior por este sistema
carregador do que pelo sistema neutro; portanto elas são sempre transportadas por ele
mais do que pelo sistema neutro, pois podem ser bloqueadas no carregador neutro por
outros aminoácidos neutros sempre presentes no intestino. Este sistema é sugerido por
estudos realizados na iminoglicinúria.
4. Os aminoácidos ácidos ou ácidos dicarboxílicos parecem ser transportados por um
outro sistema independente.
Quando existem defeitos nos mecanismos de transporte dos aminoácidos, eles são
absorvidos normalmente se forem oferecidos à mucosa intestinal na forma de
dipeptídios, através do transporte dos dipeptídios. Este fato deve ser aproveitado para a
terapêutica das doenças das deficiências do transporte dos aminoácidos, quando, então,
uma das propostas é oferecer dipeptídios.
Durante a absorção, os aminoácidos acumulam-se nas células mucosas, pois eles entram
na face mucosa mais rapidamente do que deixam o lado basal para entrar no sangue. A
entrada nas células é o passo mais rápido da absorção. Quando a absorção de um
aminoácido é competitivamente inibida por outro, sua concentração dentro das células
mucosas é mais baixa do que a que prevalece durante a absorção não inibida. Uma vez
dentro das células mucosas, a maioria dos aminoácidos não é extensivamente
metabolizada, com exceção dos ácidos glutâmico e aspártico. Uma fração destes dois
sofre transaminação com ácido pirúvico, através da qual a alanina é formada e liberada
dentro do sangue portal.
A acumulação intracelular dos aminoácidos neutros, como a acumulação de glicose e
galactose, requer gradiente de concentração de sódio da luz para a célula. O transporte
de aminoácidos pode ser explicado pela suposição de que o sódio e os aminoácidos
combinam-se com um só carregador na borda em escova das células e que a energia
para acumulação de aminoácidos é provida pelo gradiente de sódio. Os aminoácidos,
tendo sido acumulados em altas concentrações dentro das células, deixam-nas por
difusão passiva.
3.3.2.2. Absorção dos peptídeo
Existem duas hipóteses das formas de absorção dos dipeptídios: a) os aminoácidos são
absorvidos após a hidrólise dos dipeptídios e b) todo o dipeptídeo é transportado intacto
para o interior da célula e hidrolisado pelas peptidases citoplasmáticas. O mais provável
é que ocorram as duas formas de acordo com a afinidade dos dipeptídios com as
peptidases do enterócito. Também os tripetídeos são assimilados como os dipeptídios,
com vantagem cinética sobre a absorção dos aminoácidos.
O transporte dos di e tripetídeos através da membrana celular do enterócito é um
processo independente dos aminoácidos. O número de sistemas transportadores de
peptídios é desconhecido, mas existe pelo menos um que requer energia. O transporte
de peptídios tem grande significado nutricional porque os produtos da digestão incluem
grande quantidade de pequenos peptídios. Além disso, muitos aminoácidos são
absorvidos mais rápida e eficientemente na forma peptídica 27. Este sistema é de
especial importância nos defeitos do transporte de aminoácidos (doença de Hartnup e
cistinúria), nos quais a assimilação dos aminoácidos só ocorre na forma peptídica. Do
mesmo modo, na nutrição enteral, a mistura de peptídios é mais eficaz do que a de
aminoácidos 26.
Para suportar que os dipeptídios são transportados intactos, existem várias evidências.
Primeiro, a conhecida competição entre aminoácidos livres para a absorção está ausente
ou muito reduzida quando soluções de dipeptídios são apresentadas à mucosa intestinal
ao invés da correspondente mistura de aminoácidos. Segundo, no transporte de
dipeptídios observa-se que a velocidade de absorção de pelo menos um dos
aminoácidos é maior quando se oferece o dipeptídeo contendo o respectivo aminoácido
do quando se oferece uma solução de aminoácidos livres. Terceiro, a acidificação do
meio abole a hidrólise de dipeptídios pelas peptidases da borda em escova, mas reduz a
apenas 40% a absorção de dois peptídios contendo glicina.
A maioria dos autores defende que um único sistema mediado por um único
transportador é o responsável pela translocação de todos os dipeptídios não hidrolisados
através da membrana do enterócito.
Após o uptake intracelular, muitos dipeptídios e provavelmente todos os tripetídeos são
rapidamente hidrolisados em seus aminoácidos constituintes, pelas peptidases presentes
no citoplasma dos enterócitos. Elas incluem uma dipeptidase com especificidade para o
substrato, a prolina dipeptidase e uma aminotripeptidase. Somente dipeptídios muito
resistentes como glicil-prolina conseguem escapar à hidrólise intracelular, chegando ao
sistema porta na sua forma intacta. As hidrolases citoplasmáticas para peptídios não são
idênticas às da membrana da bordadura em escova. Elas diferem em suas propriedades
biológicas e químicas e em sua resposta à composição da proteína da dieta 5.
