BIOLOGIA
MOLECULAR
Hipertrofia cardíaca e
treinamento físico
Aspectos moleculares
Autores:
Edilamar Menezes de Oliveira
Laboratório de Bioquímica, Departamento de Biodinâmica
do Movimento do Corpo Humano, Escola de Educação
Física da USP
José Eduardo Krieger*
Laboratório de Genética e Cardiologia Molecular
Departamento de Clínica Médica – FMUSP, Instituto do
Coração (InCor) HC-FMUSP
Resumo
A hipertrofia cardíaca constitui-se num dos principais mecanismos de adaptação do músculo cardíaco à sobrecarga de trabalho. Essa resposta, que é fundamental para o atleta atingir alto desempenho, é também um importante fator de risco de morbi-mortalidade em situações patológicas, como a hipertensão arterial. A compreensão dos mecanismos moleculares associados a resposta
adaptativa e mal-adaptativa é um dos principais desafios da atualidade. Adventos na área de genética-genômica estão abrindo perspectivas importantes para a realização dessa tarefa, que permitirá o
desenvolvimento de novas estratégias para diminuir o impacto da
resposta adversa da hipertrofia cardíaca.
A hipertrofia cardíaca constitui-se num dos principais mecanismos de adaptação do coração em face de uma sobrecarga de trabalho, de pressão ou de volume imposta ao coração em determinadas condições. Esse aumento da massa ocorre em decorrência de
alterações genéticas isoladas, como a cardiomiopatia hipertrófica,
em resposta a condições fisiopatológicas, tais como a hipertensão
arterial, infarto do miocárdio ou de hiperatividade simpática ou como
*Endereço para correspondência:
Laboratório de Genética e Cardiologia Molecular
Departamento de Clínica Médica – FMUSP,
Instituto do Coração (InCor) HC-FMUSP
Av. Dr. Enéas C. Aguiar, 44
05403-000 – São Paulo – SP
Tel.: (11) 3069-5579
E-mail: [email protected]
resposta fisiológica devido à sobrecarga de trabalho imposta pelo
treinamento físico dinâmico e estático realizado de forma crônica
por atletas.
A expressão “coração de atleta” tem sido amplamente empregada na literatura para caracterizar as adaptações que ocorrem no
sistema cardiovascular causadas pelo exercício físico de longa duração em atletas. Como revisto por Rost, um dos primeiros relatos dos
efeitos da atividade física sobre o coração foi feito por Bergmann
em 1884, ao notar que a relação peso do coração / peso corporal de
animais selvagens era muito maior quando comparada com os mesmos ao serem domesticados. Henschen, em 1899, foi quem primeiro
descreveu o coração de atleta através de técnica diagnóstica simples
de exame físico. O tamanho do coração foi determinado através de
percussão torácica cuidadosamente realizada nos esquiadores de campo antes e após uma corrida. Mais tarde, tais resultados foram confirmados pelo emprego da radiografia e por evidências de autópsia.
Posteriormente, o uso de técnicas como ecocardiografia e tomografia computadorizada veio facilitar os estudos com o atleta vivo. As
dimensões internas do coração e a espessura da parede puderam ser
determinadas com mais detalhes, tornando-se possível fazer estimativas da massa ventricular esquerda do coração, mostrando diferenças em relação a indivíduos sedentários. Portanto, o coração de atleta é um dos assuntos mais antigos e bastante estimulantes na pesquisa com exercício físico.
Tipos de hipertrofia
As hipertrofias cardíacas resultantes da adaptação do miocárdio a uma sobrecarga fisiológica ou patológica apresentam características fenotípicas e funcionais diferentes e podem ser classificadas, de modo geral, como concêntricas ou excêntricas1.
Em estados patológicos, dois tipos de sobrecarga crônica podem levar à hipertrofia cardíaca de maneiras diferentes. Uma sobrecarga de volume, como ocorre na insuficiência aórtica ou mitral,
leva a um aumento do diâmetro interno do ventrículo esquerdo e a
um aumento proporcional da espessura da parede. Esse tipo de adaptação é chamado de hipertrofia ventricular esquerda excêntrica. Uma
sobrecarga de pressão, como ocorre na estenose aórtica ou na hipertensão arterial, está associada a um espessamento da parede ventricular esquerda e a uma diminuição da dimensão interna, ou hipertrofia ventricular esquerda concêntrica. De modo geral, as hipertrofias patológicas apresentam-se de forma irregular ou assimétricas e
estão associadas a um maior índice de morbidade e mortalidade.
Em condições fisiológicas, como no exercício físico, dois tipos de sobrecarga intermitente podem levar à hipertrofia cardíaca
de maneiras diferentes, porém desenvolvidas de forma simétrica no
coração. No exercício estático ou isométrico (ex. levantadores, arre-
Volume 5 / Número 2/ 2002
Sem título-26
73
26/06/03, 14:57
73
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
messadores de peso e martelo, luta romana e fisiculturistas), a força
é desenvolvida com pouco ou nenhum movimento. Esse tipo de exercício apresenta como conseqüência hemodinâmica uma ligeira elevação do débito cardíaco (DC), resultante do aumento da freqüência
cardíaca (FC) e uma grande elevação da pressão arterial (PA), levando a uma sobrecarga de pressão no coração, que resulta em um espessamento da parede ventricular esquerda sem diminuição da dimensão interna da cavidade, desenvolvendo-se uma hipertrofia ventricular esquerda concêntrica. Têm sido demonstrados aumentos
como 480/350 (pressão sistólica e diastólica) em fisiculturistas durante a realização do exercício2. No exercício dinâmico, em que os
atletas realizam exercícios isotônicos (por exemplo, nadar, pedalar,
correr e andar) os principais padrões hemodinâmicos são um aumento na FC e no volume sistólico (VS), os dois componentes do
DC. Portanto, a carga sobre o coração é predominantemente
volumétrica, levando ao desenvolvimento de uma hipertrofia ventricular esquerda excêntrica. O exercício físico dinâmico, realizado de
forma crônica, mostra-se eficiente em proporcionar adaptações no
sistema cardiovascular3,4. Os principais parâmetros cardiovasculares
que sofrem adaptações com esse tipo de treinamento físico são a
freqüência cardíaca e a pressão arterial. Durante o repouso, observase uma queda de freqüência cardíaca após o treinamento físico, que
ocorre tanto em humanos5 como em animais6–8. A bradicardia de repouso tem sido utilizada como marcador dos efeitos do treinamento
físico aeróbio sobre o sistema cardiovascular.
