1
Modelagem Molecular e Docking de Proteína - Ligante
Bruno Cesar S. Novaes1 e Luis Paulo Barbour Scott1
1
UFABC - Universidade Federal do ABC
Centro de Matemática Computação e Cognição
Santo André, São Paulo, Brasil
[email protected]; [email protected]
Este trabalho apresenta uma análise da modelagem molecular, mais especificamente do “folding” de proteínas através do programa
Chimera, auxiliado por bibliotecas de rotameros, embora o maior enfoque seja o “docking” de proteínas através do programa
ArgusLab4.0, por diferentes métodos de cálculo de energia, sendo um deles os algoritmos genéticos. Diversas simulações foram
executadas, por diferentes mecanismos seguindo um protocolo próprio para se efetuar analises comparativas dentre os resultados
obtidos e então encontrar uma melhor maneira de testar diferentes ligantes em suas proteínas alvo.
Palavras-Chave: Modelagem Molecular, “Folding” e “Docking” de Proteínas, Algoritmos Genéticos, Biblioteca de Rotâmeros.
I.INTRODUÇÃO
A
Síndrome da Imunodeficiência Adquirida (SIDA),
comumente conhecida como AIDS (sigla do termo
Acquired Immunodeficiency Syndrome), é provocada pelo
Virus da Imunodeficiencia Humana (VHI, ou HIV do inglês
Human Immunodeficiency Virus), seu agente etiológico
possuidor de grande caráter mutante, caracterizado
primeiramente pelo Dr. Luc Montagnier. [1] A ausência da
cura ou imunização profilática para a AIDS até os dias atuais
(embora já tenham sido desenvolvidas drogas antivirais que
administradas de forma terapêutica, atuam na inibição de
proteases, alem de fármacos atuantes na transcriptase reversa),
torna a doença um alvo do estudo de novas técnicas na
concepção de novas drogas e uma possível cura. Uma área
crescente é a predição computacional de estruturas protéicas e
ate mesmo a descoberta de novos fármacos que inibam a ação
das enzimas do vírus, especificamente a HIV Protease (HIV
PR) responsável pela maturação das proteínas de um novo
vírus e que servira de base para este estudo.
As proteínas são as moléculas orgânicas mais abundantes e
importantes das células, logo são encontradas por todas as
células realizando tarefas fundamentais, seja na estrutura, seja
nas funções celulares.A função de uma proteína conhecida
esta diretamente relacionada à estrutura espacial nativa. As
forças responsáveis pelo seu enovelamento podem estar na
seqüência de aminoácidos que compõe a sua cadeia e nas suas
interações com o meio. São estas forças que, “in vivo”, tornam
a busca por esta conformação não aleatória e muito mais
espontânea (Paradoxo de Levinthal), dado o grande numero de
conformações possíveis em uma proteína.
Conhecer a estrutura tridimensional de uma proteína passa a
ser um grande interesse para biólogos, químicos e até mesmo
cientistas da computação, que passam a desenvolver modelos
e paradigmas para tal estudo. Surge então a Modelagem
Molecular (MM), que compreende um número de ferramentas
e métodos computacionais e teóricos que tem como objetivo
entender e prever o comportamento de sistemas reais; usando
para descrever e prever estruturas moleculares, propriedades
do estado de transição e equilíbrio de reações, propriedades
termodinâmicas, entre outras.
II.ALGORITMOS GENÉTICOS
Uma das áreas técnicas da Inteligencia Artificial, inspirada
na Teoria da Evolução Natural e na Genética, é conhecida
como Computação Evolutiva, ou seja, trata-se de sistemas
para resolução de problemas que utilizam modelos
computacionais baseados na teoria da evolução natural e
propõe o agrupamento dos sistemas de Computação Evolutiva
em três grandes categorias, sendo uma destas os GA's
(Algoritmos Genéticos).
Algoritmos genéticos (GA´s) é um ramo dos algoritmos
evolucionários e são técnicas heurísticas de otimização global
(ou seja, nem sempre representam a melhor solução, mas
tendem a ficar bem próximo dela), onde as populações de
indivíduos são criadas e submetidas a operadores genéticos:
seleção, recombinação (crossover) e mutação. Ou seja,
combinam a sobrevivência entre os melhores com uma forma
de troca de informações genéticas entre os melhores
indivíduos para formar estruturas heurísticas melhores. [2]
Os G.A.´s fazem uso de uma terminologia própria, cuja
apresentação se faz necessaria para que termos futuros seriam
devidamente entendidos. Assim, pode-se dizer que um
indivíduo, de uma população num programa, é visto como um
CROMOSSOMO, cujos GENES são os termos característicos
deste (por exemplo, características inteiras, flutuantes,
caractere, etc.). Como pode ser visto na Figura 1.