3.3.2.3. Absorção das proteínas in natura
Apesar da grande eficiência dos sistemas de hidrólise, que formam uma barreira
química na luz intestinal, algumas macromoléculas protéicas podem atravessar, na sua
forma intacta, a barreira mucosa sob condições fisiológicas normais. Este fenômeno,
que ocorre provavelmente por endocitose 19, não parece ter significado nutricional, mas
tem papel importante no desenvolvimento da reação imunológica à proteína alimentar
19,25.
As imunoglobulinas do colostro parecem ser absorvidas intactas pelo recém-nascido,
conferindo-lhe imunidade passiva, entretanto isto só ocorre nas primeiras 48 horas de
vida.
A absorção de quantidades muito pequenas de proteínas in natura é bem demonstrado
em bebês que nunca ingeriram clara de ovo. A injeção intradérmica de albumina de ovo
provoca a sensibilização. A ingestão subseqüente desta proteína, ocasiona uma placa
urticariforme no lugar da sensibilização, mostrando que ocorreu absorção de umas
poucas moléculas de albumina de ovo in natura. Em crianças normais alimentadas com
albumina de ovo purificada na quantidade de 1g por quilo, a dosagem
imunohistoquímica mostra 2 a 7 micro g/ml de albumina no sangue uma hora mais
tarde. Do mesmo modo, muitos adultos podem absorver quantidades imunologicamente
detectáveis de todas as proteínas, mas as quantidades absorvidas são negligenciáveis do
ponto de vista nutricional.
Normalmente as proteínas não são absorvidas in natura, mas algumas situações
patológicas dão uma maior permeabilidade da mucosa às proteínas íntegras, permitindo
sua absorção: desnutrição, diarréias de diferentes etiologias (rotavírus, infecções
bacterianas), alergias alimentares. As conseqüências da passagem das moléculas de
proteínas são o estímulo antigênico, com conseqüente alergia alimentar 19.
4. Fisiopatologia da digestão e absorção das proteínas
Muitas condições patológicas podem alterar o processo de assimilação das proteínas,
envolvendo digestão, absorção ou ambos, com conseqüências deletérias para a nutrição
4.
As deficiências de digestão mais comuns são causadas pela insuficiência pancreática
(pancreatite crônica, neoplasias pancreáticas, fibrose cística e ressecção pancreática).
Além destas existem defeitos raros genéticos que provocam diminuição da atividade
proteolítica.
Existem muitas doenças do intestino delgado que levam à destruição das células da
mucosa intestinal com diminuição da superfície absortiva (doença celíaca, sprue
tropical, etc), causando conseqüentemente malabsorção generalizada. Além disto, há
deficiências isoladas do transporte de aminoácidos que estão relacionadas aos
mecanismos fisiopatológicos da digestão-absorção das proteínas.
4.1. Alterações gástricas
As gastrectomias totais ou subtotais podem se seguir de perda de peso, desnutrição e
malabsorção de proteínas. Isto é devido, em parte, à redução de ingestão, à digestão
péptica reduzida pelo estômago residual e pela mistura inadequada dos alimentos com
as secreções digestivas 10. São encontradas concentrações baixas de tripsina no
intestino proximal de pacientes com gastrectomias BI ou BII durante a primeira hora
após a ingestão, talvez por esvaziamento gástrico acelerado. Há também assincronia
pancreática ou diminuição da estimulação duodenal pós-prandial, com níveis séricos
reduzidos de colecistocinina.
A deficiência primária de pepsinogênio ou de pepsina não provoca defeito da
assimilação de proteínas. Em vários casos de gastrite atrófica com ou sem acloridria não
há malabsorção de proteínas.
4.2. Alterações pancreáticas
O pâncreas tem reserva funcional muito grande, tanto que a creatorréia ocorre somente
quando há destruição de mais de 90% do tecido pancreático.
A fase pancreática é muito mais importante do que a gástrica para a absorção de
proteínas. Podem ocorrer deficiências simples ou generalizadas, mas, com exceção das
deficiências de tripsinogênio-tripsina e de enteroquínase, as deficiências simples não
são clinicamente significantes 10.
A insuficiência pancreática exócrina provoca deficiência generalizada das enzimas
pancreáticas. As doenças que reduzem a massa pancreática (fibrose cística, pancreatite
crônica, carcinoma pancreático, ressecção e desnutrição) e as obstruções das vias
pancreáticas podem causar malabsorção de todos os nutrientes.
