1
I – PARTE
Aminoácidos
Estrutura dos aminoácidos:
Da análise de um número vasto de proteínas, chegou-se à conclusão de que
todas as proteínas são compostos por 20 aminoácidos, ditos fundamentais.
Os aminoácidos mais comuns são os conhecidos por α-aminoácidos, pois
possuem um grupo amino (-NH2) e um grupo carboxílico (-COOH):
NOTA: A excepção dos α-aminoácidos é a prolina, pois possui um segundo
grupo amino (-NH-).
Todos os aminoácidos diferem entre si no grupo R.
Propriedades Gerais:
Os grupos amino e carboxílicos tendem a ionizar-se. Assim, como o pK 1 (pK
para o grupo carboxílico) roda o valor 2.2 e o pK 2 (pK para o grupo amino)
ronda o valor 9.4, isto vai implicar que no pH fisiológico o grupo amino esteja
protonado e o grupo carboxílico esteja na sua forma ionizada COO-. Conclui-se
assim que um aminoácido pode comportar-se tanto como ácido como base.
Moléculas como estas, que possuem grupos com cargas de sinal oposto são
chamadas de zwitteriões ou iões dipolares.
Os aminoácidos, tal como outros compostos iónicos, são mais solúveis em
solventes polares do que em solventes apolares. Estas propriedades iónicas
dos aminoácidos vão influenciar as propriedades químicas e físicas de um
aminoácido livre e dos aminoácidos em complexos proteicos.
Ligações peptídicas:
Os aminoácidos podem ser polimerizados para formar cadeias; este processo
pode ser representado como uma reacção de condensação (em que há a
eliminação de uma molécula de água). A resultante ligação CO-NH é a
chamada ligação peptídica.
2
Polímeros compostos por dois, três, poucos ou vários aminoácidos são
chamados dipeptídeos, tripeptídeos, oligopeptídeos e polipeptídeos,
respectivamente. Depois de estarem incluídos num peptídeo, os aminoácidos
individuais são designados por resíduos aminoácidos.
Polipeptídeos são polímeros lineares, em que cada residuo aminoácido
participa em duas ligações peptídicas; os resíduos das duas pontas do
polipeptídeo apenas participam numa ligação peptídica. O resíduo que tem o
grupo amino livre (por convenção, a ponta esquerda do aminoácido) é
chamado N-terminal, assim como o resíduo com um grupo carboxílico livre (à
direita) é designado por C-terminal.
Proteínas vão ser moléculas que contêm uma ou mais cadeias polipeptídicas.
Propriedades ácido-base:
Os α-aminoácidos possuem dois ou três grupos ácido-base. A valores baixo de
pH, ambos os grupos estão totalmente protonados, predominando assim a
forma catiónica. Ao longo de uma titulação com uma base forte, estes vão
perder dois ou três protões, formando uma curva característica de um ácido
diprótico ou triprótico. Os valores de pH dos vários grupos podem ser
calculados pela equação de Henderson-Hasselbalch:
pH = pK + log [A-]/[HA]
O pH a que a molécula adquirir carga electrónica neutra é conhecido com
ponto isoeléctrico (pI):
pI = ½ (pKi + pKj)
“Os valores de pK dos grupos ionizáveis dependem de grupos próximos”:
Os valores de pK 1 dos aminoácidos são muito menores do que o valor de pK de
um simples ácido carboxílicos. A grande diferença é causada pela influencia
electrostática do grupo carregado amónia. O grupo NH3+ estabiliza mais
electrostaticamente o grupo COO- do que o grupo COOH. Igualmente o grupo
NH3+ possui um pK mais baixo do que uma amina normal, devido à tendência
electrónica negativa do grupo carboxílico. Assim, ambas as características
electrónicas e electrostáticas influenciam o pK do grupo NH3+.
3
Estereoquímica:
Todos os aminoácidos que derivam de proteínas (resíduos) têm uma
configuração estereoquímica L.
Uma molécua com n centros quirais possui 2n diferentes possíveis
estereoisómeros.
Todos os aminoácidos L nas proteínas são S aminoácidos excepto a cisteína.
Outros aminoácidos para além dos 20 fundamentais:
O código genético universal especifica apenas 20 aminoácidos. No entanto,
muitos outros fazem parte de certas proteínas. Estes aminoácidos, na maior
parte resultam de uma modificação específica de um resíduo aminoácido,
depois de uma cadeia polipeptídica ter sido sintetizada. Também os grupos
amino e carboxilíco terminais de um polipept ídeo podem ser modificados,
modificação esta que é muito importante para a função da proteína.
Aminoácidos D:
Os resíduos D-aminoácidos são componentes de relativamente pequenos
polipeptídeos bacterianos. Estes polipeptídeos são, talvez, grande parte
integrante das paredes celulares bacterianas, para assim impedir o “ataque”
das enzimas produzidas pelos outros organismos para digerir as bactérias.
São também componentes dos antibióticos antibacterianos.
Aminoácidos biologicamente activos:
Muitos aminoácidos são sintetizados não para serem resíduos de
polipeptídeoss, mas para funcionarem como independentes; muitos
organismos usam aminoácidos para transportar azoto na forma de grupos
amino.
Também podem ser oxidados de maneira a produzirem energia, assim como
funcionar como mensageiros químicos para comunicação entre células.
Proteínas: Estrutura primária:
As proteínas são o centro da acção dos processos biológicos. Quase todos os
processos do metabolismo celular são catalisados por proteinas; as proteínas
também regulam as condições extra e intracelulares, assim como são parte
essencial da estrutura celular; enfim, uma lista de todas as funções proteicas
teria milhares e milhares de alíneas.
Um dos passos para decifrar a função de uma proteína é compr eender a sua
estrutura: tal como as outras biomoléculas, as proteínas são polímeros de
pequenas unidades, só que não possuem uma estrutura regular, em parte
devido aos 20 aminoácidos seus constituintes não possuirem todas as
mesmas propriedades físicas e químicas.
4
Diversidade polipeptídica:
Como os outros polímeros, as proteínas podem descrever-se em termos de
níveis de organização, neste caso em estrutura primária, secundária, terciária
e quaternária.
A estrutura primária é a sequência de aminoácidos da sua cadeia ou cadeias
polipeptídicas, onde cada resíduo está ligado ao outro por uma ligação
peptídica.
Com os 20 diferentes aminoácidos é possivel obter um número astronómico de
diferentes proteínas: para uma proteína de n resíduos, há 20n possibilidades
de os sequenciar.
Em geral, uma proteína contém pelo menos 40 resíduos, levando a que
polipeptídeos mais pequenos que isso sejam apenas chamados de peptídeos;
embora haja maiores, a maior parte dos polipeptídeos contêm entre 100 e
1000 resíduos.
As proteínas vão depender mais da sua sequência de resíduos do que dos seus
resíduos constituintes; podem também formar complexos com iões metálicos,
como o Zn2+ e o Ca2+; podem ligar-se por ligações covalentes ou não covalentes
a outras pequenas moléculas orgânicas e podem ser modificadas pela ligação
covalente de grupos como fosfatos e hidratos de carbono.
Purificação proteica:
Felizmente, variações nos tamanhos e na composição química dos
polipeptídeos tornam mais fácil separar proteínas umas das outras e doutras
moléculas biológicas. Normalmente, quando uma substância perfaz <0.1% do
peso seco de um tecido, então tem de ser convertida a uma pureza de ≈98%, o
que implica certas técnicas de purificação:
-
O primeiro passo para isolar uma proteína ou qualquer outra molécula
biológica, é retirá-la da célula para uma solução: a maior parte dos
processos usa uma variação de esmagamento e trituramento das
células, seguido por uma filtração e centrifugação para remover as
partículas grandes insolúveis. Por outro lado, se a proteína desejada
está dentro de uma membrana lipídica, um detergente ou um solvente
orgânico podem ser usados para dissolver os lípidos e recuperar a
proteína.
Processos de estabilização:
Depois de uma enzima ter sido removida do seu ambiente natural, fica exposta
a vários agentes que a podem danificar irreversivelmente:
Ø pH:
Existe uma gama onde cada material é estável. Violando essa propriedade
pode levar à desnaturação proteica, ou mesmo à degradação química (lise).
5
Ø Temperatura:
A estabi lidade térmica das proteínas varia. Algumas apenas desnaturam a
elevadas temperatura, outras a temperatura alguns graus acima do seu
ambiente natural; normalmente, a purificação é feita a temperaturas perto dos
0ºC, de maneira a superar esta barreira.
Ø Presença de enzimas degradantes:
Quando os tecidos são destruídos para libertar moléculas que nos interessam,
também são libertadas enzimas, as quais podem ser proteases (estas enzimas
degradam, como o próprio nome indica, as proteínas). Estas podem ser
inibidas pelo ajustamento do pH ou da temperatura para valores que as
inactivam ou por compostos que inibam a sua acção (inibidores).
Ø Adsorção a superfícies:
Muitas proteínas são desnaturadas pelo contacto com o vapor de água do ar
ou com superfícies de vidro ou plásticos. Assim, as soluções proteicas são
feitas de forma a minimizar a espuma e mantidas a elevadas concentrações.
Ø Soluções proteicas guardadas há demasiado tempo:
As soluções proteicas são, muitas vezes, guardadas envoltas em gás de azoto
ou árgon e congeladas a –70ºC ou a –196ºC, de maneira a evitar uma lenta
oxidação (oxigénio do ar) ou contaminação por agentes microbianos.
Técnicas de separação:
As características das proteínas e de outras biomoléculas, que são usadas nos
vários processos de separação são: solubilidade, carga iónica, tamanho
molecular e especificidade da ligação com outras moléculas biológicas.
