Farmacodinâmica
É o que a droga, que com fins terapêuticos nós chamamos de fármaco,
efetivamente faz. Quando a gente usa o ácido acetil salicílico, geralmente é para
febre e dor. É um inibidor da enzima ciclooxigenase, não seletivo para as
lipooxigenases (?). A aspirina consegue controlar os processos febris e inflamatórios
porque ela inibe essa enzima, que é o seu alvo farmacológico. Exceto os fármacos
que tem a sua ação devido a alterações no potencial, na osmolaridade do meio por
exemplo, todos agem sobre um alvo farmacológico.
Quando se pensa num fármaco agindo sobre o sistema enzimático, ele pode
ser um inibidor da enzima que pensa que ele é o seu ligante, o seu substrato, porém
não é, pois o fármaco não é metabolizado por essa enzima. Logo, vai haver a
diminuição da formação do produto daquela enzima. É o que acontece com a aspirina
e a ciclooxigenase, o ácido acetil salicílico inibe a ciclooxigenase, acetilando-a.
O fármaco que age sobre sistemas enzimáticos também pode se comportar
como um falso susbtrato. Tanto inibidores enzimáticos quanto falsos substratos vão
competir com o substrato normal, porém o falso substrato ele não só entra no sítio
ativo da enzima como é metabolizado, porém o que é gerado não é o produto comum
da catálise da enzima.
Alguns fármacos são administradas sob a forma de pró drogas, que não
possuem atividade precisando serem metabolizadas para adquirir atividade. São
metabolizadas muitas vezes por enzimas hepáticas, por exemplo pelo sistema P450.
Temos então três maneiras diferentes de modular a atividade enzimática:
Inibidor da atividade enzimática
Falso substrato da enzima
Utilizar a atividade enzimática para produzir a droga ativa
Canais iônicos, Canais de sódio voltagem dependente. Xilocaína é um
anestésico local porque ela bloqueia os canais de sódio voltagem depende, com isso
impede a propagação de estímulos algésicos que levam a dor. Então bloqueadores
são como rolhas dos canais iônicos, eles não deixam que os canais iônicos funcionem
da maneira adequada. Eles se tornam impermeáveis aos íons que passariam
normalmente pelo canal no estado aberto. Os canais iônicos podem ser controlados
por moduladores, ou seja por drogas que mantenham ele por mais tempo aberto, ou
mais tempo fechado, logo temos moduladores positivos e negativos.
Transportadores normalmente estão envolvidos no processo de difusão
facilitada. Pegam o substrato do lado extracelular e passam para o intracelular, e vice
versa. Como eu posso modular a atividade do transportador? Posso ter inibidores,
drogas que simplesmente vão inibir o transporte. Drogas que são falsos substratos,
que serão transportados para o intracelular mas não possuem a mesma função no
compartimento aonde serão entregues. A glicose, por exemplo, tem transportadores
específicos. Se eu coloco um análogo da glicose que não é fosforilado pela
hexoquinase, essa molécula não estará comprometida com a geração de energia,
logo estará sendo um veneno metabólico para a célula, mudando então a atividade
final do transportador. O que vai acontecer com o falso susbtrato? Enquanto o
transportador estiver comprometido com a passagem do falso substrato, o substrato
nativo não vai conseguir passar, tendo assim um meio de competição. Além disso,
uma vez dentro da célula, aquele sistema que teria início com o susbtrato não
acontecerá, ou acontecerá de forma diferente. A alfa metildopa é um anti
hipertensivo porque o nosso organismo, neurônios adrenérgicos acham que a alfa
metil dopa é uma molécula de dopa que é convertida em dopamina que é convertida
em noradrenalina, só que não é a dopa, então todos os substratos que serão gerados
serão alfa metilados, eles não tem a mesma utilidade que os produtos nativos,
adrenalina e noradrenalina. Vocês vão ver que a alfa metildopa tem ações
farmacodinamicas bastante distintas que a noradrenalina normal, ela tem afinidade
por diferentes receptores, o que contribui ainda mais para o seu papel anti
hipertensivo. Lembrando então que os falsos substratos agem em duas frentes, a
primeira competindo com o substrato normal e a segunda é impedindo que ocorra
normalmente os processos biológico subseqüentes.