Geralmente, a maioria dos tipos de exercícios ou programas de
treinamento físico consiste em uma associação entre exercício dinâmico e estático. Portanto, a hipertrofia fisiológica que ocorre normalmente é uma combinação de diferentes graus de ambas, hipertrofia concêntrica e excêntrica, levando a uma hipertrofia cardíaca
mista, como a observada em triatletas9. Além disso, o grau de hipertrofia fisiológica que ocorre está relacionado com a intensidade e
duração do exercício, assim como ao programa de treinamento físico, e está diretamente relacionado ao nível de “fitness” ou VO2
máx10,11. Schaible e Scheuer mostraram que também o aumento no
fluxo coronário é proporcional ao grau de hipertrofia induzido pelo
treinamento físico, resultante de um aumento do leito vascular coronário. Às vezes, a hipertrofia fisiológica desenvolvida pelos atletas
de força de alto nível assemelha-se à hipertrofia patológica, podendo ser incorretamente interpretada como patológica.
Controle e aspectos histológicos da
hipertrofia cardíaca
O mecanismo proposto para essas alterações estruturais é a hipertrofia induzida por carga direta. Alguns trabalhos têm demonstrado que a própria carga cardíaca é um fator suficiente e possivelmente a causa primária, responsável pela regulação do crescimento
no miocárdio do mamífero adulto12,13.
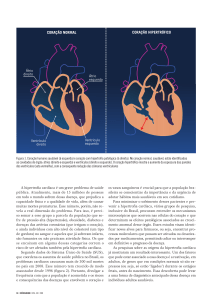
O crescimento induzido por carga parece ser mediado pela regulação da expressão gênica em resposta ao efeito físico direto do
estresse e esforço celular. Conforme ilustrado na figura 1, o padrão
de crescimento pode ser conceitualmente dividido em aumento no
número de miofibrilas por sarcômero, em série, levando a aumento
no comprimento dos miócitos (aumento no volume da câmara), e em
paralelo, levando a aumento da área tranversa do miócito (aumento
74
Sem título-26
na espessura da parede)14. Nesse modelo, o estímulo para o crescimento em série se deve ao aumento de pré-carga (estresse parietal
diastólico), enquanto o estímulo para o crescimento em paralelo se
deve ao aumento de pós-carga (estresse parietal sistólico). Portanto,
a partir desse ponto de vista conceitual, pode-se dizer que uma elevação no estresse parietal estimula o crescimento, até que o estresse
volte ao normal15,16. Os padrões de hipertrofia observados em atletas
encaixam-se nessas duas hipóteses. Atletas que experimentam grande elevação no DC sem grandes cargas de pressão (atletas de resistência) apresentam maior carga diastólica e manifestam maior volume ventricular com a razão massa/volume normal. Por outro lado,
atletas que experimentam uma acentuada elevação na PA com pequena ou nenhuma elevação no DC (levantadores de peso) mostram
um padrão de massa aumentada com pouca elevação no volume e a
razão massa/volume aumentada. A maioria dos atletas está entre esses dois extremos15,16.
Figura
1
FLUXO DE EVENTOS NOTADOS NA HIPERTROFIA CARDÍACA
EXCÊNTRICA E CONCÊNTRICA INDUZIDA POR CARGA
Hipertrofia patológica vs.
hipertrofia fisiológica
Resumidamente, poderíamos dizer que a hipertrofia cardíaca
pode ser decorrente de um processo patológico, como o aumento de
pós-carga secundário a hipertensão arterial ou a sobrecarga localizada pós-infarto do miocárdio, situação em que uma parte do músculo
saudável assume o trabalho do segmento que foi perdido devido a
morte celular ou processo reparativo. Essa resposta inicialmente é adequada, mas aos poucos evolui para a disfunção do órgão, com degeneração de miofibrilas, levando a insuficiência cardíaca17. Por outro lado,
o aumento de trabalho cardíaco pode estar associado a maior demanda fisiológica, como mostrado acima pelo treinamento físico dinâmico, representando uma resposta adaptativa fisiológica do organismo.
Analisando-se esses dois quadros e considerando-se que o treinamento físico aeróbio tem se mostrado eficiente em provocar adaptações
cardiovasculares benéficas em pacientes que desenvolvem hipertrofia cardíaca em decorrência de hipertensão arterial ou mesmo na insuficiência cardíaca, várias perguntas deverão ser respondidas em futuro próximo: qual a semelhança entre os mecanismos moleculares
associados à hipertrofia compensatória e à patológica? Trata-se de
vias comuns que num determinado momento divergem ou são vias
independentes desde o princípio? A resposta a essas perguntas pode
HIPERTENSÃO
74
26/06/03, 14:57
gerar novas oportunidades de intervenção, especialmente nos quadros
de hipertrofia patológica que estão associados a aumento substancial
de morbi-mortalidade na população.