Característica
Carga
Processo de separação
Cromatografia de permuta iónica;
Electroforese;
Polaridade
Cromatografia de interacção hidrofóbica;
Cromatografia de gel;
Tamanho
SDS-page;
Ultra-centrifugação;
Especificidade da ligação
Cromatografia de afinidade;
6
Solubilidade proteica:
A solubilidade proteica em soluções de baixa concentração iónica vai
aumentando à medida que se adiciona um sal, pois os iões vão formar um
“escudo” à molécula proteica, devido aos seus múltiplos grupos carregados,
levando a que as forças atractivas entre as várias moléculas proteicas seja
menor, forças essas que podem levar à precipitação. No entanto, com a
contínua adição de sal vai-se dar uma diminuição da solubilidade, como
resultado da competição entre os iões do sal adicionado e outros dissolvidos
no solvente.
Como cada proteína precipita a concentrações de sal diferentes, esta
característica é aproveitada como um meio de purificação proteica. O sal mais
usado é o sulfato de amónia (NH4) 2 SO4, pois é muito solúvel.
Cromatografia:
Ø Cromatografia de permuta iónica:
Na cromatografia de permuta iónica, moléculas carregadas ligam-se a grupos
de carga oposta que se situam na matriz. A matriz pode ser catiónica (quando
fixa catiões)ou aniónica (quando fixa aniões). Para proteínas, a matriz mais
usada são resinas celulosas.
Ø Cromatografia de interacção hidrofóbica:
Na cromatografia de interacção hidrofóbica, o material da matriz é levemente
substituído por grupos octil e fenil; grupos apolares da superfície das
proteínas interagem com os grupos hidrofóbicos, isto é, ambos os grupos são
excluídos pelo solvente polar.
Ø Cromatografia em gel:
Na cromatografia em gel, as moléculas são separadas de acordo com o seu
tamanho e forma; a fase estacionária (matriz) consiste num gel com poros que
atingem um certo tamanho. Ao fazer passar uma solução através da coluna,
as moléculas que são maiores não vão passar pelos poros, logo, atravessam a
coluna mais rapidamente, enquanto que as moléculas mais pequenas, ao
terem de atravessar toda a coluna, demoram mais tempo.
Ø Cromatografia de afinidade:
Uma característica de muitas proteínas é a sua habilidade de se manterem
perto de certas moléculas. Esta propriedade é usada para purificar proteínas
por cromatografia de afinidade. Nesta técnica, um ligando que tem uma
afinidade específica a uma certa proteína é ligado a uma matriz inerte por
ligação covalente. Quando uma solução impura dessa proteína é passada
através da matriz, a proteína vai ligar-se ao ligando, ficando retida, enquanto
que as outras substâncias passam pela coluna. Depois, a proteína desejada
pode ser recolhida mudando as condições do eluente, de modo a libertar a
7
proteína da matriz, por exemplo, soluções de grande concentração de ligando
livre ou soluções de diferente pH ou força iónica.
Electroforese:
Ø Electroforese de gel de poliacrilamida:
Similar à cromatografia por gel, só que a mobi lidade das grandes moléculas é
menor que a mobilidade das moléculas pequenas, com a mesma carga. Como
o pH do gel ronda o valor de 9, quase todas as proteínas têm cargas negativas
e movem-se assim para o ânodo através do gel.
Ø SDS-page:
O detergente sulfato-dodecyl de sódio (SDS) é usado para desnaturar
proteínas. Estas assumem assim uma forma linear na presença de SDS e
algumas até se ligam a este numa proporção de 1.4g SDS/1g proteína. Este
tratamento leva a que as proteínas fiquem com forma e carga por massa
similares. Assim, são separadas por filtração por gel, de acordo com a sua
massa molecular. A mobilidade relativa das proteínas no gel varia linearmente
com o logaritmo das suas massas moleculares.
Ø Electroforese de capilaridade:
Esta técnica é executada em muito finos tubos capilares. Tais capilares, como
dissipam rapidamente o calor, permitem o uso de campos eléctricos fortes, que
reduzem o tempo de separação em relação à electroforese por gel. No entanto
tem a limitação de separar apenas pequenas quantidades de material.
Ultracentrifugação:
Numa solução de proteínas precipitadas, estas não se sedimentam por acção
da gravidade, como soluções de água e areia. Assim, para separar soluções de
proteínas saturadas usa-se a ultracentrifugação.
Na ultracentrifugação atingem-se velocidades de 80 000 rpm.
Sequenciação de proteínas:
As sequencias de aminoácidos de centenas de milheres de polipeptídeos são
agora conhecidas. Esta informação é valiosa para:
-
Determinar a estrutura tridimensional da proteína, de modo a entender
o seu mecanismo de acção molecular;
Comparação das sequencias de proteínas de espécies diferentes de
forma a observar relações em termos de evolução;
Muitas doenças são causadas por mutações que têm como base a
mudança de um aminoácido numa proteína. Logo, a análise da
sequencia de aminoácidos pode desenvolver um diagnóstico e a sua
apropriada terapia.
8
Primeiros passos:
Ø Achar N ou C terminais:
Cada cadeia polipeptídica tem um resíduo N-terminal e C-terminal. Logo,
sabendo quantos N-terminais há numa proteína, podemos saber quantas
cadeias polipeptídicas a proteína possui; os N-terminal de um polipeptídeo
podem ser determinados por vários métodos, tal como por um composto
fluorescente que se vai “agarrar” ao N-terminal. Por hidrólise separam-se os
vários aminoácidos, em que por cromatografia se vai identificar os de cor
fluorescente. Também há a possibilidade de através da enzima
carboxipeptidase separar o aminoácido C-terminal, que posteriormente pode
ser isolado e identificado.
Ø Encontrar pontes de dissulfureto:
As ligações por ponte de dissulfureto entre as cisteínas têm de ser partidas, de
forma a garantir que a cadeia polipeptídica é totalmente linear. Estas ligações,
podem ser partidas por oxidação ou redução, através do ácido pe rfórmico ou
por “mercaptans”, respectivamente. A primeira reacção tem a desvantagem de
também oxidar a metionina e parcialmente destruir a cadeia da tripsina.
Ø A combinação aminoácida de um polipeptídeo pode ser
determinada:
A composição aminoácida de um polipeptídeo é determinada pela sua
completa hidrólise, seguida pela análise dos aminoácidos libertados. Visto que
a separação por ácido ou base destrói alguns aminoácidos e a separação por
enzimas não é completa e, sendo enzimas também elas proteínas, estas podem
interferir na análise dos aminoácidos obtidos. Conclui-se assim que nenhum
dos processos é totalmente satisfatório. A análise quantitativa vai ser feita
num instrumento que separa os vários aminoácidos hidrolisados por
cromatografia e identifica-os em termos de % de volume, por técnicas de
absorvância e fluorescência.
Ø Clivagem polipeptídica:
Polipeptídeos com mais do que 40 a 100 resíduos não podem ser identificados
por degradação de Edman e logo têm de ser partidos em fragmentos mais
pequenos.
Ø Degradação de Edman:
Depois das reacções de clivagem, a sequência de aminoácidos dos polipetídeos
resultantes pode ser determinada através de vários ciclos da degradação de
Edman:
9
Proteínas: Estrutura tridimensional:
Como vimos, a estrutura primária de uma proteína é a sua sequência linear de
aminoácidos. No entanto, no estudo da estrutura de proteínas, mais três
níveis de complexidade estrutural são invocados:
-
Estrutura secundária: é o arranjo espacial dos átomos de um
polipeptídeo, sem ter em conta conformações ou cadeias laterias;
Estrutura terciária: refere -se à estrutura tridimensional de um
polpeptídeo completo;
Estrutura quaternária: como a maior parte das proteínas são
compostas por duas ou mais cadeias polipeptídicas, então a sua
estrutura quaternária será o arranjo espacial das várias cadeias.
10
Estrutura secundária:
Ø O grupo dos péptidos:
Estudos indicam que o grupo dos péptidos tem uma rígida e planar estrutura
como resultado de interacções de ressonância que dão à ligação pe ptídica um
carácter duplo.
Os grupos de péptidos assumem a conformação trans, na qual sucessivos
átomos C α estão em lados opostos da ligação peptídica que os liga. No entanto,
a conformação cis no caso da prolina tem uma ocorrência média de cerca de
10%.
A cadeia principal de uma proteína são os átomos que participam nas ligações
peptídicas, ignorando-se as cadeias laterais dos resíduos aminoácidos. Assim,
na cadeia principal vai haver ângulos de torsão para cada resíduo, de modo a
adquirirem a conformação trans. Esta torção pode provocar colisões entre
átomos adjacentes e mesmo entre átomos de resíduos que estão afastados da
sequência. Assim, os valores de φ e ψ podem ser calculados de modo a que os
átomos distem da distância mínima de contacto, sem haver ligação entre eles
(diagramas de Ramachandran).
Estruturas secundárias regulares:
Estas estruturas são consideradas regulares, pois os seus valores de φ e ψ são
repetitivos.
Ø A hélice α :
A hélice α é a única hélice polipeptídica que possui uma ligação de hidrogénio
favorável e os seus valores de φ e ψ estão dentro da região regulamentar do
diagrama de Ramachandran.
A hélice α roda na direcção na qual os dedos da mão direita dobram. As
ligações de hidrogénio vão dar-se entro o oxigénio do grupo C=O e o hidrogénio
do grupo N-H entre os resíduos n e (n+4).
Ø Folha β :
Neste caso, a ligação de hidrogénio vai ocorrer entre polipéptideos vizinhos,
formando duas variantes:
-
A folha β antiparalela: em que as duas cadeias polipeptídicas se
encontram em direcçõe s/posições opostas;
A folha β paralela (menos estável, talvez devido à distorção das
ligações por ponte de hidrogénio): em que as duas cadeias
polipeptídicas estão em posição/direcção igual, ou seja, “paralelas”.