Vamos nos concentrar na aula de hoje nos receptores. Quatro deles são de
muita importância para a farmacologia. De maneira suscinta, os fármacos podem ser
agonistas ou antagonistas dos receptores. Os agonistas geram a resposta enquanto
os antagonistas são capazes de se ligar, mas não de gerar reposta.
Canais iônicos regulados por ligantes (Receptores Ionotrópicos)
São buraquinhos presentes na membrana da célula que se abrem na presença
de um ligante. São proteínas integrais transmembranas com um poro no meio que se
abre ou fecha na presença ou ausência do agonista. Os agonistas são capazes de
gerar uma mudança conformacional terciária e quaternária do receptor. Só existe
estrutura quaternária quando a proteína tem subunidades e ela mostra a relação
entre essas subunidades.Esse canal pode gerar despolarização ou hiperpolarização da
célula ou do compartimento envolvido. O que vai determinar um ou outro? Depende
das concentrações dentro e fora da célula do íon envolvido no canal. Se for de sódio,
por exemplo, que está mais concentrado do lado de fora, quando o canal abrir o
sódio vai entrar, causando então despolarização. Se for um canal iônico ligante
dependente permeável a potássio. Em condições fisiológicas o potássio está mais
concentrado do lado de dentro da célula, logo a presença do agonista vai abrir o
canal levando a saída de potássio e consequentemente à hiperpolarização da célula.
Receptores ionotrópicos não são capazes de agir contra o gradiente de concentração,
a saída ou entrada de íons vai sempre depender da concentração dentro e fora da
célula. A resposta do receptor é muito rápida, sem gasto energético pois o que ocorre
é o efluxo ou influxo de íons a favor do gradiente de concentração.
Exemplos: Receptores nicotínicos para acetil colina, Receptores para
Glutamato, Receptores para Gaba.
Os receptores que estão ligados a atividade quinase, ou tem atividade
intrínseca ou se acoplam a enzimas que são quinases. Isso depende de processos
tanto de (?) quanto de transcrição gênica. Já os receptores nucleares, também
chamados de receptores intracelulares tem seus efeitos quase que exclusivamente
dependentes da transcrição gênica. Eles precisam translocar para o núcleo, ou o
ligante precisa chegar até o núcleo para que ocorra o efeito esperado. O agonista ou
antagonista vai interagir com o receptor na membrana celular, salvo no caso dos
nucleares. Uma característica farmacocinética importante do fármaco nesse caso é a
lipossolubilidade.
Existem também os canais voltagem dependentes que nesse caso não
precisam de agonistas para abrir. A interação do agonista com o receptor é apenas
uma das maneiras de modular a atividade desses canais iônicos.
Outra forma de modulação são os moduladores positivos e negativos:
Tanto os moduladores positivos quanto os negativos que serão exemplificados irão
modular o receptor GABA A.O receptor GABA A é um canal de cloreto ligante
dependente. O cloreto passa por ele e como tem sempre mais cloreto do lado de fora
do que do lado de dentro ocorre a entrada de cloreto na célula causando
hiperpolarizacao. O GABA A é uma substancia endógena.
Moduladores Positivos: Ex: DIAZEPAM
O Diazepam é uma substancia exógena, um ansiolítico. Ele interage (se ligando a
sítios na estrutura dos canais específicos) nesse canal modulando-o positivamente
mesmo não sendo se agonista. Seu mecanismo de ação age fazendo com que esse
canal fique mais permeável ao cloreto. Ele não é considerado um agonista desse
canal, pois não causa a abertura do canal, apenas faz com que na presença do seu
agonista fique mais tempo aberto. Por não ser capaz de agir diretamente na abertura
do canal o diazepam constitui uma classe de agente ansiolitico seguros.Essa
segurança existe desde que ele não seja usado junto com álcool ou outras drogas
psicotrópicas.
