1.
O que são xenobióticos
Compostos químicos estranhos a um organismo ou sistema biológico. Podem
ser encontrados num organismo mas não são normalmente produzidos ou
esperados nesse organismo. O corpo humano é exposto diariamente a
inúmeros xenobióticos:
Componentes alimentares
Toxinas ambientais
Fármacos / drogas
O corpo humano está exposto ao longo da vida a uma grande quantidade de
xenobióticos provenientes de alimentos, toxinas ambientais, ou medicamentos,
entre outros, tendo desenvolvido complexos mecanismos enzimáticos para
proceder à destoxificação dessas substâncias. Estes mecanismos apresentam
grande variabilidade de indivíduo para indivíduo e são afectados por
variadíssimos factores, entre eles o estilo de vida e as influências genéticas.
Variabilidade individual
Afectados por diversos factores: ambiente, estilo de vida, constituição
genética
Evidências da literatura mostram correlação entre a destoxificação e
algumas doenças: cancro, doença de Parkinson, fadiga crónica,
alterações do sistema imunitário
Os conhecimentos sobre os sistemas de destoxificação e os mecanismos
da sua regulação sugerem que a eficiência na destoxificação de
xenobióticos pode influenciar o aparecimento de doenças crónicas.
A disfunção hepática prejudica a destoxificação
2. Metabolismo dos xenobióticos
O órgão onde ocorre o metabolismo dos fármacos é o fígado, por
apresentar várias enzimas ou complexos enzimáticos especializados. Dentre
elas, destacam-se as mono-oxigenases do complexo enzimático CYP, as
redutases, as esterases e as transferases. Outros locais do metabolismo dos
fármacos são as células epiteliais do tracto gastrointestinal, pulmões, rins e
pele.
O metabolismo de xenobióticos envolve a absorção, a distribuição, a
biotransformação e a excreção. A absorção ocorre após a inalação ou a
ingestão da substância tóxica. A menos que um fármaco actue topicamente ou
seja, no seu próprio local de aplicação, ele deve inicialmente penetrar no
sangue para depois ser distribuído para o seu local de acção. A mera presença
do fármaco no sangue, contudo, não provoca uma resposta farmacológica; para
que seja eficaz, o fármaco deve deixar o espaço vascular e penetrar nos
espaços intracelulares e/ou extracelulares (distribuição).
Após ser absorvido na corrente sanguínea o fármaco distribui-se para os
líquidos extra e intracelular. Os xenobióticos lipossolúveis interagem muito
prontamente com os lípidos componentes das biomembranas e não podem ser
excretadas, a não ser que passem por um processo de aumento da sua
solubilidade em água - A biotransformação, que é a transformação enzimática
dos fármacos em metabólitos com características mais hidrofílicas, tendo como
objectivo facilitar a excreção
destes pelo organismo. Está
dividida em 2 fases , fase I e
fase
II,
podendo
considerar-se
fase
uma
(exportação).
ainda
terceira
Os
processos das fases I e II são
independentes, ou seja, o
fármaco pode sofrer apenas
reacções de fase I ou de fase
II, ou as duas, sequencialmente.
A biotransformação de xenobióticos na fase I ocorre através de reações de
redução, hidrólise e/ou oxidação ocasionando sempre uma modificação
estrutural do fármaco, o que na maioria das vezes pode levar à sua inactivação.
No caso de administração de pró-fármacos, a fase I vai ser fundamental para
gerar a substância farmacologicamente activa.
Essas reações são responsáveis por expor ou introduzir grupos funcionais
relativamente reactivos no xenobiótico, principalmente o grupo hidroxilo, que
servirá, então, como ponto de ataque para o sistema conjugador, que fixa a ele
um substituto maior, como um grupo glucuronil, sulfato ou acetil.
A principal reação da fase I é a oxidação, catalisada principalmente pelos
citocromos P-450, e também realizada pelas monoamino oxidases (MAO) e
pelas flavinas monooxigenase (FMO). As reações de hidrólise são catalisadas
principalmente pelas epóxido hidrolases, peptidases e A-esterases. As
reduções são catalisadas por várias enzimas, dentre elas as carbonil redutases
e as glutatião redutases, como também por processos não enzimáticos através
de agentes redutores.
Os produtos da fase I, mais hidrossolúveis, podem ser excretados ou podem,
ainda, sofrer biotransformação na fase II cujas etapas frequentemente
envolvem reações de conjugação na tentativa de conjugar o fármaco com
substâncias endógenas, /*para facilitar a sua excreção. Estas reações de
conjugação são catalisadas pelas glutation S-transferases (GST), uridina
difosfoglicuronosil transferases (UGT) e sulfotransferases (ST).