Também possui rotação dada segundo a regra da mão direita.
11
Proteínas fibrosas:
Historicamente, as proteínas são classificadas como sendo fibrosas ou
globulares.
Ø α -queratina (um “rolo enrolado”):
A queratina é uma proteína mecanicamente duradora e que não reage
quimicamente. Encontra-se presente em todos os vertebrados superiores. É a
principal constituinte do cabelo, do “corno”, das unhas e das penas. A
queratina pode ser classificada de α-queratina, que aparece nos mamíferos, e
β-queratina, que se encontra nos pássaros e répteis.
A α-queratina vai ser o resultado do enrolamento “left-handed” de duas
hélices-α, de maneira a formar um “rolo”; as duas hélices estão inclinadas
cerca de 18º uma em relação à outra.
Ao N e C terminais pode ligar-se outras queratinas (dímeros), de maneira a
formar um protofilamento. Dois protofilamentos constituem uma protofibra e
quatro protofibras dão origem a uma microfibra. As microfibras associam-se a
outras microfibras de forma a formarem macrofibras.
A α-queratina é rica em resíduos de cisteína, logo, forma ligações de
dissulfureto. Estas ligaçoes podem ser partidas (por meio de um “mercaptan”)
e o “rolo” ser assim “esticado”, assumido uma conformação do género da da βqueratina.
Ø A fibroína da seda (uma folha β ):
As fibras das sedas consistem em folhas β antiparalelas cujas cadeias se
extendem paralelamente ao eixo da fibra. As folhas β empilham-se de modo a
formar um microcristal.
Ø Colagénio (a hélice tripla):
O colagénio, que aparece em todos os animais multicelulares, é composto por
fibras fortes e insolúveis. É constituinte dos ossos, tendões, pele e vasos
sanguíneos.
Uma molécula de colagénio é constituída por três cadeias polipeptídicas. Cada
uma destas cadeias forma uma hélice “esquerda”, e as três hélices paralelas
enrolam-se de forma “right-handed”, formando assim a estrutura da triplahélice de uma molécula de colagénio.
No colagénio as ligações entra as hélices não são pontes de dissulfureto, pois
quase não existem resíduos de cisteína. Assim, as ligações vão dar-se na lisina
e na histidina da cadeia lateral.
12
Estruturas proteicas não repetitivas:
Ø Estruturas irregulares:
Segmentos de cadeias polipeptídicas cujos resíduos sucessivos não possuem
um φ e ψ similar são muitas vezes chamados de “rolos”. Estes “rolos” não
devem ser confundidos com a estrutura de “rolo” aleatório que adquirem as
proteínas desnaturadas.
Ø Variações na estrutura secundária:
A hélice-α frequentemente se desvia da sua conformação ideal nas suas curvas
finais e iniciais da hélice. Similarmente, uma cadeia de polipeptídeos de uma
folha β pode conter um resíduo extra que não “realiza” a ponte de hidrogénio
com a cadeia vizinha, produzindo assim distorções.
Ø Voltas e loops:
Segmentos de estruturas secundárias regulares, como hélices α ou folhas β
são tipicamente unidas por cadeias polipeptídicas que abruptamente mudam
de direcção. Estas “voltas repentinas” quase sempre ocorrem na superfície da
proteína. Podem ser de dois tipos, ambos estabilizados por uma ponte de
hidrogénio. Quase todas as proteínas com mais de 60 resíduos têm uma ou
mais loops, chamadas Ω-loops; estas loops são entidades globulares
compactas, pois as suas cadeias laterais tendem a ser preservadas nas suas
cavidades internas.
Estrutura terciária:
A estrutura terciária das proteínas descreve o envolvimento das suas
estruturas secundárias e especifica a posição de cada átomo na proteína.
Ø Determinando a estrutura proteica:
A estrutura proteica pode ser determinada por cristalografia de raios X. Nem
todas as proteínas formam cristais, mas as que formam podem adquirir várias
formas, formas essas que possuem 40% a 60% de água no seu volume total.
Na cristalografia de raios X, uma resolução de poucos ºA não é suficiente para
revelar a posição de átomos individuais, mas chega para observar a cadeia
polipeptídica principal, logo, as cadeias laterais são deduzidas, obtendo-se
assim o conhecimento da estrutura primária.
Para determinar a estrutura de proteínas que não cristalizam, usam-se
técnicas de ressonância magnética nuclear (NMR).
13
Estruturas supersecundárias e domínios:
Ø A posição das cadeias laterais varia com a polaridade:
As estruturas primárias de proteínas globulares geralmente não comportam
uma sequência regular. No entanto, as cadeias laterais de aminoácidos das
proteínas globulares são espacialmente distribuídas de acordo com as suas
polaridades:
-
-
-
Os resíduos apolares aparecem quase sempre no interior da proteína,
fora do contacto com os solventes aquosos. Este efeito hidrofóbico é um
dos grandes responsáveis pela estrutura tridimensional das proteinas.
Os resíduos polares com carga estão normalmente localizados na
superfície da proteína, em contacto com os solventes aquosos; tal facto
é devido a um ião num meio virtualmente anidro se encontrar num
estado energeticamente desfavorável.
Os resíduos polares sem carga estão normalmente à superfície da
proteína, mas também aparecem no seu interior, pois são eles que
normalmente fazem as pontes de hidrogénio com os outros grupos.
Ø Hélices e folhas podem ter várias combinações:
As principais estruturas secundárias das proteínas (folhas β e hélices α),
ocorrem nas proteínas globulares em variadíssimas proporções e combinações.
Certos grupos de estruturas secundárias, chamados estruturas
supersecundárias, ocorrem na maioria das proteínas globulares:
-
A estrutura βαβ é a mais comum, na qual uma hélice α se liga a duas
cadeia paralelas de folhas β.
A estrutura “β-hairpin”, consiste em cadeias antiparalelas ligadas por
voltas.
A estrutura αα é consiste em duas hélices α antiparalelas sucessivas
encostadas uma à outra, segundo um eixo inclinado.
A estrutura barril β consiste em folhas β extendidas e enroladas de
maneira a formar um cilindro.
Ø Grandes polipéptidos formam domínios:
Cadeias polipeptídicas com mais de 200 resíduos, usualmente juntam-se em
dois ou mais empilhamentos globulares, chamados domínios. Estes domínios
conferem à proteína uma aparência bi ou multilobal. Uma cadeia polipeptídica
movimenta-se dentro de um domínio, mas domínios vizinhos estão apenas
ligados por um ou dois segmentos polipeptídicos. Assim, muitos domínios são
unidades estruturalmente independentes, que têm características de
pequenas proteínas globulares.
14
Ø Família proteica:
Os milhares de estruturas proteicas conhecidas, revem ainda um maior
número de domínios existentes. Estes podem se agrupados em famílias, tendo
em conta o caminho seguido pelas suas cadeias polipeptídicas, sem ter em
conta a sua sequência de aminoácidos. Comparando as estruturas dos vários
domínios, chega-se à conclusão da existência de apenas algumas centenas de
domínios. Surpreendentemente, poucas dúzias delas perfazem praticamente
metade das estruturas proteicas conhecidas. Este facto leva a pensar que as
estruturas proteicas estão, de algum modo, ligadas evolucionariamente.
Estrutura quaternária e simetria:
A maior parte das proteínas consiste em mais do que uma cadeia
polipeptídica. Estas cadeias estão associadas a uma geometria específica. O
arranjo espacial destas subunidades é conhecido como a estrutura
quaternária de uma proteína.
Ø As subunidades geralmente ligam-se não-covalentemente:
Uma proteína de várias subunidades pode possuir idênticas ou não-idênticas
cadeias polipeptídicas. Proteínas com mais de uma subunidade são chamadas
oligómeros e as suas unidades protómeros; um protómero pode assim possuir
uma ou várias cadeias polipeptídicas. A região de contacto entre duas
subunidades próximas assemelha-se ao interior de uma proteína de uma só
unidade: possui cadeias laterais apolares, pontes de hidrogénio envolvendo as
cadeias principais e laterais do polipeptídeo e, por vezes, pontes de
dissulfureto.
Ø As subunidades formam arranjos simétricos:
Na vasta maioridade de oligómeros, os protómeros encontram-se
simetricamente arranjados. Como as proteínas não podem ter inversão ou
simetria especular, para fazer um protómero coincidir com sua imagem no
espelho seria necessário converter os resíduos L em D. Assim, as proteínas só
podem ter simetria rotacional.
Pode existir simetria rotacional cíclica, onde os protómeros estão relacionados
a um único eixo de rotação: C1, C3, Cn (n- número de subunidades). Há
também a simetria rotacional diedral, em que um eixo intersecta dois eixos de
rotação: D2, Dn (n- 2n subunidades). Para além destes dois tipos de simetria
rotacional, há ainda a considerar a ocorrência de simertrias rotacionais
tetraédricas, octaédrica e icosaédricas.
Dobramento e estabilidade proteica:
Por incrível que pareça, medidas termodinâmicas indicam que a maior parte
das proteínas apenas são relativamente estáveis em condições fisiológicas. São
então os seus efeitos hidrofóbicos, as suas interacções electrostáticas e as
suas pontes de hidrogénio que as vão estabilizar.
15
Forças que estabilizam uma proteína:
Ø O efeito hidrofóbico:
O efeito hidrofóbico, que faz com que substâncias apolares minimizem o seu
contacto com a água, é o efeito mais determinante de uma estrutura proteica.
A agregação de cadeias laterais apolares no interior da proteína é favorecida
pelo aumento da entropia das moléculas de água que em caso contrário se
organizariam ordenadamente à volta dos grupos hidrofóbicos.