Diferente do diazepam existem os barbitúricos.Eles são drogas que tem uma faixa
terapêutica limitada,ou seja,em doses baixas eles ajudam o GABA A abrir o canal
(agem como o diazepam) mas em doses elevadas eles promovem ,mesmo na
ausência de agonistas do GABA A,a abertura do canal.
Moduladores negativos:
São drogas que diminuem o desempenho/facilidade de abertura do canal, levando a
uma menor quantidade de cloreto entrando na célula. Esse efeito é contrario ao dos
benzodiazepínicos que são moduladores positivos do canal iônico cloreto dependente.
Modulador negativo NÃO é antagonista do canal.
Resuminho:
Gaba +modulador negativo = Menos cloreto entrando
Gaba + modulador positivo= Mais cloreto entrando
Receptores acoplados a proteína G (Receptores Metabotropicos):
Esses receptores têm uma porção extracelular e uma porção intracelular:
- Porção extracelular: Responsável pela interação com ligante
- Porção intracelular – Porção citosolica
Esses receptores são chamados de serpentinas (?) 7 alças,porque eles
serpenteiam(?) a membrana plasmática em 7 alças.
Nesse caso, a interação do agonista gera uma modificação na estrutura terciaria do
receptor (cauda intracelular que estava virada para esquerda vai para
direita=desenho do quadro).Isso vai gerar uma exposição do sitio fazendo com que
essa cauda do receptor interaja com uma proteína ,que é a proteína G.Essa ptn G
esta perto do receptor e ela tem 3 subunidades,alfa beta e gama.A interação da
cauda intracelular do receptor com a ptn G faz com que o seguinte fenômeno
aconteça : O receptor que é 7 alfa transmembrana quando ativado recruta a ptn
G.Nesse recrutamento , a subunidade alfa permanece ligada ao receptor e as
subunidades beta e gama(juntas) se soltam da região alfa.Dentro da subunidade alfa
existe uma molécula de GDP.A interação dessa subunidade alfa com a cauda do
receptor faz com que esse GDP (nucleotídeo difosofatado que esta em estado de
repouso) seja trocado por GTP.A partir disso essa molécula alfa passa a ser capaz de
interagir com uma grande quantidade de enzimas e outros alvos
farmacológicos.Assim ela pode tanto ativar uma outra enzima da vizinhança quanto
inibir uma enzima.Essa ativação e inativação enzimática que vai gerar a resposta
biológica.
Resuminho:
- Receptor+agonista = modificação devido à mudança conformacional do receptor
- Receptor passa interagir com a molécula de proteína G que tem 3 subunidades:
- Beta e Gama – Elas se distanciam e até podem ativar outras enzimas também.
- Alfa – Fica com o receptor. Nessa molécula ocorre a troca do nucleotídeo
difosfatado (GDP) por um trifosfatado (GTP).Com isso,a alfa não quer ficar perto do
receptor e então passa a interagir com outras proteínas que são as enzimas.Essas
enzimas que estavam com uma atividade basal passam a ter sua atividade
aumentada ou diminuída quando essa subunidade alfa interage com ela.É isso que
vai gerar a resposta mediada pelos receptores acoplados a ptn G.
Exemplos: Mediador metabólico que utiliza esse tipo de interação: Glucagon
O glucagon é o hormônio secretado quando se está com fome. Ele aumenta a
atividade lipase fazendo beta oxidação aumentando a concentração de acido graxo
livre. O glucagon é o agonista que vai interagir com os receptores acoplados a ptn
G, ativando a enzima adenilato ciclase,aumento assim os níveis de AMP
cilico,ativando a proteína quinase A,desencadeando uma cascata de fosforilacao e
resultando no aumento da lipólise,diminuição da síntese de glicogênio e aumento da
quebra de glicogênio liberando glicose 6 fosfato.
Como um agonista pode modular negativamente uma resposta biológica?
A subunidade alfa pode ativar ou inativar uma determinada enzima. Isso vai depende
do tipo de ptn G que se tem.Existem receptores que vão interagir com proteínas G do
tipo estimulatoria e outros irão interagir com proteínas do tipo inibitória.
