METABOLISMO DE PROTEÍNAS
Hidrólise das proteínas da dieta e absorção dos aminoácidos livres
Tripsina
Quimiotripsina
Carboxipeptidase
Aminopeptidase
As proteínas ingeridas na dieta são hidrolisadas por proteases digestivas (pepsina, tripsina, quimotripsina, etc.) até aminoácidos.
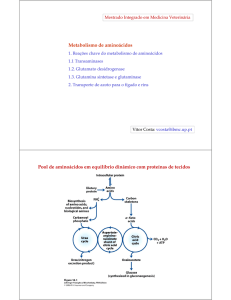
Pool de aminoácidos em equilíbrio dinâmico com proteínas de tecidos
Proteínas
da dieta
Síntese de:
~ 30 g / dia
purinas
pirimidinas
neurotransmissores
glucose
Pool de
amino ácidos
Ureia (urina)
~ 300 g / dia
Proteínas
de tecidos
Aminoácidos
1
Essenciais
Não essenciais
arginina1
fenilalanina
histidina
isoleucina
leucina
lisina
metionina
treonina
triptofano
valina
alanina
aspartato
asparagina
cisteína
glutamato
glutamina
glicina
prolina
serina
tirosina
apenas durante períodos de crescimento rápido ou doença
Biossíntese de aminoácidos não essenciais
Os esqueletos carbonados
vêem de intermediários
da glicólise, da via das
pentose-­fosfato
ou
do
ciclo de Krebs
Os aminoácidos glutamato e glutamina são sintetizados por assimilação de NH4+
D-­cetoglutarato
glutamato
desidrogenase
NH4+ + NADPH
H2O + NADP+
glutamato
glutamina
sintetase
NH4+ + ATP
ADP + Pi
glutamina
O glutamato é sintetizado a partir de NH4+ e D-­cetoglutarato pela enzima
glutamato desidrogenase
A glutamina é sintetizada a partir de NH4+ e glutamato pela enzima glutamina
sintetase
Os aminoácidos alanina e aspartato são sintetizados por reacções de
transaminação ± transferência de um grupo amina de um aminoácido para um
cetoácido
D-­cetoglutarato
glutamato
piruvato
D-­cetoglutarato
glutamato
oxaloacetato
alanina
alanina transaminase
aspartato
aspartato transaminase
A asparagina é formada por aminação do aspartato
Aspartato + NH4+ + ATP Asparagina + AMP + PPi + H+
A tirosina é sintetizada
fenilalanina hidroxilase
por
hidroxilação
da
fenilalanina
pela
Fenilalanina + O2 + NADPH Tirosina + NADP+ + H2O NOTA: A tirosina é um a.a. essencial em pessoas com carência em fenilalanina hidroxilase
(fenilcetonúria) acumulação de fenilactato e fenilpiruvato e deficiência de tirosina
Degradação de aminoácidos
Os aminoácidos que excedem as necessidades da síntese proteica não
podem ser armazenadas (ao contrário dos ácidos gordos e glicose) e
não são excretados
O esqueleto carbónico é convertido em intermediários metabólicos.
O grupo amina é utilizado para síntese de aminas biológicas e o
excesso excretado sob a forma de ureia (em mamíferos).