Figura 1: Representação binária de um cromossomo.
2
A codificação da informação em cromossomos é de vital
importância dentro de um G.A., e é junto com a função
avaliação o que une o algoritmo genético ao problema a ser
resolvido. A representação cromossomial mais comum de um
G.A. é a BINÁRIA (como visto na figura 1), isto é, um
cromossomo nada mais é que uma seqüência de bits (e cada
gene somente um bit), embora existam hoje melhores
representações, tais como números reais, permutações de
elementos e listas de regras, dependendo do problema. [2]
A seleção deve ser feita de tal forma que os indivíduos mais
aptos sejam selecionados com maior freqüência que aqueles
menos aptos, garantindo assim que somente as características
necessárias predominem na nova população. A função de
avaliação é outra parte fundamental em um algoritmo
genético, pois é a maneira utilizada para determinar a
qualidade de um individuo como solução do problema em
questão, calculando um valor numérico que reflete quão bons
são os parâmetros representados nos cromossomos para
resolver o problema. Na verdade é a ligação verdadeira do
programa com o problema real. [3]
Dois principais operadores num G.A responsáveis pela
diversificação dos indivíduos no decorrer das gerações são os
operadores de CROSSOVER (ou cruzamento entre
cromossomos) e de MUTAÇAO (mimese ao processo de
mutação molecular, em nucleotídeos), vistos na figura 2.
maximizar sua extensibilidade e prepará-lo para os avanços de
“hardware”. O resultado destas modificações é um programa
de visualização, analise molecular e dados relacionados,
incluindo mapas de densidade, montagens supramoleculares,
alinhamento de seqüências, trajetórias e montagens
conformacionais, e uma das imagens relacionadas a HIV
Protease, enzima fundamental para o vírus HIV, pode ser vista
na figura 3. [5]
Figura 3: Representação em Chimera da HIV Protease,
complexada com o fármaco DMP-323.
Mas o objetivo principal ao se trabalhar com o Chimera é a
possibilidade da manipulação de rotâmeros, podendo
visualizar novas conformações e analisar uma proteína ainda
mais profundamente. O Chimera associado á uma biblioteca
de rotâmeros poderá mostrar quais os ângulos mais prováveis
para um determinado resíduo com base, por sua vez, nos
arquivos PDB´s existentes. Assim, ao selecionar um resíduo,
desejando saber seus ângulos mais prováveis, é possível
visualizá-los como um todo na cadeia, e analisar se com um
determinado ângulo não há uma incompatibilidade física, ou
se um ângulo menos provável não venha a se tornar
energeticamente mais favorável no enovelamento de uma
proteína. Figuras 4 e 5.
Figura 2: Representação dos operadores de “crossover” e
mutação.
III.CHIMERA E BIBLIOTECA DE ROTÂMEROS
No estudo da predição de conformações protéicas ou em
seu desenvolvimento, um dos empecilhos para a construção de
modelos computacionais válidos reside na flexibilidade das
cadeias laterais cujo empacotamento exerce um dos principais
papeis no enovelamento das proteínas. A partir da
determinação das estruturas cristalográficas de algumas
proteínas em meados do século XX, as conformações das
cadeias laterais passam a ser um grande alvo de estudos. E
com o aumento do número de estruturas para uma mesma
proteína, começou a ser possível definir-se a conformação
mais comum por meio de analises estatísticas, resultando nas
bibliotecas de rotâmeros (sendo rotâmero a abreviação de
isômero rotacional – “rotational isomer”). [4]
UCSF Chimera, mais conhecido simplesmente como
Chimera, é um programa desenvolvido para o uso interativo
de visualização de estruturas moleculares. É altamente
extensível, ou seja, permite que seus usuários possam
desenvolver novas ferramentas que estendam sua capacidade,
e na sua ultima versao foi completamente redesenhado para
Figura 4: Em verde, um dos aminoácidos da cadeia da HIV
Protease que possui a maior probabilidade segundo a
biblioteca de rotameros; em branco e vermelho estão outros
rotameros menos prováveis.
Figura 5: HIV Protease indicando a posição do aminoácido
em sua conformação mais provável.
3
O estudo de “docking” de proteínas, no qual se testa a
afinidade de um “ligante” a um determinado substrato ou sítio
de ligação, também se fez necessário para um completo
entendimento computacional nesta área. O “software”
utilizado foi o ArgusLab4.0, distribuído gratuitamente pela
Planaria Software. Rapidamente se tornou o pacote acadêmico
preferido em modelagem molecular, principalmente devido a
sua interface amigável e seus menus intuitivos para cálculos.