Exemplo: Receptor Beta 1 adrenérgico:
Essa ativação de beta 1 frente ao agonista adrenalina ou noradrenalina promove a
taquicardia.Esse receptor é ligado a ptn G do tipo estimulatorio (PTNGs),ou seja,sua
atividade alfa é estimulatoria.Com isso ocorre a estimulação da adenilato ciclase
resultando no aumento dos niveis de AMP ciclico nos cardiomiocitos promovendo
aumento da atividade da ptn quinase A,aumento na abertura dos canais de cálcio…..o
que resulta da taquicardia final.
Resuminho:
- Classes de Ptn G:
Estimulatoria (Ptn Gs) –aumentam os níveis de AMP ciclico porque estimulam a
adenilato cilcase
Inibitória (Ptn Gi) – diminuem os níveis de AMP ciclico porque inibem a adenilato
ciclase
Existe também uma terceira classe de proteína G: Ptn Gq
A Ptn Gq tem como função ativar a fosfolipase C.
Exemplo de PtnGq: Receptores Alfa 1
A noradrenalina age como agonista ativando os receptores alfa 1.Dessa forma ocorre
todo o processo já citado antes (subunidade alfa se separa....)cujo resultado final é
a ativação da fosfolipase C.A fosfolipase C pega o fosfolipídio de membrana e
transforma em ip3 e diacil glicerol no meio extracelular.O ip3 mobiliza cálcio do
reticulo sarcoplasmático ,aumentado os níveis de cálcio intracelular e
então,promovendo a vasoconstricção.
Exemplo ligado a fisiologia: os leitos vasculares que irrigam a musculatura
esquelética são ricos em receptores do tipo BG2 adrenérgicos. O que acontece
quando se leva um susto? O sangue sai da periferia e vai principalmente para
musculatura esquelética pois se tem muito receptor alfa 1 na vasculatura da periferia
e muito receptor beta 2 na vasculatura que irriga a musculatura esquelética. Na
periferia ela fará vasocontricção pois o receptor alfa 1 está ligado a proteína GP. Já
beta 2 está acoplado a proteína GF que aumentam os nucleosídeos cíclicos em
células musculares lisas, causando vaso dilatação. Logo, um mesmo agonista
(adrenalina) pode levar a respostas totalmente diferentes, dependendo do tipo de
proteína acoplada a seu receptor.
Obs: agonista muscarinico faz bradicardia pois seu receptor, que está no
coração, está acoplada a proteína GI.
Qual é a novidade evolutiva dos receptores acoplados à ptn G? É a
amplificação do sinal. Uma única molécula de agonista interagindo com um único
receptor pode levar a ativação de um série de cascata de ativação. O que aconteceria
se uma via dessas estivesse sendo persistentemente ativada? Ex: um descarga
adrenérgica; tenho muita noradrenalina. A célula morre? Não. Pq? Ocorrerá a
ativação de diversas quinases que fosforilarão outras substancias; a principal quinase
ativada é a pKa, que fosforila principalmente proteínas e residos de serina,
compostos que estão presentes inclusive em alguns receptores acoplados a proteína
G; logo os receptores podem ser fosforilados, o que leva a uma alteração de sua
função, perdendo sua capacidade de interagir com a proteína G, o que “desliga” o
estímulo, evitando resposta exacerbada. O período de inativação dos receptores é
chamado de desensibilização heteróloga (receptores diferentes que recebem
agonistas diferentes, mas a ativação de um pode interferir negativamente na
ativação do outro. Esse efeito pode ser revertido por fosfatases.
A ativação persistente do receptor ativa várias quinases e esse estímulo leva a
ativação de uma quinase muito específica: quinase do receptor acoplado a proteína G
(GRK). Ela fosforila o receptor, em sítios muito específicos, que está evocando essa
resposta. A fosforilação recruta a proteína arrestina que irá reconhecer essa cauda
fosforilada do receptor e irá englobá-lo (endocitose do receptor) através da
mobilização das proteínas do citoesqueleto. Isso protege o receptor de ser fosfatado,
o que prolonga o efeito do agonista. A partir disso, o receptor pode voltar á superfície
plasmática ou ser degradado.