Ø Interacções electrostáticas:
No interior das proteínas, as forças de Van der Waals, embora fracas, são uma
importante influência estabilizadora, pois actuam apenas a pequenas
distâncias e desaparecem quando a proteína é “desenrolada”. Por incrível que
pareça, as pontes de hidrogénio contribuem de uma forma minoritária para a
estabilidade da molécula, porque também à formação de pontes de hidrogénio
entre a proteína e a água.
A associação de dois grupos iónicos de cargas opostas é chamada de par
iónico ou ponte salina. Este caso ocorre na maior parte dos resíduos
carregados e contribui também para a estabilização de uma proteína, embora
de forma minoritária, pois a energia livre de um ião não compensa a perda de
entropia e de energia livre aquando da formação de um par iónico.
Ø Ligações químicas cruzadas:
As ligações de dissulfureto não são essenciais na estabilidade, pois uma
proteína sem estas ainda é estável, mas têm um importante papel sob o ponto
de vista estrutural.
Os iões metálicos que aparecem em muitas proteínas podem interagir com
ligandos presentes em muitos resíduos aminoácidos, contribuindo assim para
alguma estabilização.
Desnaturação e renaturação de proteínas:
A baixa estabilidade conformacional das proteínas torna fácil a sua
desnaturação:
-
-
O aquecimento faz com que propriedades das proteínas se modifiquem
abruptamente, levando ao desenrolamento ou à lise da proteína;
Variações de pH alteram os estados de ionização das cadeias laterais
dos aminoácidos, modificando a distribuição das cargas na proteína e
pontes de hidrogénio.
Detergentes associados com resíduos apolares da proteína vão interferir
com as interacções hidrofóbicas, grandes responsáveis pela estrutura
proteica;
Os agentes ião guanidina e ureia, que em elevadas concentrações
aumentam a solubilidade de substâncias apolares na água;
16
Ø Proteínas desnaturadas podem ser renaturadas:
As proteínas podem enrolar-se espontaneamente para as suas conformações
habituais em condições fisiológicas. Isto implica que a estrutura primária de
uma proteína dita o seu arranjo tridimensional.
Passos do enrolamento proteico:
No início pensava-se que proteínas desnaturadas, ao voltar ao seu estado
habitual, iriam experimentar todas as conformações possíveis até encontrar a
certa. Após muitos anos de estudo, chegou-se à conclusão de que a
renaturação por este processo levaria imenso tempo, enquanto se verifica que
as proteínas renaturam em apenas alguns segundos. Assim, concluiu-se que
as proteínas voltam à sua conformação habitual por passos directos, de
maneira à sua estabilidade conformacional aumentar, ou seja, diminuindo a
energia livre: começa com a formação das estruturas secundárias, que
estabiliza, começando a formar a estrutura terciária. De seguida sofre as
transformações estruturais necessárias de modo a produzir as estruturas
terciária e quaternária.
Isomeria dissulfídrica proteica:
No renaturamento das proteínas, muitas vezes há formação de pontes de
dissulfureto que na conformação nativa da proteína não existem; estas vão
então formar as ligações nativas; a enzima PDI catalisa este processo.
“Chaperons” moleculares:
“Chaperons” moleculares são proteínas essenciais que se unem a
polipeptídeos desenrolados ou parcialmente enrolados, para assim prevenir a
associação imprópria de segmentos hidrofóbicos que possam levar ao não
enrolamento ou à precipitação do polipeptídeo. Permitem também que
proteínas mal enroladas se re-enrolem na sua conformação nativa.
Dinâmica proteica:
De facto, as proteínas são moléculas flexíveis e rapidamente variáveis, cuja
mobilidade estrutural é funcionalmente significante.
17
II - PARTE
Catálise Enzimática:
•
Propriedades Gerais:
A catálise enzimática difere de uma catálise química tradicional em vários
aspectos importantes:
ü Grande aumento da velocidade de uma reacção (de 106 a 1012
vezes superior à velocidade da mesma reacção não-catalisada);
ü Condições mais suaves de reacção, ou seja, as condições em que
reacções catalisadas por enzimas são médias: temperaturas abaixo
dos 100ºC, pressão atmosférica e pH próximo do fisiológico;
ü Especificidade muitissimo elevada, que tem a ver com a natureza
do substrato e dos produtos finais da reacção; a enzima “reconhece” o
seu substrato e catalisa a sua reacção de tal forma que raramente
são formados produtos laterais (indesejados) da reacção;
ü Capacidade de regulação: a actividade catalítica de muitas
enzimas varia de acordo com a concentração de substrato. Os
mecanismos de regulação incluem controlo alostérico, modificação
covalente de enzimas e variação da quantidade de enzima
sintetizada;
Especificidade do substrato:
As forças não-covalentes pelas quais substratos e outras moléculas se ligam
às enzimas, envolvem forças de Van der Waals, electrostáticas, pontes de
hidrogénio e interacções hidrofóbicas. Em geral, o local de ligação do
substrato consiste num “buraco” na superfície da enzima que tem
complementaridade com a forma geométrica do substrato; este modelo tem
como base o modelo da “chave -fechadura” proposto por Emil Fisher em 1894 e
já abandonado, visto que a ligação enzima-substrato não se resume apenas à
complementaridade geométrica entre ambos. Como veremos mais adiante,
esta complementaridade geométrica é uma condição necessária mas não
suficiente para uma catálise eficiente.
Ø As enzimas são estereospecíficas:
As enzimas são altamente específicas na sua ligação a substratos quirais e na
catálise das suas reacções. Esta estereospecificidade tem origem na
quiralidade inerente a todas as proteínas (as proteínas possuem apenas Laminoácidos), que formam centros activos assimétricos. Como veremos, quase
todas as enzimas que participam em reacções quirais são absolutamente
estereospecificas.
Ø As enzimas variam na sua especificidade geométrica:
Em adição à sua estereospecificidade, muitas enzimas são muito selectivas
quanto à identidade dos grupos químicos nos seus substratos. Assim sendo,
uma substância com a quiralidade errada não encaixará no centro activo da
enzima pela mesma razão de que não conseguimos encaixar a nossa mão
direita numa luva esquerda. Muito poucas enzimas são específicas
18
unicamente para um substrato, catalisando geralmente uma pequena porção
de compostos quimicamente relacionados: o YADH catalisa a reacção de vários
pequenos álcoois primários e secundários nos seus respectivos aldeídos e
cetonas, mas nenhum tão eficazmente como o entanol. Esta especificidade
geométrica é bastante mais restrita do que a estereoespecificidade.
Cofactores e Coenzimas:
Os grupos funcionais das proteínas podem facilmente participar em reacções
ácido/base, formando certos tipos de ligações covalentes e tomam parte nas
interacções carga/carga. São assim piores para catalisar reacções redox mas,
apesar disso, as enzimas catalisam estas reacções, graças à associação nestas
de pequenas moléculas, cofactores, que agem como “dente químico” da
enzima.
Os cofactores são geralmente iões metálicos, Cu2+, Fe 3+ ou Zn2+. A natureza
essencial destes compostos explica porque os organismos necessitam deles
nas suas dietas (de outra forma não os conseguiriam produzir e as catálises
enzimáticas dentro dos próprios organismos não seriam eficazes). Explica-se
da mesma forma da mesma maneira a toxicidade do Hg2+ e do Cd2+ que são do
mesmo grupo da Tabela Periódica, substituindo o Zn2+ e o Cu2+ como
cofactores, inactivando as enzimas. Os cofactores também podem ser
moléculas orgânicas, sendo nesse caso denominados como coenzimas e
funcionam essencialmente como cosubstratos. Um exemplo de uma molécula
deste tipo é o NAD+ (nicotinamida). Outros cofactores chamados grupos
prostéticos estão geralmente associados à enzima por ligações covalentes. Um
complexo enzima-confactor cataliticamente activo é chamado holoenzima;
Quando se remove o cofactor da holoenzima, resulta uma proteína
cataliticamente inactiva, denominada apoenzima.
Ø As coenzimas têm que ser regeneradas:
As coenzimas são alteradas quimicamente pela reacção enzimática em que
participam. Assim, para completar o ciclo catalítico, a coenzima tem de voltar
ao seu estado inicial. Nos grupos prostéticos a regeneração ocorre numa fase
separada da sequência da reacção enzimática.
Ø Muitas vitaminas são coenzimas:
Muitos organismos não são capazes de sintetizar certas coenzimas. Assim,
essas substâncias têm de estar presentes na dieta desse organismo. Certas
vitaminas estão assim nesta lista de coenzimas e a sua carência pode provocar
uma série de doenças provocadas por catálises enzimáticas incompletas.
Mecanismos catalíticos:
As enzimas, tal como outros catalisadores, vão baixar a energia de activação
duma determinada reacção. O que as torna tão eficientes é o facto de terem
uma enorme especificidade de ligação ao substrato, combinada com o arranjo
dos seus grupos catalíticos e com a combinação de vários mecanismos
catalíticos que iremos agora descrever. Existem assim seis grupos de catálises
empregues pelas enzimas.
19
Ø Catálise ácido-base:
A catálise ácida é, geralmente, um processo no qual protões parciais se
transferem de um ácido e vão baixar a energia de transição de uma reacção.
Uma reacção pode também ser estimulada por catálise básica se a sua
velocidade for aumentada por remoção parcial de protões por uma base.
Algumas reacções podem ser simultaneamente sujeitas aos dois processos.
Muitas reacções bioquímicas são susceptíveis a catálise ácido-base; as cadeias
laterais de alguns resíduos proteicos possuem pK’s perto do pH fisiológico, que
vai assim permitir que ajam como catalisadores ácidos ou básicos. Assim, a
habilidade das enzimas em arranjarem vários grupos catalíticos em volta do
seu substrato, faz com que a catálise ácido-base seja um mecanismo de
catálise enzimática bastante comum. Logo, vem que a actividade catalítica
destas enzimas é sensível ao pH, já que o pH influencia o estado de
protonação das cadeias laterais do centro activo.