Em múltiplas reações metabólicas de transferência de electrões em células
aeróbicas são formadas espécies reativas de oxigênio que causam peroxidação
de lipídios, alterações em proteínas e em ácidos nucléicos, produzindo danos às
células. A proteção contra estas ROS é feita pelas enzimas superóxido
dismutase, catalase, GST, glutation peroxidase selênio-dependente, aldo-ceto
redutase e enzimas de reparo do DNA.
3. Qual a função do Citocromo P450?
Citocromo P450 designa uma superfamília de enzimas, as mono-oxigensases,
que cataliza o metabolismo oxidativo de uma grande variedade de substratos
(reacções nas quais um átomo de oxigénio da molécula de O2 é incorporado na
molécula do substrato orgânico, o outro átomo é reduzido a H2O). O CYP é
uma proteína (tem grupo prostético heme ou grupo ferro-porfirina), e o seu
cofactor é o NADPH. É um sistema que está subdividido em três famílias (CYP1,
CYP2 e CYP3) e sete isoenzimas, sendo as mais importantes as CYP2C9, 2CA9 e
3A4.
Muitas formas de citocromo P450 catalizam a biotransformação de uma vasta
gama de agentes químicos ou xenobióticos, de entre os quais, fármacos de uso
terapêutico, nutrientes, produtos naturais e da indústria química, poluentes
ambientais, corantes, aditivos, conservantes, etc. No seu conjunto, os
citocromos P450 constituem o grupo mais versátil e abundante das chamadas
enzimas metabolizadoras de fármacos (drug metabolizing enzymes) e estão
presentes a níveis particularmente elevados no fígado.
Os citocromos P450 são produto de um vastíssimo nº de genes existente na
natureza, cuja expressão é condicionada pelo estado de desenvolvimento da
célula ou do organismo, pela identidade ou fenótipo de tecido em causa, bem
como pela acção de agentes reguladores, tanto endógenos como exógenos.
Com efeito, a expressão destes genes está sujeita a complexos mecanismos de
regulação. Os citocromos P450 actuam ao nível da chamada fase I ou fase de
activação do metabolismo de xenobióticos. Os progressos da tecnologia do DNA
recombinante aplicada ao estudo dos citocromos P450, a par da purificação e
análise da estrutura destas proteínas, têm tornado possíveis os mais recentes
avanços na identificação da diversidade de formas que compõem esta muito
vasta família. Durante os últimos anos, tem-se verificado igualmente uma
intensificação da investigação dos mecanismos que regualm a actividade dos
genes CYP. Pretende-se explicar a base molecular da especificidade de
expressão diferencial de cada gene citocromo P450, que se sabe variar em
função da natureza do tecido, do estádio de desenvolvimento, dos estados
hormonal e nutricional e ainda em muitos casos, da acção de agentes químicos,
indutores e/ou repressores. Estes genes CYP são caracterizados por um
acentuado polimorfismo com consequentes variações dos fenótipos de
expressão individuais.
Um dos aspectos intrigantes do CYP é que algumas das suas isoformas são
indutíveis e outras, não. A indução (aumento de produção de enzimas,
ocasionado pela administração de fármacos), no caso do CYP, é um processo
regulatório lento que pode reduzir a concentração de alguns fármacos no
plasma e aumenta a actividade de desintoxicação.
O uso de etanol e o tabagismo também podem causar a indução do CYP2E1 e
CYP1A,
respectivamente,
toxicológicas importantes
o
que
pode
ter
implicações
terapêuticas
e
INIBIÇÃO DO METABOLISMO DE XENOBIÓTICOS
Inibição por formação de complexos inactivos com o citocromo P450
Inibição por co-substratos competitivos/não competitivos
Inibição através da destruição do citocromo P450 hepático
Diminuição da síntese
Falta de cofactores
4. O que são interacções medicamentosas? Quais
os mecanismos?
Interação medicamentosa pode ser definida como a influência recíproca de
um medicamento com outra substância. Ou seja, quando um medicamento é
administrado isoladamente, produz um determinado efeito. Porém, quando este
é associado a outro medicamento, a alimentos ou a outras substâncias (como o
tabaco , drogas de abuso, ou mesmo substâncias que o paciente possa entrar
em contato, como inseticidas, produtos de limpeza, cosméticos etc.) ocorre um
efeito diferente do esperado, caracterizando uma interação.