O estímulo fisiológico induzido pelo exercício físico pode ser
um importante regulador da expressão gênica de proteínas estruturais do músculo cardíaco. Essa modulação do exercício físico sobre
a expressão de proteínas do coração pode ser processada por efeitos
diretos sobre o coração, a partir de sobrecarga de volume e/ou pressão, ou por efeitos indiretos, através de fatores hemodinâmicos ou
neuro-humorais. É importante salientar que diferentes sinais (mecânico, neuro-humoral etc.) são traduzidos no interior da célula como
alterações bioquímicas que levam à ativação de segundos mensageiros (citosólicos) e terceiros e quartos mensageiros (nucleares) que
irão agir no núcleo da célula, interagindo com o DNA, promovendo
a reprogramação da atividade celular.
Modificação de proteínas contráteis
Na hipertrofia patológica o aumento no volume dos miócitos
provocado pelo aumento do número de miofibrilas em paralelo é
acompanhado de aumento dos componentes do estroma, que geralmente apresentam-se de forma desproporcional à resposta dos miócitos1, principalmente aumentando o conteúdo de colágeno, podendo levar a deficiência no processo de relaxamento do miocárdio18.
Na hipertrofia fisiológica, o aumento de volume dos miócitos
se faz pela síntese de novos componentes, tais como aumento no conteúdo das proteínas contráteis e indução de suas isoformas, que levam
ao aumento predominante no comprimento das miofibrilas, não ocorrendo grandes alterações nas características do estroma, portanto sem
prejuízo funcional do órgão1,18. Em paralelo, ocorre aumento do retículo sarcoplasmático e do número e tamanho das mitocôndrias para
manter um estado funcional adequado ao aumento dos componentes
contráteis. Esse é um aspecto bastante evidenciado nas hipertrofias
fisiológicas. Além disso, são observadas alterações nas proporções
dos diferentes tipos de actina e miosina produzidos com o objetivo de
adequar a velocidade e a força de contração necessárias ao processo
de adaptação em face do estímulo que gerou a hipertrofia19,20.
O sarcômero, unidade contrátil do miocárdio, é formado por
proteínas que estão organizadas em filamentos grossos de miosina e
finos de actina. As principais proteínas que constituem o sarcômero
são a miosina de cadeia pesada (MCP), miosina de cadeia leve (MCL1
e MCL2), a tropomiosina, o complexo troponina (TnT, TnI e TnC) e
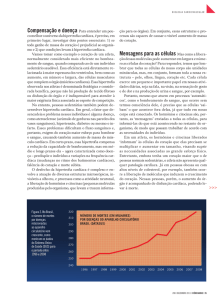
a actina. No ventrículo da maioria das espécies de mamíferos, inclusive do homem, foram identificadas três isoformas de miosina (V1,
V2 e V3). Essas três isoformas são constituídas de somente dois tipos de MCP, α e β. V1 e V3 são homodímeros α/α e β/β respectivamente, enquanto a V2 é um heterodímero α/β (figura 2). A α-MCP
apresenta uma maior atividade ATPásica e maior velocidade de encurtamento dos sarcômeros, enquanto a β-MCP apresenta menor
velocidade de encurtamento dos sarcômeros20–24.
As alterações hemodinâmicas que ocorrem após o nascimento
representam um estímulo para a regulação dessas isoformas. Durante a vida fetal, a grande maioria dos mamíferos expressa a β-MCP
no ventrículo. Nos mamíferos pequenos (rato e coelho) a α-MCP
aumenta de forma rápida imediatamente antes do parto e corresponde à isoforma dominante durante toda a vida adulta. Portanto, do
ponto de vista molecular ocorre uma “downregulation” da expressão do gene da β-MCP ventricular e uma “upregulation” do gene α-
MCP. Nos mamíferos maiores (cão, porco e homem), ao contrário, a
α-MCP é dominante somente transitoriamente após o nascimento,
sendo o gene da β-MCP expresso de forma dominante durante toda
a vida. Nos átrios a situação é diferente, uma vez que a isoforma α é
expressa de forma dominante durante toda a vida, em todos os mamíferos. Entretanto, a distribuição das isoformas da MCP pode ser
modificada em resposta a uma sobrecarga de trabalho, tanto em condições fisiológicas como em condições patológicas19,20,25. A α-actina
esquelética é encontrada no músculo esquelético de animais adultos,
enquanto na espécie humana ela está presente na fase fetal e no miocárdio submetido a hipertrofia patológica.
Uma atenção considerável tem sido dada aos efeitos do treinamento físico sobre a composição das proteínas contráteis. Por exemplo, ratos treinados com natação demonstraram um aumento na atividade da ATPase miosínica no ventrículo esquerdo, que foi secundário ao aumento da expressão da isoforma V1 (α-MCP), melhorando a função sistólica do animal treinado26. Nessa espécie, com predomínio da isoforma V1 e alta atividade da ATPase miosíca, a imposição de uma sobrecarga patológica como coarctação da aorta ou
hipertensão, resulta em uma rápida mudança (dois a três dias) no
padrão das isoenzimas para a isoforma V3 (β-MCP), que está associada com a diminuição da atividade da ATPase miosínica. Essas
alterações impostas por condições patológicas levam, na realidade,
a uma mudança fisiológica importante, que implica a expressão de
um fenótipo fetal, ou seja, uma reprogramação gênica. Interessante
ainda é o fato de que a hipertrofia cardíaca induzida pelo treinamento físico com natação reverte a isoforma da miosina de V3 para V1
em ratos espontaneamente hipertensos (SHR), levando a uma melhora da função sistólica, normalização da atividade da ATPase
miosínica e retorno para o fenótipo V1 predominante26. Nessa condição também foi observado um aumento na ligação e captação do
cálcio pelo retículo sarcoplasmático25,27. Em modelos de treinamento físico com esteira, em ratos, podem ocorrer alterações na função
contrátil do miocárdio sem que ocorram mudanças significativas na
atividade da ATPase miosínica, bem como na composição das isoformas da miosina ventricular28. Retornando às nossas questões levantadas anteriormente, vale lembrar que em animais experimentais
Figura
2
ISOMIOSINAS
Miosina de cadeia pesada (MCP): V1 e V3 são homodímeros
α e β respectivamente e V2 é um heterodímero α/β.