O tratamento é feito com reposição de enzimas pancreáticas para manter a digestão
normal e conseqüentemente a absorção e o balanço nitrogenado positivo. O controle das
doses é feito tendo-se sempre a preocupação da avaliação da esteatorréia. A
suplementação com enzimas pancreáticas é mais eficiente em reduzir a azotorréia do
que a esteatorréia.
4.3. Deficiência de mistura das secreções com o alimento
As gastrectomias totais ou subtotais podem se seguir de perda de
peso, desnutrição e malabsorção dos nutrientes. Isto pode ser devido à mistura
inadequada dos alimentos com as secreções digestivas 10. A passagem rápida dos
alimentos para o intestino delgado leva à diminuição da estimulação duodenal pósprandial, com níveis séricos reduzidos de colecistocinina. Também há assincronia
pancreática, ocorrendo liberação de enzimas após a passagem do alimento pelo duodeno
30. Como conseqüência, não há digestão eficaz dos nutrientes.
Este mecanismo fisiopatológico pode ocorrer, também, na presença de fístulas gastroentéricas.
4.4. Alterações intestinais múltiplas não seletivas
As doenças que causam atrofia da mucosa intestinal causam malabsorção dos nutrientes
por vários mecanismos: diminuição do número dos enterócitos, diminuição da
superfície absortiva, diminuição da produção de hormônios tróficos para o pâncreas,
com conseqüente diminuição da digestão e da absorção. Nestas lesões da mucosa a
absorção de aminoácidos livres está reduzida, enquanto a absorção jejunal de
dipeptídios está menos afetada 22. Quanto mais grave a alteração da mucosa proximal
mais está diminuída a absorção de aminoácidos.
A doença celíaca é o exemplo clássico da atrofia da mucosa intestinal. Foi aventada a
hipótese de que o mecanismo da doença celíaca se deve à deficiência da peptidase
envolvida na hidrólise do glúten. Mas aceita-se que a redução desta enzima não parece
ser fator etiológico na patogênese desta doença, mas sim reflete o dano à mucosa. O
tratamento é a dieta isenta de glúten, que faz com que a mucosa retorne ao normal,
assim como a atividade da peptidase.
Neste grupo pode-se incluir o espru tropical, a desnutrição, a dermatite herpetiforme, a
enterite por irradiação, a doença imunoproliferativa do intestino delgado, a doença de
Whipple, a gastroenteropatia eosinofílica e também a ressecção intestinal.
As doenças que afetam outros órgãos, que não o intestino delgado, podem alterar
também a absorção dos aminoácidos. Pacientes com insuficiência pancreática e
insuficiência renal parecem ter diminuição da absorção de aminoácidos pelo intestino
27.
Algumas drogas podem induzir a defeitos na absorção: neomicina, biguanidos, ácido
etacrínico, medicamentos antineoplásicos, probenicida, fenilbutazona e salicilatos, e
seus mecanismos não estão totalmente esclarecidos.
4.5. Alterações únicas seletivas
Existem alterações seletivas de algumas enzimas proteolíticas e do transporte dos
aminoácidos que podem levar a várias doenças, as quais serão descritas a seguir e estão
resumidas no quadro 11.1.
Nunca foi descrito defeito genético de transporte de peptídios. Considerando a
importância do transporte dos peptídios na assimilação das proteínas, tais defeitos
seriam incompatíveis com a vida1.
4.5.1. Deficiência congênita de enteroquínase (doença de Hadorn)
A deficiência congênita de enteroquínase foi descrita por Hadorn et al em 1969 18. É
defeito genético autossômico recessivo e raro. Até 1983 haviam sido descritos oito
casos 15.
O quadro clínico é de desnutrição protéica precoce na infância, com retardo do
crescimento, diarréia, vômito, hipoproteinemia e edema desde o nascimento,
decorrentes da falta de digestão das proteínas. O quadro clínico se assemelha ao da
deficiência de tripsinogênio. A suspeita é feita quando, além da ingestão adequada de
proteínas, a dosagem de cloretos no suor é normal. O suco duodenal ou pancreático
mostra ausência de tripsina, mas presença de amilase e lipase.
O diagnóstico é feito quando a adição de enteroquínase ao suco duodenal leva ao
aumento das atividades da tripsina, da quimotripsina e da carboxipeptidase. Outras
distúrbios pancreáticos e da mucosa intestinal (por exemplo, doença celíaca) não
respondem a este teste, que é específico e deve ser realizado sempre que houver
suspeita.
A deficiência secundária de enteroquínase pode ocorrer por doença da mucosa intestinal
e no supercrescimento bacteriano do intestino delgado24, mas sua atividade não está tão
reduzida como as dissacaridases. Talvez seja porque a enteroquínase esteja presente
tanto nos enterócitos jovens quanto nos bem diferenciados.