Ø Catálise covalente:
A catálise covalente acelera a reacção através da formação de uma ligação
covalente catalisador-substrato. Usualmente, esta ligação covalente é formada
pela reacção de um grupo nucleófilo catalizador com um electrófilo no
substrato.
A catálise covalente pode ser decomposta em três partes:
1. A reacção nucleófila entre o catalisador e o substrato para formar uma
ligação covalente;
2. A troca de electrões do centro da reacção com o agora electrofílico
catalisador;
3. A eliminação do catalisador, uma reacção que é essencialmente a inversa
de 1.
Um aspecto importante da catálise covalente é que quanto mais estável for a
ligação covalente formada, menos facilmente se decompõe a reacção nos seus
passos finais. Assim, vem que uma boa catálise covalente é aquela que
combina o poder nucleófilo com a habilidade de reverter formação dessa
mesma ligação, tal como o fazem certas coenzimas.
Ø Catálise metal-iónica:
Perto de um terço de todas as enzimas conhecidas necessitam da presença de
iões metálicos para a actividade catalítica. Este grupo de enzimas inclui as
metaloenzimas que contêm como cofactores iões metálicos (como o próprio
nome indica). As enzimas metal-activadas, em contraste, ligam metais iónicos
de soluções, usualmente metais alcalinos ou alcalino-terrosos.
Os iões metálicos participam no processo catalítico de três formas principais:
1. Ligando-se aos substratos, de maneira a orientá-los adequadamente
para a reacção;
2. Permitindo reacções redox, através de mudanças reversíveis nos seus
estados de oxidação;
3. Através de estabilização electrostática ou “blindando” cargas negativas.
20
Em muitas das reacções catalisadas por iões metálicos, estes vão funcionar
tal qual um protão, neutralizando uma carga negativa. No entanto, estes
iões têm a vantagem de poderem existir em mais altão concentrações a pH
neutro e de possuirem cargas superiores a +1.
Ø Catálise electrostática:
A ligação de um substrato geralmente exclui a água do centro activo duma
enzima. Assim, pode dizer-se que o centro activo tem as características polares
dum solvente orgânico, onde as interacções electrostáticas são muito mais
fortes que numa solução aquosa. Depois de muito estudo, verificou-se que as
distribuições de carga à volta do centro activo de uma enzima estão arranjadas
de maneira a estabilizar os estados de transição das reacções catalisadas. Por
outro lado, em muitas enzimas as distribuições das cargas vão,
aparentemente, guiar substratos polares aos sítios da ligação, aumentando
assim a velocidade da reacção.
Ø Catálise através de proximidade e efeitos de orientação:
Embora os mecanismos catalíticos das enzimas se assemelhem aos modelos
das reacções orgânicas, são muito mais eficientes do que estes últimos. Tal
eficiência deve advir das condições físicas específicas nos centros activos das
enzimas, que promovem a correspondente reacção química.
Os efeitos mais óbvios são proximidade e orientação: os reagentes têm de se
“unir” à enzima com a relação espacial própria, de forma a poder dar-se a
reacção. Assim, por simplesmente ligarem os seus substratos, as enzimas
facilitam a reacção em três aspectos:
1. As enzimas levam os substratos ao contacto com os seus grupos
catalíticos;
2. As enzimas ligam os seus substratos na orientação adquada para a
reacção;
3. As enzimas param as deslocações de translacção e rotação dos
substratos e grupos catalíticos. Este aspecto é importante pois favorece o
aparecimento do estado de transição, onde os movimentos relativos aos
compostos são mínimos.
Ø Catálise por preferência de ligação do estado de transição:
Até agora ainda não se considerou um dos mais importantes mecanismos de
catálise enzimática: um enzima pode ligar o estado de transição da reacção
que catalisa com maior afinidade que os substratos ou produtos. Assim, vem
que as enzimas que se ligam preferencialmente ao estado de transição
aumentam a concentração deste, aumentando assim proporcionalmente a
velocidade da reacção.
Por este facto, os estados de transição análogos são inibidores da reacção,
uma vez que a enzima os “agarra” como se fossem a molécula a catalisar,
21
inactivando-a. Acontece por vezes que estes análogos tenham maior afinidade
com a enzima do que a molécula que pretendemos catalizar.
22
Propriedades cinéticas de enzimas ( Modelo de Michaelis – Menten)
Geralmente, a velocidade de catálise V varia com a concentração do
substrato [S]. De tal forma que, para uma concentração fixa de enzima, V é
quase linearmente proporcional a [S], quando [S] é pequena. Por outro lado,
quando a concentração [S] tem valores elevados, a velocidade de catálise é
practicamente independente de [S].
O modelo proposto por Michaelis-Menten explica as propriedades
cinéticas das enzimas.
E + S ↔ ES → E + P
Figura 1 – Progressão das curvas para uma reacção simples catalizada por uma enzima. Com
excepção da fase inicial da reacção, os declives das curvas de [E] e [ES] são essencialmente zero
enquanto [S] >> [E].
Uma enzima E combina-se com o substracto S para formar o complexo
ES, com uma constante de velocidade K 1. O complexo ES pode dissociar-se em
E e S, com uma constante de velocidade K 2, ou pode prosseguir para formar o
produto P, com uma constante de dissociação K3.
A equação que explica as propriedades cinéticas das enzimas é a
equação de Michaelis-Menten :
V=Vmáx* [S]/( [S] + Km )
( 1 ),
sendo Km=(K2+K1)/K3 a constante de Michaelis.
De acordo com a equação, verifica-se que para concentrações muito
baixas de S, quando [S] muito menor que KM, V=[S] V máx/KM; ou seja , a
velocidade é directamente proporcional a [S]. Para concentrações muito
elevadas de substrato, quando [S] é muito maior do que K M, V=Vmáx; ou seja, a
velocidade máxima é independente da concentração do substrato. Quando a
concentração de substrato S é igual ao valor de K M, a velocidade de reacção é
metade da sua velocidade máxima; isto é, V=½ Vmáx.
23
Figura 2 – O gráfico de V0 (velocidade inicial) de uma reacção enzimática simples vs. [S]
Os valores de V máx e KM podem ser determinados fazendo variar a
concentração de S a partir da linearização de Lineweaver-Burk, a qual
transforma a equação de Michaelis-Menten num gráfico em linha recta de 1/V
em função de 1/[S] , o qual intersecta o eixo de 1/V no ponto 1/Vmáx com uma
inclinação de KM/Vmáx.
1/V=1/Vmáx+(1/[S])*KM/Vmáx ( 2 )
Figura 3 – Gráfico de reciprocidade dupla (Lineweaver-Burk). As barras de erro representam ±
0.05Vmax .
Influência do pH na actividade enzimática
A actividade catalítica aumenta, á medida que o pH aumenta. No
entanto, ao atingir um determinado valor de pH, a actividade catalítica atinge
o seu máximo - pH óptimo. A partir deste valor de pH, a actividade catalítica
das enzimas começa a diminuir, dado que valores pH muito elevados originam
a desnaturação das proteínas.
24
Figura 4 – Gráfico da influência do pH na actividade enzimática
Influência da Temperatura na actividade enzimática
À medida que a temperatura aumenta, a actividade catalítica
aumenta também, até atingir um determinado valor - Temperatura Óptima. A
partir desta tempe ratura, que corresponde ao valor máximo de actividade
enzimática, a actividade catalítica começa a diminuir, pois, a temperaturas
elevadas inicia-se a desnaturação térmica das proteínas.
-Figura 5 – Gráfico da influência da Temperatura na actividade enzimática
Inibição enzimática
As enzimas podem sofrer dois tipos de inibição: a inibição
irreversível, na qual o inibidor se dissocia muito lentamente da “enzima-alvo”;
25
e inibição reversível, que é caracterizada por uma dissociação rápida do
complexo enzima-inibidor.
A inibição reversível pode ser de três tipos :
Inibição competitiva : a enzima pode ligar-se ao substrato ( formando
um complexo ES ) ou ao inibidor ( formando o complexo EI ), porém nunca se
pode ligar aos dois ( ESI ).
Um inibidor competitivo é semelhante ao substrato, ligando-se ao
centro activo da enzima, impedindo assim que o substrato se ligue ao centro
activo da enzima. Portanto, este tipo de inibidor diminui a velocidade de
catálise, reduzindo a proporção de moléculas de enzima ligadas a um
substrato.
- Figura 6
-
-Figura 7-
-Figura 81 -
-Figura 92 -
Inibição não competitiva : o substrato e o inibidor podem ligar-se à
enzima em simultâneo. Então, um inibidor não competitivo age pela
diminuição do número de renovação, em detrimento da diminuição da
quantidade de complexos ES.
1
Gráfico de V0 em função de [S] para a reacção Michaelis -Menten na presença de diferentes
concentrações de inibidor competitivo.
2
Gráfico Lineweaver-Burk correspondente à Fig. 8.
26
- Figura 10 -
- Figura 11-
-Figura 12 – Gráfico Lineweaver-Burk da
enzima Michaelis-Menten na presença de
um inibidor não -competitivo.
Inibição mista : um inibidor tanto afecta a ligação do substrato,
quanto altera o número de renovação.
-Figura 13-
-Figura 14 – Gráfico Lineweaver-Burk da
enzima Michaelis-Menten na presença de
um inibidor misto.
As inibições competitivas são cinéticamente distinguíveis das não
competitivas, ou seja, na presença de um inibidor competitivo a equação ( 2 ) é
substituída por
1/V = 1/ Vmáx +(KM/Vmáx) * (1/[S]) * (1+ [I]/ KI )
(3)
27
Na qual [ I ] é a concentração do inibidor e KI é a constante de
dissociação do complexo E-I ( KI = [E] * [S] / [EI] ).