Modificado de Nadal-Guinard, 1988
Volume 5 / Número 2/ 2002
Sem título-26
75
26/06/03, 14:57
75
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
o treinamento físico com natação induz hipertrofia excêntrica, enquanto o treinamento físico com esteira não provoca hipertrofia cardíaca, ou quando ocorre é em pequena magnitude.
No homem e em animais maiores em que a freqüência cardíaca
de repouso é relativamente menor do que no rato e a isoforma predominante, no ventrículo, da MCP é exclusivamente a V3, o treinamento físico melhora a função ventricular sem modificar a atividade da
ATPase miosínica ou a composição das isoformas da miosina. Entretanto, em condições de sobrecarga patológica, o padrão de miosina
nos ventrículos não é modificado, uma vez que já predomina a isoforma
V3 da miosina, compreendendo aproximadamente 95% do total de
miosina expressa. O que se modifica na hipertrofia cardíaca patológica é a quantidade de proteína expressa, a isoforma V3 aumenta para
quase 100%21,29. Nos átrios, na estenose ou insuficiência valvular, ocorre aumento na expressão da isoforma V3, que passa a ser a forma
predominante, em detrimento da isoforma V1, que predomina em condições normais. Na hipertrofia cardíaca também ocorrem alterações
de outras proteínas sarcoméricas, como as isoformas da troponina T
cardíaca (TnTc). Normalmente são expressas duas isoformas da TnTc
no coração, enquanto em condições patológicas passam a ser expressas de quatro a seis isoformas da TnTc30,31.
Reprogramação gênica
Nas hipertrofias patológicas, alguns aspectos relacionados à
resposta hipertrófica são semelhantes às etapas em que ocorre o desenvolvimento do coração durante o período fetal ou perinatal, sendo, por isso, denominadas “reprogramação fetal”. São observados,
em animais experimentais, padrões de expressão gênica característicos do período fetal, como o reaparecimento de miosina de cadeia
pesada do tipo beta (β-MCP) e alfa actina esquelética (α-actina esquelética) no ventrículo32. Mudanças na composição das proteínas
contráteis do ventrículo determinam alterações na capacidade contrátil do miocárdio, podendo levar à diminuição na velocidade de
encurtamento dos sarcômeros, observada no miocárdio hipertrofiado,
aspectos que já mencionamos anteriormente. Além da indução de
proteínas contráteis, a hipertrofia cardíaca patológica é também caracterizada por alterações na expressão de genes de proteínas nãocontráteis, como Fator Natriurético Atrial (ANP), genes relacionados com o metabolismo energético (subunidade M da lactato desidrogenase e subunidade β da creatina quinase) e da enzima conversora da angiotensina I (ECA)24,29,32.
O gene do ANF é utilizado como um marcador de hipertrofia
cardíaca patológica. Durante o desenvolvimento embrionário, o gene
que codifica o ANF é expresso tanto no átrio como no ventrículo.
Logo após o nascimento, a expressão do ANF é diminuída no ventrículo, permanecendo o átrio como o sítio primário de síntese do ANF
no miocárdio adulto. Em resposta a uma variedade de estímulos (hormonal, sobrecarga de pressão e volume, hipertensão, genéticos etc.),
que induzem hipertrofia do ventrículo, ocorre a reexpressão do ANF
nas células ventriculares, o que representa a reprogramação de um
gene embriogênico na hipertrofia cardíaca. A expressão do gene do
ANF e de outros marcadores genéticos embrionários pode ser considerada um, entre vários critérios que podem distinguir estímulos que
são simplesmente tróficos (aceleram o crescimento normal) de estímulos que são fundamentalmente hipertróficos, indicando o começo de uma resposta patológica. Portanto, supõe-se que o ANF pode
participar da gênese das transformações fenotípicas observadas na
76
Sem título-26
hipertrofia, embora o significado fisiológico desse aumento de expressão do ANF no ventrículo ainda permaneça desconhecido17,32,33.
Trabalhos com treinamento físico com esteira mostram que não ocorre
aumento da expressão do ANF no ventrículo34. Embora, ainda seja
desconhecida, é de grande curiosidade a possibilidade de que atletas
de elite possam ter um aumento da expressão desse gene. Também é
difícil postular que o exercício poderia atenuar a re-expressão de
genes fetais em condições patológicas.
Uma estratégia que está sendo amplamente utilizada para identificar, no genoma, genes que possam interagir com o exercício é o
estudo de “genes candidatos”35. Essa estratégia está focada sobre genes envolvidos em vias metabólicas e sistemas fisiológicos que sabidamente interagem com determinadas características de interesse, que
estão relacionadas ao exercício. Através dessa estratégia – estudos de
associação de variantes de um ou múltiplos genes –, foi identificado
um limitado número de genes que parecem influenciar fenótipos relacionados com exercício, um desses genes é o gene da ECA.