CATABOLISMO DE AMINOÁCIDOS
1) TRANSAMINAÇÃO
2) DESAMINAÇÃO OXIDATIVA
1) TRANSAMINAÇÃO
Reacção de transferência de um grupo amina de um aminoácido para um
cetoácido catalisada por transaminases ou aminotransferases
Transaminases:
Existem em elevada quantidade no músculo e no fígado
actividade facilmente quantificada interesse clínico: concentração sérica proporcional ao grau de lesão do fígado (muito rico em transaminases)
usadas para síntese / degradação de aminoácidos
durante a degradação de aminoácidos, os grupos amino são transferidos para o D-­cetoglutarato
Transaminação de aminoácidos
PLP = piridoxal fosfato (derivado da vitamina B6): necessário para a actividade da enzima
Exs:
alanina transaminase
alanina + D-­cetoglutarato
piruvato + glutamato
aspartato transaminase
aspartato + D-­cetoglutarato
oxaloacetato + glutamato
Alanina transaminase ou alanina aminotransferase ± catalisa a transferência do grupo amina da alanina para o D-­cetoglutarato
Aspartato transaminase ou aspartato aminotransferase ± catalisa a transferência do grupo amina do aspartato para o D-­cetoglutarato
2) DESAMINAÇÃO OXIDATIVA DO GLUTAMATO
CICLO DA UREIA
Local: mitocôndria
Níveis energéticos baixos Glu desidrogenase fornece D-­KG para ciclo de Krebs
Transaminação Desaminação
oxidativa
(mamíferos)
Pássaros ± grupo amina excretado sob a forma de ácido úrico
Peixes ± Grupo amina excretado sob a forma de ião amónio
CICLO DA UREIA
HEPATÓCITOS
2 reacções na mitocôndria
3 reacções no citosol
Um átomo de N vem do ião amónio
O outro átomo de N vem do aspartato
Dieta rica em proteínas
Jejum prolongado
n UREIA
Transporte de azoto inter-órgãos a
seguir a proteólise muscular:
A ureia é excretada pelos rins. A
glutamina é transportada para o
rim e aí por acção da glutaminase
origina glutamato e amónia. O
glutamato pode ser utilizado para
gluconeogénese.
RIM
Provoca tremores
Afecta fala/visão
Provoca danos cerebrais irreversíveis
Amónia é extremamente tóxica
Toxicidade da amónia ± cérebro e SNC
NADH
D-­cetoglutarato
Ï NH4+
NAD+
glutamato
GDH
Ï NH4+
Ð D-­cetoglutarato
Ð ciclo de Krebs
Ð NADH/NAD+
Ð ATP
Ï glutamina
glutamina
danos (mec. desconhecido)
Ðglutamato (Ð GABA -­ neurotransmissor)
ÏNH4+ -­ Alteração do pH
Ð ATP
DEFEITOS ENZIMÁTICOS DO CICLO DA UREIA CAUSAM
HIPERAMONÉMIA E PODEM PROVOCAR LESÕES CEREBRAIS
Interrupção do ciclo da ureia provoca sintomas graves como atraso mental, coma e morte prematura
Deficiência na enzima N-­acetilglutamato sintetase I também provoca os mesmos sintomas (p N-­acetilglutamato o activador da carbamoil fosfato sintetase)
É verificada hiperamonémia e hiperaminoacidémia. O tratamento consiste numa dieta pobre em proteínas (mas contendo os aa essenciais) e suplementos do substrato em falta
Deficiência em ornitina transcarbamoilase
(deficiência mais comum, acumulação de ácido orótico)
NH4+
HCO3-­
carbamoil fosfato
ácido orótico
pirimidinas
ornitina
citrulina
Correlação de todos os defeitos enzimáticos do ciclo
da ureia
¾A amónia acumula-­se no sangue
¾Intoxicação por amónia
¾Danos cerebrais, coma, morte: falta de ATP
¾Defeitos neurológicos: no cérebro a amónia
combina-­se com o D-­cetoglutarato, o que leva à
queda dos níveis de oxaloacetato e à paragem do
ciclo de Krebs resultando em danos celulares
irreversíveis
¾O glutamato formado combina-­se com a amónia
levando à formação de glutamina e ao esgotamento
do glutamato necessário à formação de GABA
(neurotransmissor)
Tratamento de doenças genéticas apresentando
deficiências em enzimas do ciclo da ureia
Limitar a ingestão de proteínas
Dieta pobre em proteínas: Substituir com D-­
cetoácidos que são transaminados in vivo,
refeições ligeiras frequentes
Remover o excesso de amónia
Utilização de antibióticos que actuam contra
bactérias produtoras de amónia , administração
de benzoato de sódio
Reposição dos intermediários do ciclo em
falta
Suplemento de arginina ou citrulina
OS ESQUELETOS CARBONADOS DOS 20 AMINOÁCIDOS SÃO DESVIADOS PARA 7 MOLÉCULAS:
aminoácidos puramente cetogénicos: leucina, lisina
aminoácidos cetogénicos e gluconeogénicos: isoleucina, fenilalanina, triptofano, tirosina
aminoácidos puramente gluconeogénicos: restantes 14 aminoácidos
CICLO DA ALANINA
FFA
Metabolismo de amino ácidos: estado alimentado
excesso de
aminoácidos
ATP
glicogénio
ácidos gordos
Metabolismo de amino ácidos: jejum
cortisol
+
mobilização de
amino ácidos
dos músculos
OS AMINOÁCIDOS SÃO PRECURSORES DE MUITAS BIOMOLÉCULAS
(Para além de péptidos e proteínas, são precursores de biomoléculas
como purinas, pirimidinas, esfingosina, histamina (vasodilatador),
epinefrina
(hormona),
melanina,
serotonina
(neurotransmissor),
acetilcolina (neurotransmissor), GABA (neurotransmissor), NAD+...