EX: paciente com insuficiente cardíaca: necessita de um medicamento que
aumente a força de contração do coração. Como? Administrando aminas
simpaticomiméticas, que são agonistas dos receptores alfa 1. Há uma melhora. Ao
aumentar a dose, o paciente passa a não responder ao fármaco. A curva de ação do
fármaco é caracterizada como “em sino” (atinge uma atuação máxima e regride).
Quando o gráfico (efeito x concentração) que se observa tiver essa
característica (curva em sino), o fenômeno que está sendo apresentado é uma
desensibilização de receptores acoplados a proteína G, provavelmente
desensibilização do tipo homóloga.
Então, o que ocorre, em etapas, é o seguinte:
1º passo: fosforilação do receptor
2º passo: externalização do mesmo (?)
3º passo: endocitose do receptor
4º passo: reciclagem do receptor (retorno à membrana) e degradação do
receptor.
Revendo o que foi falado:
Gr aumenta a atividade da adenilato ciclase; Gi diminui a atividade da
adenilato ciclase; Gq aumenta a atividade da fosfolipase C. Nesse momento, isso é o
que precisamos saber. Existem outros tipos de proteínas G e outros alvos para essas
proteínas, mas não discutiremos agora.
Receptores acoplados a proteína quinase: proteínas integrais da membrana.
- Receptores tirosina quinase: tem na sua cadeia polipeptídica tanto a porção de
reconhecimento quanto a porção efetora (que é a tirosina quinase). Ele existe na
forma de monômero. Quando o agonista é introduzido ao meio há interação com a
porção de reconhecimento. Isso vai fazer com que haja a dimerização (cada uma das
subunidades fosforila a outra) - receptor torna-se ativo. O receptor ativo não
funciona na forma de monômero, ele tem que dimerizar. A dimerização e a
associação com resíduos de tirosina vão fazer com que haja o recrutamento de
outras proteínas (podem ser proteínas adaptadoras ou outras quinases), bem como a
fosforilação direta de proteínas citoplasmáticas. Esses receptores agem via cascata
de fosforilação que podem culminar, então, com a regulação da expressão gênica
(aumento ou diminuição da síntese protéica).
Exemplo: A insulina é um fator de crescimento, um hormônio anabólico.
Temos uma série de outros hormônios com características semelhantes, como alguns
fatores de crescimento que estão envolvidos em processos tumorais. Eles agem
através desses receptores.
No caso dos receptores como o da insulina, o processo descrito acima vai contribuir
para a translocação do GLUT-4 (receptor de insulina tem efeitos tanto dependentes
da síntese protéica como não-dependentes, como é o caso da translocação do
transportador de glicose GLUT-4). O caso do GLUT-4: o GLUT 4 fica dentro de
vesículas na célula. Quando tem estimulo, essas vesículas voltam a integrar a
membrana plasmática para aumentar a quantidade de transportadores. Esse
processo não é dependente de síntese protéica, e sim dependente de uma cascata de
fosforilação - mediada pela atividade tirosina quinase - que fez com que essas
vesículas voltassem a integrar a membrana plasmática. Esse é um bom exemplo de
processo independente da expressão gênica (síntese protéica).
Observação: Alguns receptores não se encaixam nos tipos clássicos estudados até
agora, mas são bem parecidos com os receptores tirosina quinase. São receptores
cuja região de reconhecimento não tem atividade tirosina quinase. Porém, quando o
agonista se liga a esse receptor, há recrutamento de uma molécula de tirosina
quinase que vai fazer com que haja a ativação dessas cascatas de fosforilação.
Exemplos: Receptores para citocinas como IL-6.
Receptores intracelulares ou nucleares: existem 3 classes bem estudadas. Todos
os receptores nucleares são fatores de transcrição (regulam a transcrição de um
gene – interagem com regiões específicas do DNA). Então, uma vez que ele interage
com o seu ligante, vai controlar positiva ou negativamente o processo de transcrição
gênica.