A ECA é um dos componentes do sistema renina-angiotensina
(SRA) e sua expressão tem sido mostrada aumentada no miocárdio,
promovendo uma maior conversão de Ang I em Ang II na microcirculação intramiocárdica, secundária a uma sobrecarga pressórica36. A
Ang II, ao ligar-se aos seus receptores AT1 nos miócitos, resultaria
num aumento da contratilidade e da resposta hipertrófica, enquanto
em fibroblastos seria desencadeada uma resposta de hiperplasia associada a um fenótipo secretor de colágeno37–39. Tais efeitos podem ser
amplificados pela capacidade de a Ang II estimular a secreção de catecolaminas das terminações nervosas, levando a potenciação da resposta inotrópica e hipertrófica40. O aumento da expressão da ECA
cardíaca parece responder a múltiplos estímulos envolvendo fatores
físicos (por exemplo, aumento da tensão superficial da parede ventricular) e neuro-endócrinos (estimulação simpática) levando a um aumento da Ang II tecidual e, subseqüentemente, a remodelagem cardíaca. Tem sido mostrado que a função endócrina do coração desempenha um papel importante no desenvolvimento da hipertrofia cardíaca3. Entretanto, a participação do SRA na hipertrofia fisiológica
induzida pelo exercício físico não parece muito clara na literatura.
O gene da ECA está sendo um dos genes mais estudados, utilizando-se a estratégia de genes candidatos. O polimorfismo do gene
da ECA humana consiste na presença (inserção, alelo I) ou ausência
(deleção, alelo D) de um fragmento de 287 pares de bases41. O alelo
D foi associado a uma maior concentração da ECA circulante e no
miocárdio. Inicialmente os autores hipotetizaram que, se o SRA cardíaco é um regulador importante da hipertrofia do ventrículo esquerdo, portanto o alelo D está associado com maior massa do VE.
Estes achados ainda são muito debatidos, visto que outros estudos
não encontraram associação. É importante salientar que a hipertrofia de VE é um fenótipo complexo influenciado por um grande número de fatores ambientais e biológicos, como, por exemplo, exercício, idade, sexo e pressão sangüínea42.
Recentes trabalhos publicados43,44 demonstraram que o genótipo do gene da ECA está relacionado com a resposta ao exercício
físico. Esses autores mostraram que indivíduos com genótipo II ou
DI apresentam melhor performace ao treinamento físico aeróbio ou
“endurance”43. Ainda, a presença do genótipo II confere uma eficiência mecânica aumentada no músculo esquelético humano44. Outros estudos têm mostrado que o genótipo DD da ECA está relacionado com a hipertrofia do VE induzida pelo exercício45,46.
HIPERTENSÃO
76
26/06/03, 14:57
Processo de excitação-contração
Fatores neurais
Além das modificações nas proteínas contráteis em resposta à
sobrecarga fisiológica ou patológica, a expressão de genes envolvidos no processo de excitação-contração (por exemplo, SERCA, fosfolamba, calseqüestrina e genes do receptor de rianodina) também
está alterada na hipertrofia cardíaca patológica. O gene da SERCA,
através de “splicing” alternativo, produz duas isoformas: SERCA 2a
e SERCA 2b. A SERCA 2a codifica para a Ca2+-ATPase do retículo
sarcoplasmático (RS) da célula cardíaca. Tal enzima é responsável
pelo transporte do Ca2+ citosólico para o RS, resultando em menor
concentração de Ca2+ no citoplasma, levando ao relaxamento da fibra. A expressão da SERCA 2a está diminuída na insuficiência cardíaca avançada, assim como no indivíduo idoso, levando a disfunção ventricular diastólica. A atividade física atenua a diminuição da
expressão do gene da SERCA 2a no miocárdio de camundongos idosos47 e aumenta a expressão de genes mitocondriais, tais como citocromo oxidase no coração de ratos senescentes, podendo portanto,
aumentar as reservas energéticas e a função diastólica do miocárdio.
Portanto, o exercício físico pode modificar a expressão de genes cardíacos alterados, exercendo efeitos benéficos em condições patológicas. A fosforilação do fosfolamba pela proteína quinase dependente de AMPc (PKA) ativa a Ca2+-ATPase do RS. Dessa forma, o
aumento nos níveis de fosfolamba está diretamente relacionado com
o estado contrátil do miocárdio. Em estudos com camundongos
“knockout” (deleção do gene) para o gene do fosfolamba foi observado um estado hipercontrátil do miocárdio, enquanto camundongos transgênicos que superexpressam essa proteína apresentam estado hipocontrátil48,49. No estágio final da insuficiência cardíaca em
humanos, que é caracterizado por hipertrofia e dilatação do miocárdio, ocorre uma diminuição nos níveis de expressão da SERCA 2a,
do fosfolamba e dos receptores de rianodina (Rya), enquanto os níveis de expressão das calseqüestrinas permanecem inalterados. O
mecanismo pelo qual a despolarização do túbulo T ativa o retículo
sarcoplasmático ocorre via receptores de diidropiridina (DHP), que
são canais de Ca2+ voltagem-dependentes localizados na membrana
do túbulo T; funcionam como sensor de voltagem e desencadeiam a
liberação de Ca2+ do RS vizinho. Esse aumento de Ca2+ produz a
abertura dos canais de cálcio rianodina-dependentes (canais do tipo
L) da membrana do RS, fenômeno conhecido como “liberação de
Ca2+-induzida por Ca2+”50. No miocárdio, essa parece ser a forma
mais rápida de liberação do Ca2+ armazenado no RS. Recentemente,
pela observação de que na hipertrofia e insuficiência cardíaca em
ratos ocorre um acoplamento prejudicado entre os receptores de DHP
e diidropiridina, surgiu um novo conceito: “distúrbio sistólico da homeostasia do cálcio”51,52. Nesse modelo, o Ca2+ citosólico é removido para a RS pela SERCA, a qual é modulada pelo fosfolamba, e
para o espaço extracelular pelo trocador Na+/Ca2+. Na insuficiência
cardíaca, o número de transportadores e a freqüência de transporte
do DHP e Rya permanecem inalterados, uma vez que o limiar de
ativação do Rya é aumentado. A recaptação de cálcio para o RS pela
SERCA está reduzida, o que é compensado, em parte, pelo aumento
na expressão trocador52. É interessante salientar a especificidade de
resposta que diferentes estímulos podem produzir; por exemplo, o
treinamento físico com natação provoca aumento na captação de
cálcio pelo SR, enquanto o treinamento físico com esteira não produz adaptações na função intrínseca e no número de canais de Ca2+
tipo L (Rya)53.