EXS.
glutamato
J-­aminobutirato (GABA)
CO2
triptofano
serotonina
CO2 LSD compete com serotonina
tirosina
hormonas da tiróide (T3/T4)
histidina
histamina
CO2 Vasodilatador libertado na resposta alérgica
glutamato + cisteína + glicina
glicina + arginina
serina
glutationa
Protege os eritrócitos de danos oxidativos
creatina
fosfocreatina
acetilcolina
Síntese de catecolaminas e melanina
tirosina
tirosina
hidroxilase
(BH4)
tirosinase
O2
H2 O
DOPA
dopamina
dopamina
hidroxilase
(ascorbato)
O2
H2O
norepinefrina
epinefrina
melanina
Metabolismo do heme
Síntese do heme
Hepatócitos (cit. P450, citocromos)
células eritróides (hemoglobina)
85 % da síntese
PORFIRIAS
(congénitas ou adquiridas)
Defeitos na biossíntese do heme (fígado ou células eritróides)
Acumulação de porfirina e/ou precursores
.
* Porfirinas ou precursores são excretadas na urina e acumulam-­se na pele e/ou
nos dentes:
urina vermelha, depósitos nos dentes vermelho-­acastanhados,
fotossensibilidade, úlceras , cicatrizes, aumento do crescimento
capilar
* Exposição à luz produção de espécies reactivas de oxigénio
Degradação do heme
Ferro é reciclado
Eritrócitos: ~120 dias de vida
São removidos da circulação e os
seus componentes degradados
A apoproteína é hidrolisada aos
aminoácidos constituintes
O grupo prostético heme é também
degradado
(verde)
(amarelo-­alaranjado)
A alteração da cor num hematoma degradação do heme (manifestação visível) Bilirrubina:
Lipofílica
Transportada para o fígado complexada com albumina
Conjugada no fígado com glicoronato Derivados conjugados da bilirrubina secretados na bílis e degradados no intestino a
urobilinogénio:
Urobilinogénio é convertido em stercobilina, pigmento vermelho-­
acastanhado (maior pigmento das fezes) e a urobilina , pigmento amarelo (excretado na urina)
Icterícia
Quantidade excessiva de bilirrubina no sangue (insolúvel)
Deposição
Coloração de pele e olhos (amarelos)
Comum em recém-­nascidos (têm deficiência numa enzima
que degrada a bilirrubina ± tratamento com uma luz
fluorescente que converte a bilirrubina em isómeros mais
solúveis que são excretados)
Provocada por degradação excessiva de eritrócitos, por
disfunção hepática ou por obstrução biliar (medida da
proporção conjugada/ não conjugada pode esclarecer a
causa)
Acumulação de bilirrubina pode provocar encefolopatia
Doenças genéticas associadas ao metabolismo de a.a.