Exemplo: receptor para glicocorticóide, na verdade, receptores para esteróides
de uma maneira geral. Substâncias muito lipossolúveis (agonistas, como o colesterol)
interagem com os receptores de glicocorticóide – sempre estão localizados no citosol
da célula protegido por proteínas chaperonas. Os receptores de glicocorticóide têm
uma sequência na região protegida pelas chaperonas que age enviando-os para o
núcleo. O que acontece quando o agonista desses receptores interage com eles?
Ocorre uma mudança conformacional e isso libera as chaperonas e, então, a
sequência fica exposta. O receptor se encaminha ao núcleo e interage com o DNA em
regiões específicas denominadas elementos responsivos (na região promotora). No
caso do exemplo, elementos responsivos para o glicocorticóide. Esses elementos
podem ser tanto positivos quanto negativos. Quando o receptor interage com o
elemento responsivo positivo, há aumento da transcrição gênica e ao interagir com o
elemento negativo, há diminuição da transcrição. Principal ação clínica dos
glicocorticóides: Antiinflamatórios esteroidais – aumenta a síntese de proteínas que
tem importante papel na luta contra a inflamação e diminui os níveis de proteínas
pro-inflamatorias.
O receptor é a vedete da farmacologia, logo, além de saber quais são esses
receptores, é importante conhecer quais são os tipos de interações possíveis.
Agonistas:
O agonista é qualquer substância capaz de interagir com um sitio especifico do
receptor e essa interação terá uma resposta. São vários os mecanismos de interação
do agonista com o receptor. Tem várias forças moleculares que podem estar
envolvidas nesse tipo de interação. Interações iônicas, pontes de hidrogênio, ligação
de Van der Waals, interação hidrofóbica, e até mesmo interação covalente. Alguns
agonistas interagem de maneira covalente com o receptor, representando assim uma
interação difícil de ser clivada.
O grau de afinidade vai determinar a interação do agonista com determinado
receptor. Uma molécula altamente hidrofóbica vai tentar interagir com alguma
porção hidrofóbica de outra molécula. Uma coisa importante, é que a interação do
fármaco com o receptor, quando a interação é covalente, obedece a “lei das zonas K”
(?? 5:20),
A interação é uma interação estática? Não, é uma ligação dinâmica, o agonista
liga e desliga toda hora do receptor. Saber disso é importante para entender o
conceito de competição. Não se pode pensar que a interação do ligante com receptor
é para sempre, pois isso não ocorre, já que ela não é estática e sim dinâmica. A
afinidade é a medida da probabilidade do evento. O evento “droga + receptor”, pode
ser mais ou menos provável em função da afinidade.
Antagonista:
O mesmo tipo de raciocínio é usado pra interação antagonista-receptor, a
única diferença é que e antagonista não é capaz de provocar uma resposta. Portanto
é errado falar que o “Atenolol” provoca bradicardia, pois ele é um antagonista, é um
beta bloqueador. O antagonista não faz nada, a função dele é não deixar o agonista
fazer. Ele faz isso porque interage direta ou indiretamente com o receptor não
deixando que a resposta do agonista seja gerada.
Então o agonista é o que interage com o receptor formando um complexo
fármaco-receptor e aciona um mecanismo de transdução de sinal. Os antagonistas
interagem com o receptor formam um complexo, mas não ativam esse receptor, não
deixam que ocorra a transdução do sinal, não gerando a resposta.
Existem vários tipos de antagonismo. Antagonismo químico corresponde a
interação de determinadas drogas com determinados componentes da dieta. Ela fala
de um antibiótico, a Tetraciclina. É um antibiotico que “complexa” na presença de
cálcio. Por isso não deve tomar junto com o leite, pois o fármaco precipita e tem uma
ação final menor. Isso é um antagonismo químico, quando a alteração da molécula
impossibilita o processo mimético ou dinâmico. Antagonismo químico ocorre quando
duas substâncias interagem, prejudicando a ação do fármaco.
Antagonismo farmacocinetico: é quando um fármaco altera distribuição e/ou
absorção de um outro fármaco. Ex. um fármaco ácido pode alterar a absorção de um
fármaco alcalino pois altera sua dispersão no fluido intestinal.