Todos os pacientes respondem favoravelmente à reposição de enzimas pancreáticas, e a
desnutrição reverte com a administração de hidrolisado de proteínas por via oral.
4.5.2. Deficiência de tripsinogênio-tripsina (síndrome de Townes)
Descrita por Farber em 1943 e por Townes em 1965 apud 3. Há ausência de
tripsinogênio, precursor da tripsina, resultando a não ativação das outras enzimas
pancreáticas.
O quadro clínico é semelhante ao da deficiência de enteroquínase. Há desnutrição
protéica, com hipoproteinemia , edema e anemia, com início na infância. É associada a
atresia biliar.
O diagnóstico é feito quando a adição de enteroquínase ao suco duodenal não altera a
atividade de tripsina, mas a adição de tripsina ativa outras pró-enzimas pancreáticas, o
quimotripsinogênio e a procarboxipeptidase.
O tratamento é feito com extrato pancreático total.
4.5.3. Doença de Hartnup
É afecção autossômica recessiva descrita em 1956 na família Hartnup, que deu o nome
à doença. É a primeira doença genética que demonstrou um defeito isolado do
transporte intestinal de proteínas ou seus componentes.
Há anormalidade no transporte dos aminoácidos neutros (gli, ala, ser, tre, val, leu,
isoleu, tri, his, met) pelas células dos túbulos renais proximais e pela mucosa intestinal.
Este defeito abrange v rios aminoácidos, mas as conseqüências nutricionais são
discretas. Os pacientes têm peso abaixo do normal e quadro clínico de pelagra, devido à
deficiência de triptofano e, conseqüentemente de nicotinamida, pois esta é produto do
metabolismo do triptofano (60 mg deste forma 1 mg daquela). Apesar do transporte
deficiente de aminoácidos livres, há absorção dos dipeptídios contendo os aminoácidos
neutros, sendo este mecanismo alternativo responsável pela manutenção razoável da
nutrição. Com relação à alteração da síntese de nicotinamida, a explicação seria que as
proteases pancreáticas e as peptidases intestinais têm grande afinidade por hidrolisar as
ligações que envolvem o triptofano, de forma que a quantidade de peptídio contendo
triptofano é insuficiente para manter a síntese de nicotinamida6. O triptofano não
absorvido é degradado pelas bactérias e seus metabolitos (triptamina, indóis e aminas
tóxicas) são absorvidos, contribuindo para os sintomas neurológicos (ataxia) e
psiquiátricos da pelagra, que podem ser melhorados pela suplementação de
nicotinamida.
O quadro clínico é de peso baixo, ataxia cerebelar, distúrbios neuropsiquiátricos e
aminoacidúria de aminoácidos neutros 10.
Para o tratamento é necessária a suplementação com nicotinamida 28.
4.5.4. Cistinúria
É afecção autossômica recessiva cuja anormalidade básica é o sistema de transporte do
aminoácidos dibásicos (ornitina, arginina e lisina) e cistina pelos túbulos renais
proximais e pela mucosa intestinal. Quando fragmentos de biópsias jejunais de
pacientes com cistinúria são incubados com aminoácidos in vitro, eles não conseguem
absorver aminoácidos básicos, mas o fazem ativamente com relação aos aminoácidos
neutros. Os aminoácidos não absorvidos presentes no tubo digestivo sofrem ação das
bactérias intestinais, transformando-se em putrescina, cadaverina e outros produtos da
decarboxilação, os quais são absorvidos e eliminados na urina. O uso de antibióticos por
via oral diminui a quantidade destes produtos na urina. O transporte de cistina pelo
sistema de transporte de aminoácidos neutros não é bem entendido, mas a alteração do
transporte da cistina pode levar ao dano renal. O transporte pelas células epiteliais do
intestino e do rim mostram os mesmos defeitos genéticos do transporte de aminoácidos.
No rim, entretanto, o defeito para reabsorver a cistina filtrada resulta na aminoacidúria
e, por ser a cistina relativamente insolúvel na urina ácida, ela precipita nos túbulos
renais, com formação de cálculos de cistina no trato urinário e, conseqüentemente, dano
ao parênquima renal. Alguns autores 28 aventaram a hipótese de que a cisteína teria
importância na patogênese da cistinúria, mas nada foi confirmado.
O quadro clínico é de cálculos urinários recorrentes, pancreatite hereditária, distúrbio
cerebral e aminoacidúria de aminoácidos dibásicos e cistina 10. É a causa mais
freqüente de cálculos renais na idade pediátrica.
O transporte dos dipeptídios parece estar intacto como na doença de Hartnup, não
havendo em ambas as doenças desnutrição protéica aparente.
O diagnóstico é tradicionalmente feito pelo encontro de cristais hexagonais na urina e
pela homocistinúria.