A hipertrofia cardíaca também tem sido demonstrada através
de fatores neurais. São inúmeras as demonstrações de que estímulos
do sistema simpático ou a estimulação dos receptores adrenérgicos
pelos neurotransmissores naturais ou por agonistas sintéticos, como
o isoproterenol54, induzem hipertrofia cardíaca. Tanto a estimulação
dos receptores α1 como a dos receptores β1-adrenérgicos podem
induzir hipertrofia cardíaca, portanto envolvendo a geração dos sinais do sarcolema que ativam os mensageiros intracelulares que irão
regular a atividade gênica necessária ao crescimento da célula55. Um
significativo número de trabalhos tem se preocupado em investigar
se o treinamento físico altera essas vias de sinalização intracelular,
porém não existe consenso sobre seus efeitos56. Uma característica
clara demonstrada na literatura é que o “drive” simpático e os níveis
de catecolaminas circulantes estão diminuídos no indivíduo treinado quando comparado com o sedentário56–58, porém no coração isso
não é muito claro. Os níveis de catecolaminas no miocárdio têm sido
demonstrados inalterados ou aumentados em diferentes programas
de treinamento físico6. A resposta inotrópica do coração as catecolaminas encontram-se aumentadas59,60, diminuídas61 e inalteradas62 com
o treinamento físico. O número de receptores β-adrenérgicos no ventrículo e a atividade da adenilato ciclase permaneceram também inalterados63–65 ou foram diminuídos59,66 pelo treinamento aeróbio crônico, e uma similar falta de clareza existe em relação aos efeitos do
exercício físico sobre as proteínas G, que estão integralmente envolvidas no processo de acoplamento do receptor adrenérgico com o
processo de produção dos segundos mensageiros intracelulares56,63,66.
E poucos são os estudos relacionados com os efeitos do treinamento
físico sobre os receptores α-adrenérgicos do coração. Portanto, é
difícil concluir como o treinamento físico interfere nas vias de sinalização adrenérgica no coração.
Fatores vasculares
Além dos efeitos sobre a expressão de genes inerentes à fibra
cardíaca, o exercício físico pode regular também alguns fatores vasculares. Em vasos coronários, o exercício físico modula a expressão
do óxido nítrico (NO). O NO é um fator de relaxamento derivado do
endotélio (EDRF), que desempenha um importante papel vasodilatador na circulação coronária67. Durante o exercício físico, a síntese
de óxido nítrico é aumentada para níveis superiores àqueles verificados em condições basais. Esse efeito do exercício acaba, a longo
prazo, aumentando cronicamente o fluxo coronário e potencializando
a vasodilatação durante a administração de acetilcolina67.
Em síntese, a hipertrofia cardíaca é controlada de maneira complexa e multifatorial, incluindo diversos fatores físico-hemodinâmicos, neuro-humorais e genéticos. O papel desses fatores na gênese
da hipertrofia cardíaca em resposta a estímulos específicos (fisiológicos ou patológicos) permanece um desafio. Adventos na área da
genômica, que permitem a análise simultânea dos padrões de expressão de milhares de genes e proteínas em situações biológicas
complexas, vem facilitando essa tarefa. Assim, novos genes/proteínas candidatos estão sendo identificados e caracterizados com
abordagens experimentais e clínicas. O melhor conhecimento
desse processo deve gerar novas oportunidades de intervenção
terapêutica para a redução da morbi-mortalidade que acompanha a
hipertrofia cardíaca.
Volume 5 / Número 2/ 2002
Sem título-26
77
26/06/03, 14:57
77
○
○
○
○
○
○
○
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
Referências bibliográficas
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
32.
33.
34.
35.
36.
78
Sem título-26
WEBER KT, BRILLA CG et al. Myocardial remodeling and pathological hypertrophy.
Hospital Practice, v. 26, p. 73–80, 1991.
MACDOUGALL JD, TUXEN D et al. Arterial blood pressure response to heavy
resistance exercise. J Appl Physiol, v. 58, p. 785–790, 1985.
NISHIYASU T, NAGASHIMA K et al. Human cardiovascular and humoral responses
to moderate muscle activation during dynamic exercise. J Applied Physiol, v. 88, p.
300–307, 2000.
RUSSELL B, MOTLAGH D et al. Form follows function: how muscle shape is regulated
by work. J Applied Physiol, v. 88, p. 1652–1657, 2000.
KATONA PG, MCLEAN M et al. Sympathetic and parasympathetic cardiac control in
athletes and nonathletes at rest. J Appl Physiol, v. 52, p. 1652–1657, 1982.
GEENEN D, BUTTRICK P et al. Cardiovascular and hormonal responses to swimming
and running in the rat. J Appl Physiol, v. 65, p. 116–123, 1988.
NEGRÃO CE, MOREIRA ED et al. Vagal function impairment after exercise training.
J Appl Physiol, v. 72(5), p. 1759–1753, 1992.
NEGRÃO CE, RONDON MUPB. Exercício físico, hipertensão e controle barorreflexo
da pressão arterial. Rev Bras Hipertens, v. 8, p. 89–95, 2001.
CLAESSENS C, CLAESSENS P et al. Structural heart adaptations in triathletes. Acta
Cardiol, v. 54(6), p. 317–325, 1999.
BLOMQVIST CG, SALTIN B. Cardiovascular adaptations to physical training. Ann
Rev Physiol, v. 45, p. 169–189, 1983.
MILLIKEN MC, STRAY-GUNDERSON J et al. Left ventricular mass as determined
by magnetic resonance imaging in male endurance athletes. Am J Cardiol, v. 62, p.