Antagonismo fisiológico: Interação entre duas drogas cujas ações opostas
tendem a anular-se, por atuarem sobre células ou sistemas fisiológicos separados.
Ex. noradrenalina (vasoconstritor) histamina (vasodilatador).
Antagonismo não competitivo: droga que iniba a atividade da proteína C,
assim a cascata de sinalização não será ativada, não se estará interagindo com o
receptor, assim, a sinalização estará inibida e não o receptor, a ligação do agonista
com o receptor está livre, por isso não é competitivo.
No competitivo o antagonista se liga ao sitio do receptor do agonista, seja de
maneira transitória (reversível), ou permanente (irreversível). O irreversível interage
de forma covalente com o sitio ativo do receptor.
Às vezes, esses conceitos se confundem: por exemplo, a anestesia de dentista
que não pega. Há um antagonismo farmacocinético, pois a gengiva inflamada é um
meio acido e a anestesia é básica, dessa maneira o composto estará muito ionizado
prejudicando sua absorção, além disso, ele precisa chegar no seu sitio de ligação por
dentro da célula. Dessa maneira, o meio externo prejudica ainda a ligação do
fármaco com seu sitio de ligação dentro da célula, sendo esse um antagonismo
farmacodinâmico.
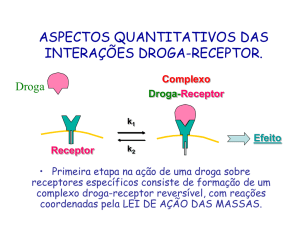
Gráficos de Afinidade e Efeito
Para determinar a interação da droga com o receptor, eu procuro um tecido
rico no receptor, faço uma preparação com meu ligante marcado radioativamente, e
isso mostra qual concentração de radioatividade tem em cada preparação. A curva
mostra a percentagem de ligação do fármaco ao receptor em função da
concentração, é uma hipérbole retangular!! Isso mostra que atinge um platô, isso é
uma característica básica da interação droga receptor, ou seja a interação é passível
de saturação. Não importa se eu coloco mais ligante no meio, vai chegar um ponto
que todos os receptores vão estar virtualmente ligados.
Essa hipérbole retangular, vira uma sigmóide simétrica. Se eu mudar o eixo do
“x”, em vez de ter uma escala linear eu passo a uma escala logarítmica. Simétrica
porque a parte de baixo é um espelho da de cima.
O que se observou para a ligação fármaco-receptor, pode ser feito para medir
seu efeito. Pode mensurar a amplitude de um efeito de acordo com o aumento da
concentração de um ligante. Antes só estava avaliando a interação do fármaco com o
receptor, então servia para agonista e para antagonista. Agora, o efeito biológico só
é possível de mensurar para o agonista, já que o antagonista não é capaz de gerar
resposta.
De novo eu vejo que o sistema é passível de saturação, o efeito é diretamente
relacionado ao número de receptores ativados, ocupados, por isso as curvas de
saturação de efeito e de interação são parecidas, pois o efeito é proporcional.
Agora se for na presença de um antagonista o gráfico do efeito tangenciaria o
eixo “x”, pois não há efeito. Ao colocar o gráfico em “log”, a curva fica sigmóide,
mais fácil de visualizar.
Mas porque as contas de efeito e de interação são feitas em cima de 50% e
não 100%? Isso é feito porque é complicado ver quando a curva chegou no 100%.
Quando há um gráfico de ligação fármaco-receptor em função da concentração eu
posso determinar a afinidade que aquele fármaco, seja agonista ou antagonista tem
pelo receptor.
Então nós temos o seguinte fluxograma, o agonista + o receptor, formando o
complexo agonista – receptor. Esse complexo não é estável.Quanto maior a afinidade
do agonista pelo receptor, maior será a formação do complexo agonista–receptor.