O tratamento se baseia no aumento da solubilidade da cistina com aumento da ingestão
de líquidos, alcalinização da urina e uso de D-penicilamina, além da diminuição da
excreção da cistina com dieta com pouca metionina.
4.5.5. Malabsorção isolada de metionina
Esta rara afecção foi descrita em 1964 e caracteriza-se por diarréia osmótica associada a
retardo mental, convulsões, olhos azuis e cabelos brancos precoces. A anormalidade
fundamental é o defeito do transporte de metionina, sendo um efeito secundário
competitivo a diminuição da absorção de outros aminoácidos. A metionina não
absorvida é metabolizada pelas bactérias intestinais em ácido alfa-cetobutírico, ácido
alfa-aminobutírico e ácido alfa-hidroxibutírico, os quais são absorvidos pelo cólon e
excretados na urina. Este último ácido é o responsável pelo odor característico que
impregna os pacientes e suas secreções. A sobrecarga oral de metionina aumenta o
conteúdo fecal de metionina, serina e aminoácidos de cadeia ramificada.
O quadro clínico é de retardo mental, episódios de hiperpnéia e de diarréia e urina com
cheiro ácido 10.
O tratamento com dieta pobre em metionina melhora o estado geral e a diarréia, mas
não o retardo mental.
4.5.6. Síndrome da fralda azul (blue-diaper syndrome) - malabsorção de triptofano
É uma variante da doença de Hartnup, que se caracteriza pela malabsorção isolada de
triptofano. É doença familiar, rara com poucos casos relatados8,28, cuja característica é
a cor azul das fraldas, retardo de crescimento e hipercalcemia8,10.
O triptofano não absorvido aparece em grandes quantidades nas fezes. Uma fração é
metabolizada pelas bactérias intestinais, produzindo excessivamente metabólitos
indólicos, que são excretados como índican urinário. A oxidação deste último gera
indigotina, ou índigo azul, que dá cor azul à urina dos pacientes, manchando a fralda8.
A sobrecarga de triptofano aumenta a absorção intestinal de cálcio, sugerindo que a
absorção de cálcio se dá pela formação de complexo cálcio-triptofano. Há hipercalcemia
e nefrocalcinose secundária, com conseqüente insuficiência renal.
Acomete recém-nascidos, que têm dificuldade para ganhar peso, apresentam febres
recorrentes inexplicáveis, irritabilidade, constipação e coloração azul das fraldas. Estão
presentes hipercalcemia e nefrocalcinose secundários ao aumento da absorção de cálcio.
As fezes contêm quantidades aumentadas de indóis e triptofano, assim como triptamina
e ácidos indólicos. Os níveis plasmáticos de triptofano se mostram baixos após
sobrecarga deste aminoácido.
A dieta pobre em cálcio provoca melhora clínica e bioquímica, persistindo a
insuficiência renal.
4.5.7. Iminoglicinéria familial (síndrome de Joseph)
É anomalia genética autossômica recessiva, onde a anomalia é a malabsorção de
iminoácidos (prolina e hidroxiprolina) e glicina. Os heterozigotos podem ter defeitos
parciais. Não está claro se há dois defeitos diferentes no transporte dos aminoácidos, ou
se há dois defeitos genéticos diferentes28. É assintomática, porque estes aminoácidos
não são essenciais. É identificada através da cromatografia de aminoácidos da urina, que
mostra aumento da excreção renal destes aminoácidos.
4.5.8. Síndrome de Lowe
A síndrome óculo-cérebro-renal é defeito recessivo ligado ao cromossomo X, onde há
inibição generalizada do transporte dos aminoácidos lisina, arginina e glicina, tanto no
rim como no intestino.
As manifestações são retardo mental, catarata, glaucoma, hipotonia, doença renal,
acidose metabólica, proteinúria e raquitismo dependente de vitamina D. A
aminoacidúria dibásica é acompanhada de fosfatúria e acidose tubular renal.
4.5.9. Síndrome de malabsorção de lisina ou aminoacidúria hiperdibásica
É uma síndrome genética autossômica recessiva e ocorre por deficiência na absorção de
aminoácidos diamino-monocarboxílicos, especialmente a lisina23.
Após a sobrecarga com os aminoácidos citados (lisina, arginina, ornitina), os pacientes
mostram níveis mais baixos que o normal, sendo esta diferença mais marcada nos
homozigotos e moderada nos heterozigotos 23. Por outro lado, a sobrecarga com
citrulina provoca níveis semelhantes nos doentes e nos normais, demonstrando absorção
normal deste aminoácido. Os níveis baixos de ornitina podem ser prevenidos com
suplementação de proteínas com citrulina, que é precursora da ornitina 23.