Na inibição não competitiva , o valor de Vmáx diminui, sendo o seu
valor dado pela equação :
V = Vmáx / ( 1+ [I]/KI ) ( 4 )
Introdução ao Metabolismo:
O metabolismo, o processo no qual sistemas vivos adquirem e usam energia
para executar as suas variadas funções, pode ser dividido em duas partes:
1. Catabolismo: ou degradação, na qual os nutrientes e os constituintes
celulares são “partidos” nos seus componentes ou para gerar energia;
2. Anabolismo: ou biossíntese, no qual biomoléculas são sintetizadas a
partir de compostos simples.
O Metabolismo:
Ø Estratégias “Trophicas”:
Os requerimentos nutricionais dum organismo refletem-se na sua fonte de
energia metabólica. Alguns procariontes são autotróficos, ou seja, conseguem
sintetizar todos os seus constituintes celulares a partir de moléculas simples
(H2O, CO2, NH3, etc.); estes seres podem assim obter a sua energia através de
duas maneiras: os chemolitotróficos, que obtêm energia através da oxidação
de compostos inorgânicos e os fotoautotróficos, que obtêm energia via
fotossíntese.
Os heterotróficos, por seu lado, obtêm a energia pela oxidação de compostos
orgânicos, dependendo assim dos autotróficos para a obtenção dessas
substâncias.
Ø Caminhos metabólicos:
Os “caminhos” metabólicos são séries de reacções enzimáticas interligadas,
que produzem produtos específicos. Em geral, processos catabólicos e
anabólicos estão relacionados da seguinte maneira: nos processos catabólicos,
complexos compostos (metabólitos) são partidos em produtos mais simples; a
energia livre libertada neste processo é conservada pela consequente síntese de
ATP a partir de ADP+Pi, ou pela redução da coenzima NADP+ para NADPH.
Assim sendo, vem que o ATP e o NADPH são as mais importantes fontes de
energia para as reacções anabólicas. A acetil-coenzima A participa na maior
parte dos processos catabólicos.
Uma das características do metabolismo degradativo é que os caminhos desse
catabolismo para um grande número de substâncias convergem em poucos
campos intermediários comuns:
28
No caso dos eucariontes, cada processo vai ocorrer num organito celular
característico; nos procariontes, como não têm organitos celulares, os
processos ocorrem num sítio característico do citosol.
Assim, nos eucariontes, para que haja a síntese dos vários metabólitos, há que
haver
mecanismos que transportem as substâncias entre os vários
compartimentos celulares. Existem assim proteínas “transportadoras” que são
responsáveis por este processo.
Ø Considerações Termodinâmicas:
1. Os mecanismos metabólicos são irreversíveis. Um reacção
altamente exergónica é irreversível; se se tratar de uma reacção
em vários passos, se um deles for irreversível, então todo o
processo se torna irreversível;
2. Todos os mecanismos metabólicos têm um primeiro passo
cometido. Embora a maioria das reacções metabólicas estejam
29
muito próximas do equilíbrio química, há quase sempre uma
reacção exergónica irreversível num dos primeiros passos do
metabolismo.
3. Os mecanismos catabólico e anabólico são diferentes. Se um
metabolito é convertido noutro metabolito por um processo
exergónico, tem que ser fornecida energia livre para converter o
segundo metabolito novamente no primeiro e assim diferentes
caminhos de reacção têm de ser tidos em conta pelo menos
nalguns passos. Se uma célula requere o metabolito 2, tem de se
“desligar” o caminho de 2 para 1 e “ligar” o caminho de 1 para 2.
Ø Controlo do Fluxo Metabólico:
Este controlo é feito no intuito de manter o organismo num estado mais ou
menos constante, o que é necessário, tendo em conta que os organismos vivos
são sistemas termodinâmicos abertos. Este controlo pode ser feito através de
vários mecanismos que contolam o fluxo através do passo determinante da
reacção:
1. Controlo alostérico: Muitas enzimas são reguladas alostericamente,
geralmente por substratos, produtos ou coenzimas.
2. Modificação covalente: Muitas enzimas controlam os fluxos de vários
“ciclos”. Possuem sítios específicos que podem ser fosforilizados ou
desforforilados enzimáticamente ou modificados covalentemente; estas
alterações vão alterar a actividade das enzimas em causa.
3. Ciclos de substrato: Se vf e vr representarem as taxas de duas reacções
opostas em não-equilíbrio que são catalizadas por diferentes enzimas, vf
e vr podem ser independentemente variadas. Por exemplo, o fluxo (vf-vr)
pode ser aumentado não apenas acelerando a reacção directa mas
também abrandando a reacção inversa. Este tipo de controlo é mais
sensível às concentrações dos efectores alostéricos do que o fluxo
através de um a reacção simples em não-equilíbrio.
4. Controlo genético: A concentração de enzimas pode ser alterada por
síntese proteica em resposta às necessidades metabólicas. Este tipo de
controlo é a resposta mais lenta de todos os que vimos até ao momento;
Ø Compostos de “alta-energia”:
Nas várias reacções oxidativas que ocorrem no organismo, muitas delas
libertam uma quantidade considerável de energia. Estes pacotes de energia
são conservados pela síntese de poucos tipos de compostos “altamente
energéticos”, que pela quebra das suas ligações químicas irão libertar essa
energia que vai ser utilizada nos processos endoenergéticos.
- ATP e grupo de transferência fosfato:
O ATP (adenosina trifosfato), que aparece em todas as formas de vida
conhecidas, consiste numa adenosina (adenina+ribose) na qual três grupos
fosforil (-PO32-) estão sequencialmente ligados por uma ligação fosfoéster e
duas ligações fosfoanídricas.
A importância biológica do ATP assenta na grande quantidade de energia que
acompanha a quebra das suas ligações fosfoanídricas. De notar que a ligação
30
fosfoanídrica não difere muito da ligação fosfoéster, se olharmos para os seus
carácteres electrónicos. Então porque é que a ligação fosfoanídrica no ATP é
tão energética? Há três factores que dão resposta a esta pergunta:
1. Os requisitos electrónicos dos grupos fosforil são menos satisfeitos
numa ligação fosfoanídrica do que nos seus produtos de hidrólise.
2. Existe também o efeito de destabilização das repulsões electrostáticas
entre os grupos carregados duma ligação fosfoanídrica comparada com
os seus produtos de hidrólise.
3. Outra influência destabilizadora é a pequena energia de solvatação da
ligação fosfoanídrica, comparada com os seus produtos de hidrólise.
De nota que, como os produtos da hidrólise do ATP são iões, o ∆G desta vai
assim também depender do pH e da força iónica.
-Reacções Conjuntas:
As reacções exergónicas de “alta energia” podem ser combinadas a processos
endergónicos e assim completá-los. Exemplo:
(1) A+B ó C+D
(2) D+E ó F+G
∆G1
∆G2
NOTA: ∆G1+∆G2<0
Seja ∆G1≥0 e ∆G2<0; como ∆G1≥0, logo o produto D vai existir em baixa
concentração; mas como ∆G2<0, então aqui D é convertido em produtos,
gastando assim o excesso em (1) levando a que a reacção (1) se dê no sentido
directo de forma a repor D(Princípio de Le Chatelier).
- A hidrólise fosfoanídrica conduz alguns processos bioquímicos:
A energia das ligações fosfoanídricas pode ser usada para conduzir reacções
ao seu equilíbrio, mesmo quando os grupos fosforil não são transferidos para
outro composto orgânico. Assim, o ATP vai-se ligar a, por exemplo, proteínas,
levando-as assim a uma alteração conformacional; após isto, a hidrólise
exergónica do ATP e a consequente libertação de ADP e Pi transforma essas
alterações conformacionais em irreversíveis, levando a que o processo avance.
-A pirofosfatase inorgânica catalisa a quebra de ligações fosfoanídricas
adicionais:
Embora muitas reacções envolvendo ATP produzam ATP e Pi, outras produzem
AMP e PPi. Neste último caso o PPi é rapidamente hidrolisado para dois Pi pela
pirofosfatase inorgânica, levando a que a quebra do ATP pela pirofosfatase
consuma duas ligações fosfoanídricas.
31
Reacções redox:
Os combustíveis metabólicos são oxidados para CO2, os electrões são
transferidos para mensageiros moleculares que, em organismos aeróbicos,
transferem os electrões para o oxigénio molecular. O processo de transporte
electrónico resulta numa trans-membrana de concentração pr otónica que,
pelo gradiente de concentração, guia a síntese do ATP.
- NAD+ e FAD:
Dois dos mais conhecidos transportadores de electrões são as coenzimas
nucleotídicas NAD+ e FAD.
A NAD+ pode transportar um electrão, resultando desse transporte um
composto sob a forma de NADH. Por seu lado, a FAD é uma coenzima que
pode transferir um ou dois electrões sob a forma de FADH e FADH2,
respectivamente. As funções metabólicas destas coenzimas levam a concluir
que a sua redução é reversível, de forma a que possam aceitar electrões,
passá-los para outro transportador electrónico e serem regenerados para
participarem em novos ciclos de oxidação e redução. De notar que a FAD não
pode ser sintetizada pelos seres humanos, o que implica que a temos de
ingerir na nossa dieta.
Catabolismo da Glucose:
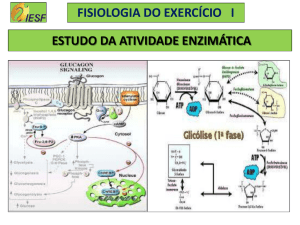
A glicólise é o processo que leva à “quebra” da glucose. É a sequência de dez
reacções enzimáticas, na qual uma molécula de glucose é convertida em duas
moléculas de gliceraldeído-3-fosfato. Dos três piruvatos carbónicos há o
aparecimento de 2 ATP’s. A glicólise é extremamente importante pois tem um
papel de destaque no fornecimento de energia sob a forma de ATP e na
preparação da glucose e outros composto para seguintes degradações
oxidativas.