301–305, 1988.
COOPER G, KENT RL et al. Load induction of cardiac hypertrophy. J Mol Cell Cardiol, v. 21 (suppl. 5), p. 11–17, 1989.
ANVERSA P, CAPASSO JM. Loss of intermediate-sized coronary arteries and capillary
proliferation after ventricular failure in rats. Am J Physiol, v. 260, p. H1552–1559,
1991.
GROSSMAN W, JONES D et al. Wall stress and patterns of hypertrophy in the human
left ventricle. J Clin Invest, v. 56, p. 56–64, 1975.
COLAN SD. Mechanics of left ventricular systolic and diastolic function in physiological
hypertrophy of the athlete heart. Cardiol Clin, v. 10, p. 227–240, 1992.
COLAN SD. The adult athlete with congenital heart disease. In: The athlete and heart
disease: diagnosis, evaluation & management. Williams RA (ed.). Philadelphia;
Lippincott Williams & Wilkins, 1999.
KRIEGER JE. Bases moleculares da hipertrofia cardíaca. J Bras Nefrol, v. 16, p. 198–
202, 1994.
MILL JG, VASSALLO DV. Hipertrofia cardíaca. Rev Bras Hipertens, v. 8, p. 63–75,
2001.
PEREIRA FEL. Hipertrofia cardíaca: aspectos morfológicos e patológicos. In: Contratilidade miocárdica. Aspectos básicos e clínicos. Editado por Vassallo, DV, Lima
EG (eds.). São Paulo: Fundo Editorial BYK, 1993.
ZARCO P. Bases moleculares de la cardiología clínica. Madrid: Editorial Médica Panamericana SA, 1996.
SWYNGHEDAUW B. Development and functional adaption of contractile proteins in
cardiac and skeletal muscles. Physiol Rev, v. 66, p. 710–771, 1986.
PETTE D. Training effects on the contractile apparatus. Acta Physiol Scand, v. 162, p.
367–376, 1998.
MARIAN AJ, ROBERTS R. The molecular biology of cardiac abnormalities in athletes.
In: The athlete and heart disease: diagnosis, evaluation & management. Williams
RA (ed.). Philadelphia; Lippincott Williams & Wilkins, 1999.
FRANCHINI KG. Hipertrofia cardíaca: mecanismos moleculares. Rev Bras Hipertens,
v. 8, p. 125–142, 2001.
MOORE R, KORZICK DH. Cellular adaptations of the myocardium to chronic exercise.
Progress in Cardiovasc Diseases, v. 6, p. 371–396, 1995.
SCHAIBLE T, MALHORTA A et al. Chronic swimming reverses cardiac dysfunction and
myosin abnormalities in hypertensive rats. J Appl Physiol, v. 60, p. 1435–1441, 1986.
SCHAIBLE T, MALHORTA A et al. Combined effects of hypertension and chronic
running program on rat heart. J Appl Physiol, v. 63, p. 322–327, 1987.
TIBBITS GF, BARNARD RJ et al. Influence of exercise on excitation-contraction
coupling in rat myocardium. Am J Physiol, v. 240, p. H472–H480, 1981.
SWYNGHEDAUW B. Molecular mechanisms of myocardial remodeling. Physiol Rev,
v. 79, p. 215–262, 1999.
ANDERSON PAW, GREIG A et al. Molecular basis of human cardiac troponin T
isoforms expressed in the developing, adult, and failing heart. Circ Res, v. 76, p.
681–686, 1995.
MESNARD L, LOGEART D et al. Human cardiac troponin T: cloning and expression
of new isoforms in the normal and failing heart. Circ Res, v. 76, p. 687–692, 1995.
CHIEN KR, KNOWLTON KU et al. Regulation of cardiac gene expression during
myocardial growth and hypertrophy: molecular studies of an adaptive physiologic
response. FASEB J, v. 5, p. 3037–3046, 1991.
IZUMO S, NADAL-GINARD B et al. Protooncogene induction and reprogramming
of cardiac gene expression produced by pressure overload. Proc Natl Acad Sci USA,
v. 85, p. 339–343, 1988.
AZIZI C, BOUISSOU P et al. Alterations in atrial natriuretic peptide gene expression
during endurance training in rats. Eur J Endocrinol, v. 133, p. 361–365, 1995.
BRAY MS. Genomics, genes, and environmental interaction: the role of exercise. J
Applied Physiology, v. 88, p. 788–792, 2000.
SCHUNKERT H, DZAU VJ et al. Increased rat cardiac angiotensin converting enzyme
activity and mRNA expression in pressure overload left ventricular hypertrophy:
Effects on coronary resistance, contractility, and relaxation. J Clin Invest, v. 86, p.
1913–1920, 1990.
37.
38.
39.
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
58.
59.
60.
61.
62.
63.
64.
65.
66.
67.
68.
69.
70.
BAKER KM, ACETO JF. Angiotensin II stimulation of protein synthesis and cell growth
in chick heart cells. Am J Physiol, v. 259, p. H610–H618, 1990.
SADOSHIMA J, IZUMO S. Molecular characterization of angiotensin II-induced
hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts: critical
role of the AT1 receptor subtype. Circ Res, v. 73, p. 413–423, 1993.
CRABOS M, ROTH M et al. Characterization of angiotensin II receptors in cultured
adult rat cardiac fibroblasts. J Clin Invest, v. 93, p. 2372–2378, 1994.
XIANG J, LINZ W et al. Ganten D, Lang RE, Scholkens B, Unger T. Effects of
converting enzyme inhibitors: ramipril and enalapril on peptide action and
sympathetic neurotransmission in isolated rat heart. Eur J Pharmacol, v. 113, p.
215–223, 1984.