O quadro laboratorial patognomônico é de aumento da excreção renal e diminuição das
concentrações plasmáticas de lisina, arginina e ornitina, além de hiperamonemia após a
ingestão de proteínas. O defeito foi previamente demonstrado nos rins e talvez esteja
presente nos hepatócitos.
Ao contrário do que ocorre com a doença de Hartnup e com a cistinúria, onde os
aminoácidos não são absorvidos quando oferecidos sob forma de dipeptídios, nesta
síndrome, o dipeptídio contendo lisina é apenas parcialmente absorvido. Parece que o
defeito na captação de lisina situa-se na membrana basolateral, que é sistema diferente
do transporte de aminoácidos através da membrana da borda em escova e sob diferente
código genético 6.
O quadro clínico é grave, com retardo do desenvolvimento físico e mental, vômitos,
diarréia, aversão a proteínas, hepatoesplenomegalia e osteoporose 10.
4.6. Perda intestinal de proteínas
O aumento na excreção de proteínas endógenas pode ocorrer em conseqüência a
algumas doenças gastrintestinais.
Diariamente, entre 10 e 30 g de proteína entram na luz intestinal através de secreções
enquanto as células descamadas contribuem com outras 25 g. Em adição, uma
quantidade variável de proteínas plasmáticas entra no trato digestivo. Alguns
investigadores estimam que 10% do catabolismo normal da albumina ocorre no
estômago e que 40% ocorre no intestino delgado, enquanto outros acreditam que não
mais do que 10% do catabolismo da proteína plasmática ocorre normalmente no trato
digestivo. Nas enteropatias perdedoras de proteínas, a velocidade da perda de proteína
pode ser maior do que aquela na qual a proteína pode ser sintetizada, resultando daí a
hipoalbuminemia. Nestes casos observa-se perda de até 50g de albumina para a luz do
tubo digestivo, em um dia.
A perda fecal de proteínas provoca a síndrome de gastroenteropatia exsudativa, cujo
quadro clínico é de hipoalbuminemia, desnutrição e aumento de proteína fecal. O
aumento da perda de proteínas endógenas pode ocorrer por 1) aumento da descamação
de células epiteliais, como na doença celíaca, 2) aumento da exsudação, como na
doença de Crohn, nos linfomas e outros tumores, 3) aumento da secreção mucípara,
como nas doenças inflamatórias (doença de Crohn, paracoccidioidomicose, tuberculose
intestinal), 4) obstrução linfática, como nas linfangiectasias intestinais tanto primária,
quanto secundária e 5) variados mecanismos, como a AIDS13.
O diagnóstico pode ser feito por testes que revelam a perda de proteína, tais como o
clareamento entérico de proteínas. Elas são marcadas com radioisótopos, dos quais a
albumina Cr51 é a mais usada no nosso meio, ou são proteínas que não sofrem hidrólise
no intestino, sendo, portanto, marcadoras da perda entérica, tais como a alfa-1antitripsina.
4.7. Supercrescimento bacteriano do intestino delgado
Nesta síndrome há perda evidente de proteínas, suficiente para causar desnutrição. Há
excreção fecal exagerada de nitrogênio, que parece estar relacionada ou à utilização das
proteínas e dos aminoácidos pelas bactérias para seu próprio metabolismo, competindo
com o hospedeiro30, ou à perda entérica de proteínas. As bactérias metabolizam os
aminoácidos em produtos não utilizáveis pelo organismo humano, por exemplo, elas
produzem enzimas que deaminam e decarboxilam os aminoácidos, formando amônia
com subseqüente utilização para a síntese da uréia. Elas provocam alteração do
enterócito, o que pode afetar o transporte de aminoácidos12 ou pode diminuir a
atividade da enteroquínase, interferindo com a digestão das proteínas24. Outras
produzem triptofanase que converte o triptofano em derivados indóis, que podem ser
absorvidos e metabolizados para índicans e subseqüentemente excretados.
O tratamento com antimicrobianos por via oral provoca diminuição da população
bacteriana, com conseqüente diminuição dos metabolitos do triptofano. Isto também é
observado na doença de Hartnup, o que salienta a importância da flora bacteriana neste
distúrbio. A perda entérica de proteínas ocorre em pacientes que apresentam inflamação
da mucosa.
O supercrescimento bacteriano do intestino delgado pode ocorrer por hipocloridria,
imunodeficiência, cujo exemplo atual é a AIDS13, fístulas entero-entéricas, estenoses,
alças cegas, perda anatômica ou funcional da válvula íleo-cecal, além de alterações
motoras do intestino delgado 30.
4.8. Fatores agravantes da malabsorção de proteínas
Alguns fatores agravam a malabsorção das proteínas. Verifica-se que alguns são
conseqüências da própria malabsorção das proteínas, levando então a um círculo vicioso
de mecanismos.