A glicólise pode ser decomposta em duas partes:
-Parte I (Investimento energético – reacções 1-5): Na sua fase inicial a
glucose é fosforilada e partida de forma a “aguentar” duas moléculas de
fosfato. Este processo consome 2 ATP’s.
-Parte II (Recuperação energética – reacções 6-10): As duas moléculas de
gliceraldeído-3-fosfato são convertidas em piruvato, com a redução de 4 ATP’s.
Assim a glicólise vai ter um balanço total positivo de 2 ATP’s, visto que na fase
inicial são gastos dois ATP’s.
32
As reacções da glicólise:
33
- Hexocinase: Primeira utilização do ATP
A reacção 1 da glicólise é a transferência de um grupo fosforil do ATP para a
glucose, formando glucose-6-fosfato (G6P), numa reacção catalisada pela
enzima hexocinase.
Há que notar que, para que a actividade da enzima seja adequada, tem que
haver a presença do ião Mg 2+, pois um ATP não complexado é um potencial
inibidor da hexocinase; assim, o Mg 2+ vai funcionar como um escudo para as
cargas negativas dos ATP’s, fazendo com que o γ-fósforo seja mais acessível ao
ataque nucleófilo do grupo C6-OH da glucose.
- Isomerase fosfoglucosídica:
A reacção 2 da glicólise é a conversão do G6P em frutose-6-fosfato (F6P),
através da acção da enzima isomerase fosfoglucosídica (transformação de uma
hexose numa pentose).
O mecanismo reaccional proposto envolve catálise ácido-base pela enzima:
1.
2.
3.
4.
O substrato liga-se.
Um ácido enzimático vai catalisar a abertura do anel da hexose.
Uma base abstrai o protão ácido formando um cis-enolato.
O protão é reposto em C1. Os protões abstraídos pelas bases
rapidamente trocam com os protões do solvente.
5. O anel fecha-se de maneira a formar o produto que é consequentemente
libertado da enzima, completando o ciclo catalítico.
-Fosfofrutocinase: segunda utilização de ATP
A reacção 3 da glicólise é catalisada pela enzima fosfofrutocinase. Esta última
fosforila o F6P, formando fructose-1,6-bifosfato (FBP ou F1,6P).
Esta reacção é semelhante à reacção 1 e também se dá na presença do ião
Mg2+.
-Aldolase:
A reacção 4 da glicólise é catalisada pela aldolase e é onde há a quebra do FBP
para
formar
duas
trioses:
gliceraldeído-3-fosfato
(GAP)
e
dihidroxoacetonofosfato (DHAP).
-Isomerase Fosfotriose:
Apenas o GAP continua o processo glicolítico. No entanto, como o GAP e o
DHAP são isómeros funcionais aldeído-cetona, podem ser interconvertidos por
uma reacção de isomerização por intermédio de um enodiol. A triosefosfato
isomerase catalise este processo: reacção 5 da glicólise.
34
-Gliceraldeído-3-fosfato desidrogenase: primeira formação energética:
A reacção 6 da glicólise é a oxidação e fosforilação do GAP pelo NAD+ e Pi,
catalisada pela enzima gliceraldeído-3-fosfato desidrogenase. Nesta reacção
(oxidação aldeídica), uma reacção exergónica conduz à síntese do altamente
energético 1,3-bofosfoglicerato (1,3-BPG).
-Fosfoglicerato-cinase: primeira formação de ATP:
Na reacção 7 da glicólise forma-se 1 ATP, sobrando 3-fosfoglicerato (3PG),
numa reacção catalisada pela enzima fosfoglicerato-cinase. De notar a ligação
Mg2+-ADP.
-Fosfoglicerato-mutase:
Na reacção 8 da glicólise 3PG é convertido em 2-fosfoglicerato (2PG) pela
enzima fosfoglicerato-mutase. Uma mutase cataliza a troca intramolecular de
um grupo funcional de uma posição para outra.
-Enolase: segunda formação energética:
Na reacção 9 o 2PG é desidratado para fosfoenolpiruvato (PEP), numa reacção
catalisada pela enolase.
-Pirovato-cinase: segunda formação de ATP:
Na última reacção da glicólise, a enzima pirovato-cinase junta a energia livre
resultante da “quebra” do PEP, sintetizando ATP e formando piruvato.
Controlo da glicólise:
Em condições constantes, a glicólise opera continuamente, embora o fluxo
glicolítico tenha de variar de acordo com as necessidades do organismo. O
controlo dos mecanismos de fluxo, como o da glicólise, envolve m três passos:
1. Identificação da velocidade de cada patamar do ciclo, medindo “in-vivo”
o ∆G de cada reacção.
2. Identificação “in-vitro” de modificadores alostéricos das enzimas
envolvidas.
3. Medição “in-vivo” dos níveis dos supostos reguladores, debaixo de
condições variadas.
35
Ciclo do ácido cítrico (Ciclo de Krebs):
O ciclo do ácido cítrico é uma engenhosa série de oito reacções que oxida o
grupo acetil da acetil-CoA para duas moléculas de CO2 de uma maneira tal
que conserva a energia libertada nos compostos reduzidos NADH e FADH2. Um
ciclo completo liberta duas moléculas de CO2, três NADH, um FADH2 e um
composto altamente energético: ATP+GTP.
36
-Síntese da acetil-coenzima A:
A acetil-coenzima A é formado do piruvato através de descarboxilação
oxidativa por um complexo multienzimático (grupo de enzimas associadas
entre si não covalentemente que catalizam dois ou mais passos sequenciais de
um processo metabólico) chamado desidrogenase pirúvica. Este complexo
possui múltiplas cópias de três enzimas: E1, E2 e E3.
O complexo desidrogenase pirúvica cataliza cinco reacções sequenciais:
- O piruvato é oxidado a acetato, com a libertação de CO2.
- Alguma energia da oxidação é conservada pela redução de NAD+ a NADH.
- Parte da energia restante é armazenada temporariamente, adicionando a
molécula de CoA ⇒ Acetil-CoA.
A sequência das reacções é a seguinte:
1.
2.
3.
4.
5.
Descarboxilação do piruvato (libertação de CO2).
O carbono hidroxietil é oxidado para um grupo acetil.
O grupo acetil é transferido para a CoA.
Oxidação da desidrolipoamida, para regenerar o grupo lipoamida da E2.
A E3 é reduzida e reoxidade, produzindo NADH.
Enzimas do ciclo do ácido cítrico:
-Reacção 1: Citrato sintase:
A enzima citrato sintase medeia a reacção da Acetil-CoA e oxaloacetato a
citrato.
-Reacção 2: Aconitase:
Mediadora da reacção de isomerização do citrato a isocitrato.
-Reacção 3: Isocitrato desidrogenase:
Esta enzima conduz à descarboxilação oxidativa do isocitrato a αcetoglutarato.
-Reacção 4: α -cetoglutarato desidrogenase:
Esta reacção é a reacção de descarboxilação oxidativa do α-cetoglutarato a
succinil-CoA.
37
-Reacção 5: Succinil-CoA sintetase:
Hidrólise da succinil-CoA a succinato por fosforilação do GTP.
-Reacção 6: Succinato desidrogenase:
Desidrogenação estereospecífica do succinato a fumarato.
-Reacção 7: Fumarase:
Hidratação estereospecífica do fumarato a L-malato.
-Reacção 8: Malato desidrogenase:
Oxidação do L-malato a oxaloacetato.
Regulação do ciclo do ácido cítrico:
A disponibilidade de substratos, a inibição pelos produtos e a inibição por
feedback por outros intermediários do ciclo, influenciam a operacionalidade
deste ciclo.
O ciclo á assim regulado por mecanismos de feedback, que coordenam a
produção de NADH com o gasto energético.
-Balanço global em termos de número de moléculas de ATP produzidas por
molécula de glucose:
Os cofactores NADH e FADH2 são reoxidados na cadeia de transporte
electrónico, com a redução de O2 a H2O.
A energia do transporte electrónico é conservada através da síntese de ATP
pela fosforilação oxidativa:
- Por cada NADH são produzidas cerca de 3 moléculas de ATP.
- Por cada FADH2 são produzidas cerca de 2 moléculas de ATP.
Assim, vem que, em condições aeróbias:
38
-Natureza Anfibólica do ciclo de Krebs:
Um trajecto metabólico ou é catabólico ou é anabólico. O ciclo de Krebs é
catabólico, pois envolve degradação e é um importante sistema de conservação
energético nos organismos. No entanto, muitos dos produtos intermediários
deste ciclo são usados em biossíntese, ou seja, em reacções anabólicas. O ciclo
de Krebs é assi anfibólico (catabólico e anabólico em simultâneo).
Reacções anapleróticas-> reacções que permitem repôr os inetermediários do
ciclo que forem sendo consumidos.
39
Transporte electrónico e fosforilação oxidativa:
Os organismos anaeróbicos consomem oxigénio e geram dióxido de carbono
num processo de oxidação dos “combustíveis” metabólicos. A completa
oxidação da glucose, por exemplo, pelo oxigénio:
C6H12O6 + 6O2 → 6CO2 + 6H2O
Esta reacção pode ser separada em duas “meias” reacções que a máquina
metabólica leva a cabo. Na primeira dá-se a oxidação dos átomos de carbono
da glucose (glicólise e ciclo de Krebs):
C6H12O6 + 6H2O → 6CO2 + 24H+ +24e Na segunda, o oxigénio molecular é reduzido:
6O2 + 24H+ + 24e - → 12H20
Assim, verificou-se que os 12 pares electrónicos libertados durante a oxidação
da glucose não são directamente transferidos para o O2, são é transferidos
para as coenzimas NAD+ e FAD, formando 10NADH e 2 FADH2. Os electrões
depois passam para a cadeia de transporte electrónico mitocondrial, um
sistema de percursos electrónicos interligados.