Vamos dizer que eu tenha uma proporção de 10 milhões de moléculas ligantes para 1
receptor. Obviamente como eu tenho muito, a probabilidade do receptor ficar vazio
diminui quando eu aumento a concentração, mas se a afininidade deles for pequena
vai haver formação de complexo mas ele não vai ser tão estável.Uma vez formado o
complexo agonista-receptor, haverá um mecanismo efetor que vai gerar uma
resposta final.
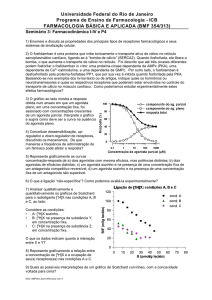
Então aqui eu tenho 2 fármacos agonistas, o que eu posso dizer em relação a
eles? A partir dessa curva eu consigo tenho 2 informaçãoes muito importantes.
Primeira, um deles é mais eficaz (eficácia é a capacidade de produção de uma
resposta). Um é um agonista total (consegue chegar a 100% de ativação), ou seja,
ele é capaz de gerar uma resposta máxima. O outro é um agonista parcial, porque
não adianta aumentar a concentração que ele nunca vai gerar uma resposta máxima.
A efiacácia de um agonista total é sempre igual a 1 porque 100/100 =1 enquanto do
agonista parcial vai ser sempre uma fração, menor que 1.
A potência, que é a quantidade de fármaco necessária para desncadear uma
resposta, está intimamente realcionada com a afinidade do agonista pelo receptor e é
medida pelo DE50(acho que é isso). Quanto mais potente for uma droga, menor será
o seu DE50. Então quanto mais para esquerda estiver a curva, mais potente é o
fármaco. O DE50 é a concentração necessrária para se alcançar 50% da resposta
máxima. Uma outra coisa importante: os 50% dessa curva aqui não é igual ao 50%
dessa outra porque cada curva tem sua resposta máxima.
Vamos falar agora dos antagonistas. Já vimos que eles podem ser competitivos
(agem no mesmo sítio de ligação do agonista) e não competitivos (agem em sítios
diferentes do agonista ou nas cascatas decorrentes da ativação do receptor).
Como antagonismo competitivo reverssível é expresso graficamente? Há o
deslocamento da curva para a direita. Em azul, vemos a resposta na presença do
agonista. Quando é introduzido um antagonista, só conseguimos perceber o mesmo
efeito quando aumentarmos a concentração do agonista. Assim, O DE50 também
aumenta. Eu continuo alcançando a resposta máxima, mas isso vai depender do
aumento da concentração.
No caso de um antagonista competitivo irreverssível na presença de agonista,
haverá allteração da potência e alteração da eficácia. Então a curva se desloca para a
direita e tem a amplitude diminuída. A amplitude diminui porque não vou mais
alcançar a resposta máxima, é como se a célula tivesse menos receptores.
Quando eu tenho o agonista na ausência e na presença do antagonista não
competitivo vamos observar a diminuição da resposta máxima. A interação está
preservada mas a resposta gerada vai ser muito menor e com isso vamos ter
diminuiçaõ do efeito máximo. Porém, eu não tenho, necessariamente, diminuição do
DE50 porque a afinidade do agonista pelo receptor não foi comprometida, só a
amplitude da resposta é menor (só eficácia é alterada).
Ela mostra um gráfico de distribuição de um fármaco com o número de
indivíduos que responde a cada dose desse fármaco. A moda desse gráfico é,
aproximadamente, 10 mg/L (grande parte das pessoas responde bem a um a dose
de 10mg/L). Podemos sobrepor esse grafico e obter uma curva sigmóide parecida
com as que vimos nas curvas de antagonismo.
Então ela compara esse gráfico com outro que mostra a concentração
necessaria para causar efeito colateral ou óbito dos pacientes. Com essas duas
curvas conseguimos gerar uma constante chamda de índice terapeutico que espressa
a distância entre essas duas curvas. O indice terapêutico é calculado dividindo-se a
dose necessária para que 50% da população apresente efeitos colaterais pela dose
necessária para que 50% da população apresente a resposta esperada desse
fármaco. Quanto maior for o indice terapêutico, maior será a distância entre essas
curvas e menor será a chance de se observar efeitos colaterais.