Alguns já foram comentados: a perda intestinal de proteínas é um deles e o
supercrescimento bacteriano do intestino delgado.
A desnutrição provoca diminuição da produção de enzimas, com conseqüente
diminuição da digestão e diminuição do número de enterócitos, com conseqüente
diminuição da absorção. O intestino delgado e o pâncreas são tecidos metabolicamente
ativos com síntese protéica elevada, sendo, conseqüentemente, muito sensíveis aos
efeitos da desnutrição. Observações experimentais e clínicas mostram hipoplasia e
hipofunção destes tecidos pela desnutrição. Algumas situações como o jejum, a falta de
ingestão de proteínas e as cirurgias de bypass para obesidade levam à diminuição da
absorção de aminoácidos, sem mudanças aparentes na morfologia intestinal. Entretanto,
a absorção de dipeptídios permanece normal, sugerindo uma maior resistência do
transporte de peptídios aos efeitos da desnutrição.
4.9. Considerações sobre a absorção/digestão das proteínas que devem ser conhecidas
quando se planeja tratamento dietético.
As proteínas são um nutriente importante a ser considerado nos planejamentos
dietéticos. Além de se calcular a quantidade delas, não se pode esquecer que sua
qualidade deve ser levada em conta para que a eficiência de sua absorção seja completa.
Tem-se questionado o uso dos aminoácidos como fonte de nitrogênio. Eles são
hiperosmóticos, de absorção mais difícil, mais caros e menos saborosos que os
peptídios. O transporte de peptídios é mais resistente às alterações fisiológicas e
nutricionais do que o transporte de aminoácidos. Em vista da vantagem relativa do
processo de transporte dos peptídios quando comparados aos aminoácidos em v rios
estados patológicos, o papel dos peptídios no suporte nutricional precoce é evidente. As
dietas à base de peptídios devem, portanto, ser usadas em pacientes com desnutrição
grave por deficiência de digestão e ou absorção 22,27.
Referências bibliográficas
1. Adibi SA. Protein assimilation. In: Berk EJ ed. Bockus - Gastroenterology.
Philadelphia, Saunders, 1985: 1530-7. 4th ed. Cap 94.
2. Campos JVM. Anatomia funcional. In: Dani R, Castro LP eds. Gastroenterologia
Clínica. 3a ed. Rio de Janeiro, Editora Guanabara, 1993: 674-704. cap 45.
3. Campos JVM. Defeitos pré-entéricos da absorção. In: Dani R, Castro LP eds.
Gastroenterologia Clínica. 3a ed. Rio de Janeiro, Editora Guanabara, 1993: 725-32.
4. Campos JVM. Mabsorção intestinal. In: Dani R, Castro LP eds. Gastroenterologia
Clínica. 3a ed. Rio de Janeiro, Editora Guanabara, 1993: 718-24. cap 47.
5. Caspary WF. Physiology and pathophysiology of intestinal absorption. Am J Clin
Nutr 1992, 55: 299s- 308s.
6. Castro LP, Campos JVM, Penna FJ, Mota JAC, Fagundes Neto U. Defeitos entéricos
da absorção. In: Dani R, Castro LP eds. Gastroenterologia Clínica. 3a ed. Rio de
Janeiro, Editora Guanabara, 1993: 733-57. cap 49.
7. Davenport HW. Digestão intestinal e absorção de proteínas. In:_______. Fisiologia
do Trato Digestivo. Rio de Janeiro, Editora Guanabara, 1978: 200-6. 3a ed. Cap 16.
8. Drummond KN, Michael AF, Ulstrom RA, Good RA. The blue diaper syndrome:
familial hypercalcemia with nephrocalcinosis and indicanuria. A new familial disease,
with definition of the metabolic abnormality. Am J Med 1964, 37: 928-48.
9. Erickson RH, Kim YS. Digestion and absorption of dietary protein. Annu Rev Med
1990, 41: 133-9.
10. Freeman HJ, Kim YS. Digestion and absorption of protein. Ann Rev Med 1978, 29:
99-116.
11. Freeman HJ, Sleisenger MH, Kim YS. Human protein digestion and absorption:
Normal mechanisms and protein-energy malnutrition. Clin Gastroenterol 1983, 12: 35778.
12. Giannella RA, Rout WR, Toskes PP. Jejunal brush border injury and impaired sugar
and aminoacid uptake in the blind loop syndrome. Gastroenterology 1974, 67: 965-74.
13. Guarino A, Tarallo L, Guandalini S et al. Impaired intestinal function in
symptomatic HIV infection. J Pediatr Gastroenterol Nutr 1991, 12: 453-8.
14. Guan D, Masahiro Y, Erickson RH et al. Protein digestion in human and rat small
intestine: role of new neutral endopeptidases. Gastrointest Liver Physiol 1988, 18:
G212-20.