RIGAT B, HUBERT C et al. An insertion/deletion polymorphism of the angiotensin Iconverting enzyme gene accounting for half the variance of serum enzyme levels.
J Clin Invest, v. 86, p. 1343–1346, 1990.
MONTGOMERY HE. Should the contribution of ACE gene polymorphism to left ventricular hypertrophy be reconsidered? Heart, v. 77, p. 489–490, 1997.
MONTGOMERY HE, MARSHALL R et al. Human gene for physical performance.
Nature, v. 393, p. 221–222, 1998.
WILLIAMS AG, RAYSON MP et al. The ACE gene and muscle performance. Nature,
v. 403, p. 614, 2000.
SCHUNCKERT H, HENSE HW et al. Association between a deletion polymorphism
of the angiotensin-converting-enzyme gene and left ventricular hypertrophy. New
Eng J Med, v. 330, p. 1634–1638, 1994.
MONTGOMERY HE, CLARCKSON P et al. Association of angiotensin-converting
enzyme geneI/D polymorphism with change in left ventricle mass in response to
physical training. Circulation, v. 96, p. 741–747, 1997.
TATE CA, HELGASON T et al. SERCA 2a and mitochondrial cytochrome oxidase
expression are increased in hearts of exercise-trained old rats. Am J Physiol, v. 271,
p. H68–H72, 1996.
HOIT BD, KHOURY SF et al. In vivo echocardiographic detection of enhanced left
ventricular function in gene-targeted mice with phospholamban deficiency. Circ
Res, v. 77, p. 632–637, 1995.
KADAMBI VJ, PONNIAH S et al. Cardiac-specific overexpression of phospholamban
alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice.
J Clin Invest, v. 97, p. 533–539, 1996.
FABIATO A. Simulated calcium current can both cause calcium loading in the trigger
calcium release from the sarcoplasmic reticulum of a skinned canine cardiac purking
cell. J Gen Physiol, v. 85(2): 291–320, 1985.
GÓMEZ AM, VALDIVIA HH et al. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science, v. 276, p. 800–806, 1997.
MITTMANN C, ESCHENHAGEN T et al. Cellular and molecular aspects of contractile
dysfunction in the failure. Cardiovascular Research, v. 39, p. 267–275, 1998.
MOKELKE EA, PALMER BM et al. Endurance training does not affect intrinsic
calcium current characteristics in rat myocardium. Am J Physiol, v. 273(42), p.
H1193–H1197, 1997.
OLIVEIRA EM, KOIKE MK et al. Isoproterenol induced hypertrophy increases left
ventricle ACE activity and ACE promoter expression in vivo. J Hypertens, v. 14
(suppl 1), p. S211, 1996.
ROCKMAN HR, KOCH WJ et al. Cardiac function in genetically engeneered mice with
altered adrenergic receptor signaling. Am J Physiol, v. 272, p. H1553–H1559, 1997.
MOORE R. Cellular adaptations of the heart muscle to exercise training. Ann Med, v.
30 (suppl 1), p. 46–53, 1998.
PERONNET F, CLEROUX J et al. Plasma norepinephrine response to exercise before
and after training in humans. J Appl Physiol, v. 51, p. 812–815, 1981.
SEALS DR, VICTOR RG. Regulation of muscle sympathetic nerve activity during
exercise in humans. Exerc Sports Sci Rev, v. 19, p. 313–349, 1991.
TAKEDA N, DOMINIAK P et al. The influence of endurance training on mechanical
catecholamine responsiveness, β-adrenoceptor density and myosin isozyme pattern
of rat ventricular myocardium. Basic Res Cardiol, v. 80, p. 88–99, 1985.
SPINA RJ, OGAWA T et al. Exercise training improves left ventricular contractile
response to β-adrenergic agonist. J Appl Physiol, v. 72, p. 307–311, 1992.
EKBLOM B, KILBOM A et al. Sympathectomy and pharmacological blockade in
trained rats. Acta Physiol Scand, v. 89, p. 283–285, 1973.
NUTTER DO, PRIEST RE et al. Endurance training in the rat. Myocardial mechanics
and biochemistry. J Appl Physiol, v. 51, p. 934–940, 1981.
HAMMOND HK, RANSAS LA et al. Noncoordinate regulation of cardiac Gs protein
and β-adrenergic receptors by a physiological stimulus, chronic dynamic exercise.
J Clin Invest, v. 82, p. 2168–2171, 1988.
MOORE RL, RIEDY M et al. Effect of training on β-adrenergic receptor number in rat
heart. J Appl Physiol, v. 52, p. 1133–1137, 1982.
BÖHM M, DORNER H et al. Effects of exercise on myocardial adenylate cyclase and
Gia expression in senescence. Am J Physiol, v. 264, p. H805–H814, 1993.
WERLE EO, STROBEL G et al. Decrease in rat cardiac beta1- and beta2-adrenoceptors
by training and endurance exercise. Life Sci, v. 46, p. 9–17, 1990.
SHEN W, ZHANG X et al. Nitric oxide production and NO synthase gene expression
contribute to vascular regulation during exercise. Med Sci Sports Exerc, v. 27, p.
1125–1134, 1995.
NATELSON BH, ZHOU Z et al. Effect of acute exhausting exercise on cytokine gene
expression in men. Int J Sports Med, v. 17, p. 299–302, 1996.
NEGRÃO CE, IRIGOYEN MC et al. Effect of exercise training on RSNA, baroreflex
control, and blood pressure responsiveness. Am J Physiol, v. 265(34), p. R365–
R370, 1993.
SCHAIBLE T, SCHEUER J. Cardiac function in hypertrophied hearts from chronically
exercised female rats. J Appl Physiol, v. 50, p. 1140–1145, 1981.
HIPERTENSÃO
78
26/06/03, 14:57