Útil (benéfica)
Interacção
medicamentosa
Respostas
desfavoráveis
não previstas no
regime
terapêutico
(adversas)
Pequeno
significado
clínico
Potencialização do
efeito terapêutico
Nenhuma modificação
no efeito desejado do
medicamento
Respostas decorrentes
da interacção
Redução da eficácia
Aparecimento de
reacções adversas
Distintos graus de
gravidade
Na prática a questão das interações medicamentosas é complexa,
pois além das inúmeras possibilidades teóricas de interferência entre os
medicamentos, factores relacionados com o indivíduo (idade, constituição
genética, estado fisio-patológico, tipo de alimentação) e a administração do
medicamento (dose, via, intervalo e seqüência da administração) influenciam
na resposta do tratamento.
Interacções físicoquímicas
•As interações físico-químicas
ocorrem fora do paciente
pois, entre drogas diferentes
podem ocorrer numerosas
incompatibilidades, que
levam a reações quando
estas são misturadas em
infusão intravenosa, frascos
ou seringas, podendo
ocasionar a inativação dos
fármacos em questão
Interacções
terapêuticas
•Interacção Farmacocinética
•Interacção Farmacodinâmica
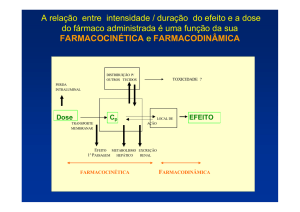
Interação Farmacocinética
As interações deste tipo interferem no perfil farmacocinético do
medicamento, podendo afetar o padrão de absorção, distribuição,
metabolização ou excreção. São interações difíceis de prever, porque
ocorrem com medicamentos de princípios activos não relacionados. Modificam a
magnitude e duração do efeito, mas a resposta final do medicamento é
preservada
Interação Farmacodinâmica
A interação farmacodinâmica causa modificação do efeito bioquímico ou
fisiológico do medicamento. Geralmente ocorre no local de acção dos
medicamentos
bioquímicos
(receptores
específicos,
farmacológicos)
sendo
capaz
de
ou
causar
(sinergismo) ou opostos (antagonismo).
GRUPOS DE RISCO
Idosos
Portadores de doenças crónicas
através
de
mecanismos
efeitos
semelhantes
Usuários de dispositivos para infusão de medicamentos intravenosos ou
de sonda enteral
Insuficientes renais, hepáticos, cardíacos ou respiratórios
Doentes com hipotireoidismo
Diabetes descompensada
5.Como é metabolizado o álcool?
Pequena molécula com grande capacidade de interacção;
Somente 2- 10% absorvido é eliminado inalterado (rins e pulmões)
90 – 98% oxidado no organismo, principalmente no fígado.
1. ÁLCOOL DESIDROGENASE (ADH)
2. SISTEMA MICROSSOMAL DE OXIDAÇÃO DO ETANOL (SMOE)
3. Catalase
Utilizada em menor quantidade, apenas quando há necessidade de
reduzir H2O2.
Acontece nos peroxissomos e não produz NADH
Interacções associadas ao alcool
Fármacos que inibem a aldeído desidrogenase (ALDH)
Os fármacos que inibem a oxidação do acetaldeído, por inibição da ALDH,
podem provocar uma reacção sistémica se administrados simultaneamente com
o álcool – reacção “tipo dissulfiram”.
O dissulfiram, usado para encorajar a abstinência em doentes alcoólicos que
decidiram parar de beber, ao ser administrado concomitantemente com o álcool
conduz a um aumento da concentração de acetaldeído, cujos efeitos adversos
são caracterizados por rubor, náuseas, vómitos, palpitações, hipotensão,
taquicardia, e cefaleias.
Mais raramente, poderá ocorrer uma reacção severa caracterizada por
colapso cardiovascular, convulsões ou morte.
Interacções farmacocinéticas ao nível do sistema hepático microssomal
O álcool pode interferir com os fármacos metabolizados pelas enzimas do
sistema hepático microssomal, interacção essa que dependerá da forma como o
álcool é consumido. No caso do consumo agudo há uma competição entre o
álcool e o fármaco ao nível microssomal, com diminuição do metabolismo do
fármaco resultando numa excreção diminuída dos produtos de metabolismo e
num aumento do risco de sobredosagem. O consumo crónico de álcool traduzse num aumento da actividade microssomal mesmo na ausência de álcool, com
aumento do metabolismo do fármaco e diminuição das suas concentrações
plasmáticas.