Fenilalanina Tirosina homogentisato fumarato + acetoacetato
1
2
Fenilcetonúria
deficiência em fenilalanina hidroxilase (1)
sintomas neurológicos graves
baixo QI
Acumulação de fenilalanina n fenilcetoácidos
Deficiência em tirosina pdopamina, pcatecolaminas, p melanina (hipopigmentação)
Dieta: pfenilalanina, ingestão de tirosina
Alcaptonúria
deficiência em homogentisato oxidase (2)
Acumulação de homogentisato, que é excretado na
urina
(fica
escura
devido
à
oxidação
do
homogentisato). O homogentisato deposita-­se nas
cartilagens e e tecidos conjuntivos levando a danos
nos ligamentos e a artrite.
Albinismo
Doença de Parkinson
Deficiência em tirosinase
Deficiência na síntese de dopaminas
tirosina
melanina
tirosinase
hipopigmentação
susceptibilidade
tirosina
L-­DOPA
Tirosina hidroxilase
Dopamina
queimaduras solares
cancro de pele
Tratamento ± L-­DOPA
Dieta rica em proteínas ou uma situação de jejum n UREIA.
PORQUÊ?
Doenças hepáticas podem provocar hiperamonémia.
Sabem porquê?
1± Uma alta concentração de ião amónia pode levar a uma diminuição da taxa de síntese de ATP mitocondrial. a) Justifique.
b) De onde provêm estes iões e qual o seu destino em condições normais? 2.Piruvato e alanina são componentes de um ciclo que envolve:
a) gluconeogénese hepática e renal
b) gluconeogénese hepática e transporte do azoto muscular para o fígado sob a forma de alanina
c) transporte de alanina para o músculo para fornecer piruvato
d) produção de alanina para uso na síntese proteica na maioria dos tecidos periféricos
3-­ Uma criança do sexo masculino nasceu após uma gravidez normal, mas depois de nascer não se
desenvolveu normalmente. Aos 10 anos, ele era incapaz de se alimentar sozinho, apenas pronunciava algumas
palavras, e tinha movimentos involuntários de membros. O seu QI era 65. Ele tinha olhos e pele claros, e o seu
cabelo era branco em alguns locais.
Essa criança provavelmente sofre de _____________, uma doença caracterizada por uma deficiência na enzima
_________________, responsável pela conversão de _______________ em tirosina. A tirosina é um aminoácido
precursor de _______________, o que justifica a falta de pigmentação da pele e dos olhos, e de
_____________, o que justifica o aparecimento do atraso mental na criança. Se o problema tivesse sido
diagnosticado à nascença, poder-­se-­ia ter optado pelo uso de uma dieta pobre em __________ e suplementada
com _____________, de modo a assegurar um desenvolvimento normal.
4-­ O grupo heme é um grupo prostético existente em proteínas como ____________, _____________ ou _____________. Tem na sua composição um átomo de _________ que permite a ligação ao oxigénio, quando se encontra no estado ___________ (reduzido/oxidado). A degradação do grupo heme origina produtos pigmentados como ____________ e _______________. As células/tecidos que apresentam maior taxa de síntese do gupo heme são ____________ e ____________. 5. As transaminases transformam aminoácidos em:
a) ácidos gordos
b) D-­cetoácidos
c) ácido úrico
d) ureia
e) amoníaco
6. V/F
O D-­cetoglutarato pode ser produzido a partir de um aminoácido por acção de uma transaminase
os grupos amina entram no ciclo da ureia são provenientes do ião amónio e do oxaloacetato
Na conversão do grupo amina de varios aminoácidos em ureia ocorre transaminação e desaminação oxidativa
Um paciente encontra-­se em balaço negativo de azoto sempre que a quantidade de azoto excretada na urina,
fezes e suor é menor do que a quantidade de azoto ingerida
A proteína muscular é uma fonte possível de glucose.
Um indivíduo com doença hepática apresenta acumulação de amónia no sangue
7-­ A proteínas ingeridas em excesso:
a)
São eliminadas já que não existe reserva de proteína ao contrário do que acontece para os hidratos de
carbono e lípidos.
b) São armazenadas numa reserva de proteínas tal como acontece para os hidratos de carbono e lípidos.
c)
São convertidos sempre em proteína activa ( ex: proteína muscular, enzimas, hormonas...) mesmo que
em excesso.
d) Nenhuma das opções está correcta-­