15. Ghishan FK, Lee PC, Lebenthal E et al. Isolated congenital enterokinase deficiency:
recent findings and review of the literature. Gastroenterology 1983, 85: 727-31.
16. Guyton AC. Digestão e absorção no tubo gastrintestinal. In:______. Tratado de
Fisiologia Médica. Rio de Janeiro, Editora Guanabara Koogan, 1992: 638-646. 8a ed.
Cap. 65.
17. Guyton AC. Metabolismo das proteínas. In:______. Tratado de Fisiologia Médica.
Rio de Janeiro, Editora Guanabara Koogan, 1992: 657-61. 7a ed. Cap. 69.
18. Hadorn B, Tarlow MJ, Wolff OH. Intestinal enterokinase deficiency. Lancet 1969,
1: 812-3.
19. Heyman M, Desjeux JF. Significance of intestinal food protein transport. J Pediatr
Gastroenterol Nutr 1992, 15: 48-57.
20. King CE, Toskes PP. Protein-losing enteropathy in the human and experimental rat
blind-loop syndrome. Gastroenterology 1981, 80: 504-9.
21. Matsumoto H, Erickson RH, Kim YS. Characterization of a proline-specific
aminopeptidase anchored to the brush border membrane (BBM) of rat small intestine by
covalent attachment to glycosyl-phosphatidyl-inositol. Gastroenterology 1989, 96:
A328.
22. Nutzenadel W, Fahr K, Lutz P. Absorption of free and peptide-linked glycine and
phenylalanine in children with active celiac disease. Pediatr Res 1981, 15: 309-12.
23. Rajante J, Simell O, Perheentupa J. Intestinal absorption in lysinuric protein
intolerance: impaired for diamino acids, normal for citrulline. Gut 1980, 21: 519-24.
24. Rutgeerts L, Mainguet P, Tytgat G, Eggermont E. Enterokinase in contaminated
small-bowel syndrome. Digestion 1974, 10: 249-54.
25. Sategna-Guidetti C. Fisiopatologia delle sindromi da malassorbimento. Min
Gastroenterol Dietolol 1991, 37: 47-57.
26. Steinhardt HJ, Adibi SA. Kinectics and characteristics of absorption from an
equimolar mixture of 12 Glycil-dipeptides in human jejunum. Gastroenterology 1986,
90: 577-82.
27. Sterner G, Lindberg T, Denneberg T. Small intestinal absorption of glycine and
glycyl-glycine in patients with chronic renal failure. Acta Med Scand 1983, 213: 375-9.
28. Thier SO, Alpers DH. Disorders of intestinal transport of amino acids. Am J Dis
Child 1969, 117: 13-23.
29. Tobey N, Heizer W, Yeh R, Huang T-I, Hoffner C. Human intestinal brush border
peptidases. Gastroenterology 1985, 88:913-26.
30. Toskes PP. Malabsorption. In: Wyngaarden JB, Smith Jr LH eds. Cecil - Textbook
of Medicine. Philadelphia, Saunders, 1988: 732-45. Cap 104.
31. Yoshioka M, Erickson RH, Kim YS. Digestion and assimilation of prolin-containing
peptides by rat intestinal brush border membrane carboxypeptidases. Role of the
combined action of angiotensin-converting enzyme and carboxypeptidase P. J Clin
Invest 1988, 81: 1090-5.
Quadro 11.1. Distúrbios por alterações seletivas da absorção de proteínas.
Distúrbio
Defeito
Quadro clínico
Doença de Hadorn
d.enteroquinase
Desnutrição, retardo de crescimento
Síndrome de Townes
d.tripsinogênio/tripsina
Desnutrição, retardo de crescimento
Doença de Hartnup
d.t.aminoácidos neutros
Pelagra, ataxia cerebelar, alterações psiquiátricas
Cistinúria
d.t.cistina
Cálculos urinários, pancreatite hereditária, aminoacidúria
Malabsorção de metionina
d.t.metionina
Diarréia osmótica, convulsões, olhos azuis, cabelos brancos precoces, retardo mental,
urina ácida.
Síndrome da fralda azul
d.t.triptofano
Urina azul, hipercalcemia, retardo de crescimento, nefrocalcinose.
Síndrome de Joseph
d.t.iminoácidos
Aminoacidúria assintomática.
Síndrome de Lowe
d.t.lisina, arginina, glicina
Retardo mental, catarata, glaucoma, nefropatia.
Malabsorção de lisina
d.t.aminoácidos dibásicos
Retardo de desenvolvimento físico e mental, diarréia, vômitos, hepatoesplenomegalia,
raquitismo, osteoporose.
d.= deficiência t.= transporte