40
-A mitocôndria:
Nos eucariontes, a fosforilação oxidativa ocorre na mitocôndria. A mitocôndria
é um organelo celular que possui uma membrana externa permeável à maioria
das moléculas pequenas e uma outra membrana interna, que contém uma
vasta área de invaginações. O número de invaginações chama-se cristae e
reflecte a actividade respiratória da célula, pois é aí que se dá o transporte
electrónico. Esta membrana interna divide a mitocôndria em dois
compartimentos, o espaço intermembranar e a matriz interna.
-Fosforilação oxidativa:
A fosforilação oxidativa é um processo pelo qual é formado ATP quando os
electrões são transferidos do NADH e do FADH2 para o O2, através de uma
série de transportadores electrónicos.
A fosforilação ocorre na membrana interna da mitocôndria. O ciclo de Krebs e
a oxidação dos ácidos gordos, que fornecem a maior parte dos cofactores
reduzidos, dão-se na matriz mitocondrial. A oxidação do NADH origina 3
ATP’s, enquanto que a oxidação do FADH2 origina 2 ATP’s.
A oxidação e a fosforilação são processos acoplados.
A transferência electrónica dá-se passo a passo, do NADH ou FADH2 para o O2,
através de uma série de transportadores electrónicos (I, III, IV), o que produz
um bombardeamento de protões para fora da matriz mitocondrial. Gera-se
assim uma força, que consiste num gradiente de pH e num potencial
electroquímico transmembranar.
O ATP é assim sintetizado quando os protões voltam a entrar na matriz
mitocondrial através de um complexo enzimático (V-ATPase).
A cadeia respiratória consiste em 3 complexos enzimáticos (I, III, IV), ligados
por dois transportadores electrónicos: CoQ e Citocromo C.
41
-O transporte electrónico:
Complexo I → NADH – CoQ reductase
NADH + CoQ (ox) → NAD+ + CoQ (red)
NOTA: O complexo II (succinato-CoQ-reductase) também participa nesta
reacção redox.
Complexo III → CoQ – Citocromo C – reductase
CoQ (red) + Cit C (ox) → CoQ (ox) + Cit C (red)
Complexo IV → Citocromo C oxidase
Cit C (red) + ½ O2 → Cit C (ox) + H2O
Fosforilação Oxidativa:
A energia livre libertada no transporte electrónico é conservada de modo a ser
utilizada pela ATP sintetase. Tal conservação energética é chamada de
acoplamento energético de transducção de energia.
Vamos então explorar este mecanismo de acoplamento e a operação da ATP
sintetase.
Ø Hipótese Quimiosmótica:
Esta teoria pressupõe que á acoplamento do transporte electrónico gerando a
síntese de ATP devido à criação de um gradiente protónico na membrana
mitocondrial interna.
Dados experimentais que comprovam a Teoria de Mitchell:
1. A fosforilação oxidativa requer membranas intactas.
2. A membrana mitocondrial interna é impermeável a iões, pois a sua livre
difusão iria descarregar o gradiente electroquímico.
3. Durante o transporte electrónico é gerado um gradiente protónico
através da membrana.
4. Compostos que aumentam a permeabilidade da membrana a H+,
dissipando assim o gradiente electroquímico (desacopladores),
permitindo o transporte electrónico, mas parando a síntese de ATP.
5. O aumento artificial da acidez no exterior da membrana estimula a
síntese do ATP.
Ø Força protomotriz:
Força protomotriz = Gradiente Químico (∆
∆ pH) + Potencial da membrana (E m)
42
∆ G= 2.3 RT ∆ pH + Z ∆ Ψ F
Z – carga do protão
∆Ψ- potencial da membrana
F- constante de Faraday
∆ pH = (pH matriz) – (pH citosol)
-Desacoplamento da fosforilação oxidativa:
Este desacoplamento é efeito de desacopladores (DNP, FCCP), que são ácidos
fracos, lipofílicos, que atravessam a membrana na forma protonada,
dissipando assim o gradiente protónico e destruindo o acoplamento entre o
transporte electrónico e a fosforilação oxidativa, libertando energia na forma
de calor.
-Desacoplamento da fosforilação oxidativa induzido hormonalmente:
1.
2.
3.
4.
5.
6.
A norepinefrina liga-se ao receptor
O receptor estimula a adenilato ciclase a produzir cAMP
O cAMP activa a cinase cAPK
A cAPK fosforila a lipase
Dá-se a hidrólise dos triglicéridos
Desacoplamento e entrada de H+.
Metabolismo do glicogénio e gluconeogénese:
O glicogénio funciona nos animais como uma reserva de glucose (polimerizada)
de fácil mobilidade, sendo uma constante fonte de glucose/energia para todos
os tecidos. O glicogénio é armazenado principalmente no fígado.
O glicogénio é assim um polímero de glicose, com ligações α-1,4 em cadeia e
ligações α-1,6 para as ramificações (cada ramificação tem de 8 a 12 resíduos).
Ø Porquê glicogénio?
1. Regulação dos níveis de glucose no sangue, pois o gerado pelo
metabolismo das gorduras é insuficiente;
2. Libertação da glucose entre refeições e durante a actividade muscular,
pois os músculos não mobilizam a gordura tão rapidamente como o
glicogénio;
43
Ø Porquê estrutura ramificada?
1. Maior solubilidade;
2. Maior número de pontos de metabolização, ou seja, vai possuir vários
extremos não redutores por onde a remoçã sequencial se dá, tendo por
seu lado apenas um extremo redutor.
Degradação do Glicogénio.
A degradação do glicogénio dá-se através de um processo que inclui 3
enzimas: glicogénio fosforilase, transferase e fosfoglucomutase.
1. Glicogénio fosforilase:
Glicogénio + Pi ó Glicogénio + G1P (Glucose 1-P)
(n resíduos)
(n-1 resíduos)
2. “Debranching enzyme” (transferase)
Esta enzima remove as ramificações do glicogénio, tornando os resíduos de
glicose acessíveis para a acção da enzima glicogénio fosforilase.
44
3. Fosfoglucomutase:
Converte G1P em G6P, que pode ter vários destinos metabólicos.
Síntese do Glicogénio:
Existem caminhos diferentes para a degradação e para a biossíntese.
Biossíntese:
1. Activação da Glucose 1-P:
G6P → G1P (reacção mediada pela enzima mutase)
G1P + UTP ⇔ UDGP (uridinadifosfato glucose) + PPi
2. Glicogénio sintetase:
Formação de ligações glicosídicas α-1,4 no glicogénio
UDP-Glucose + Glicogénio (n resíduos) → UDP + Glicogénio (n+1 resíduos)
Mas, é de notar que esta reacção apenas prolonga uma cadeia de glicogénio já
existente. Como se inicia então a sítese do glicogénio:
Ø Uma glucosiltransferase tirosina junta uma glucose a uma tirosina
duma proteína chamada glicogenina; então, a glicogenina autocataliza a
extensão da cadeia de glucose através da enziza glicogénio sintetase, até
sete resíduos.
45
3. Ramificação da cadeia de glicogénio:
Quebra de ligações glicosídicas α-1,4 e formação de ligações α-1,6, através
da enzima amilo (1,4→1,6) transglicosilase.
Controlo do metabolismo do glicogénio:
A síntese e degradação do glicogénio são exergónicos em condições fisiológicas,
logo, tal facto torna a sua regulação importante, de acordo com as
necessidades celulares. Assim, a glicogénio fosforilase entra em “competição”
com a glicogénio sintetase, consoante as necessidades do organismo em
questão.
O controlo dá-se por:
1. Regulação alostérica: controlo da concentração de cada uma
das enzimas anteriores por inibidores.
2. Modificações covalentes, por controlo hormonal:
-
Insula, situado no fígado, estimula a síntese do glicogénio.
Epinefrina (músculo) e glucagon (fígado), estimulam
degradação do glicogénio, mediada pelo AMP cíclico (cAMP).
a
3. Regulação em cascata: fosforilação/defosforilação/controlo
hormonal.
46
Assim, vem que a cAMP controla a percentagem de enzima na forma
fosforilada, o que implica uma maior velocidade de activação da enzima e a
consequente menor velocidade de desactivação.
Para uma taxa de glucose baixa no sangue, temos:
Pâncreas segrega glucanona
↓
Receptores da glucanona no fígado activam a adenilato ciclase
↓
Aumento da cAMP
↓
Aumento da ve locidade de degradação do glicogénio
↓
Aumento da G6P intra celular
↓
G6P + H2O
↓
Glucose + Pi
↓
Sangue
Para uma taxa alta de glucose no sangue, temos:
Pâncreas segrega insulina
↓...
Diminui cAMP
↓
Aumenta a velocidade de síntese do glicogénio
47
Gluconeogénese:
Questão central: Substituir as três reacções irreversíveis da glicólise.
48
Regulação da glucanogénese:
- Para níveis elevados de glucose no sangue:
Síntese de glicogénio
↓
Activação da glicólise
↓
Produção de Acetil-CoA
↓
Biossíntese de ácidos gordos e armazenamento de gorduras
- Para níveis baixos de glicose no sangue:
Estimulada a degradação do glicogénio
↓
Estimulada a gluconeogénese
↓
Maior produção de glucanona
↓
Maior produção de cAMP
↓
Fosforilação das enzimas
↓
Activação do FBP-2
(inactivação do PKF-2)
↓
Maior concentração de F2,6P
↓
Inibição da fosfofrutocinase (PFK) activada a frutosebifosfatase (FBP)
↓
Aumento da gluconeogénese