Com ele é possível realizar desde tarefas simples, como
desenho de moléculas ate os cálculos de energia mais
avançados. [6]
O objetivo principal ao utilizar o ArgusLab 4.0 nesta
pesquisa, reside em uma de suas recentes atualizações, que
adicionou a funcionalidade de “docking” a esta plataforma
através do pacote ArgusDock e também GADock, dois
métodos diferentes de busca. Neste caso, faz-se necessário que
um arquivo PDB, que contem os dados obtidos por difração de
raio-X da proteína, seja importado para o programa, cada
arquivo PDB com os quais foram feitos os estudos possuem a
HIV Protease complexada com um fármaco, e para este estudo
foram utilizados três diferentes arquivos PDB: arquivo
1BVE.pdb (fármaco DMP323), arquivo 1HSG.pdb
(L-735,524) e arquivo 1HXW.pdb (ABT-538).
Dadas as ferramentas apresentadas pelo programa ArgusLab
4.0 e a possibilidade do “docking”, optou-se explorá-lo de
forma a estabelecer os parâmetros que melhor se adaptassem
ao “docking” das proteínas citadas anteriormente. Então,
levou-se em consideração a presença e ausência de água
durante o “docking”, átomos de hidrogênio sobressalentes,
tamanho da caixa de “docking” e, o mais importante, as duas
diferentes formas de “docking” disponíveis: ArgusDock e
GADock; e as simulações seguiram a ordem determinada pelo
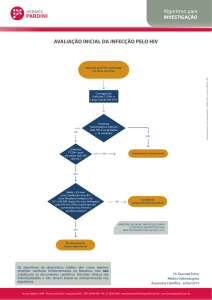
fluxograma da figura 6
Como foram feitas diversas simulações, com parâmetros
diferentes, optou-se por analisá-los um a um, seguindo uma
ordem inversa à qual os experimentos foram realizados. Para
que fique mais claro, seguiu-se comparando e analisando caixa
de “docking” (um dos últimos parâmetros a serem mudados),
posteriormente foi feita a analise do desempenho entre
simulações padrões e avançadas, “docking” rígido ou flexível,
com ou sem o hidrogênio, na presença ou ausência de água e
por último e mais importante o desempenho das ferramentas
de “docking”: ArgusDock e GADock. Embora na realidade as
simulações se cruzem, como pode ser observado na figura 7,
na qual é possível visualizar os dados da simulação mais
importantes, ou seja, as simulações relacionadas ao
mecanismo de “docking”.
"Docking" Flexível do Fármaco L-735,524
RMSD (Ǻ)
IV.Docking de fármacos utilizando ArgusLab 4.0
2,5
2
1,5
1
0,5
0
Parâm etro
s
Figura 7: Gráfico mostrando a convergência dos valores do
RMSD ao longo das simulações.
IV.CONCLUSÕES
O Chimera se mostrou um “software” eficaz na predição de
estruturas associadas à manipulação de arquivos PDB de
forma fácil e também a utilização de bibliotecas de rotâmeros
enriquecendo as possibilidades de sucesso em uma simulação.
O ArgusLab4.0, por sua vez, mostrou-se um software de
grande importância tanto na predição de estruturas, quanto na
utilização da ferramenta de “docking”, que se mostra melhor
quando maior é a complexidade da simulação. Os dois
mecanismos apresentados pelo “software”, ArgusDock e
GADock, são de maneira geral muito precisos, embora o
GADock apresente uma acurácia ainda mais notável,
conseqüência de sua ferramenta de busca, os Algoritmos
Genéticos, o que auxilia, de certa forma, o entendimento e
estudo desta ferramenta de uma forma prática.
V.REFERÊNCIAS BIBLIOGRÁGICAS
[1]
[2]
[3]
[4]
[5]
[6]
Figura 6 – Fluxograma da ordem das simulações.
Silva, Alan. Estudo por Modelagem e Dinâmica Molecular da
Protease de variantes do Vírus da Imunodeficiência Humana Tipo
1 resistentes a drogas antivirais, Dissertação de Doutorado,
UFRJ, Rio de Janeiro, 2003;
Linden, R. Algoritmos Genéticos - Uma importante ferramenta da
Inteligência Computacional.
Rezende, S. O. Sistemas Inteligente - Fundamentos e Aplicações.
Editora Manole, Barueri – SP. 2003.
Dunbrack, Roland L. Jr. Rotamer Libraries in the 21st Century;
http://www.cgl.ucsf.edu/chimera/, acessado em 17/02/09;
Joy, Saju et al. - Detailed comparison of the protein-ligand docking
efficiencies of GOLD, a commercial package and ArgusLab, a
licensable freeware, artigo científico.