CAPÍTULO 1
PRINCÍPIOS GERAIS
DA AÇÃO DE
PSICOFÁRMACOS
CLARICE GORENSTEIN
TANIA MARCOURAKIS
Os psicofármacos distinguem-se dos outros tipos de medicamentos por obrigatoriamente atuarem no sistema nervoso central (SNC). Isso implica a necessidade
de que eles e/ou seus metabólitos atravessem uma barreira adicional – a barreira
hematencefálica. Os princípios básicos que determinam os demais processos, ou
seja, absorção, distribuição, biotransformação e excreção, são essencialmente os
mesmos que para os demais fármacos (Fig. 1.1). Alguns desses conceitos básicos
de farmacologia serão brevemente revistos, com ênfase na sua aplicação em
psicofarmacoterapia.
ABSORÇÃO
Todos os processos que ocorrem desde a administração de uma droga até sua
eliminação envolvem a passagem através de barreiras representadas pelas membranas celulares. A absorção refere-se à passagem da droga do seu sítio de aplicação para a corrente sangüínea.
Os principais mecanismos por meio dos quais os psicofármacos atravessam
membranas são difusão aquosa e difusão lipídica. A difusão aquosa consiste na
passagem através dos poros aquosos, o que ocorre principalmente em função do
tamanho da molécula. As moléculas grandes difundem-se mais lentamente do
que as pequenas. Como o peso molecular da maioria das drogas não varia muito,
em geral é a passagem pelas barreiras não-aquosas que determina a velocidade
de absorção.1,2
Para que ocorra a difusão lipídica, é necessário que a molécula seja lipossolúvel
e esteja na forma não-ionizada. Como muitas drogas são ácidos ou bases fracas
(os psicotrópicos em geral são bases fracas), elas se apresentam sob duas formas
em equilíbrio dinâmico: não-dissociada (ou não-ionizada), e dissociada (ou ioni-
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
zada). Para a maioria das drogas, a lipossolubilidade da fração não-dissociada é
suficiente para permitir considerável absorção.
A proporção de moléculas que se encontra na forma não-ionizada, ou seja, o
grau de dissociação, depende das propriedades físico-químicas da droga (que
determinam seu pKa) e do pH do meio em que está dissolvida. Em conseqüência
dessas propriedades, sabe-se que drogas de caráter ácido são mais bem absorvidas
no estômago, enquanto as básicas tendem a ser absorvidas no intestino.3
Destaca-se, entre os vários fatores que modificam a velocidade de absorção,
a influência da via de administração. A via endovenosa, embora possibilite um
melhor controle da quantidade administrada e seja, sem dúvida, a via mais rápida
para a obtenção de efeitos, apresenta risco de efeitos adversos ou de superdosagem relativa muito maior do que as demais vias. Efeitos tóxicos autonômicos e
cardíacos podem ser observados com drogas como clorpromazina ou amitriptilina,
cuja administração endovenosa deve ser feita com cautela.2
A via intramuscular, utilizada para sedação de pacientes agitados e para administração de neurolépticos de ação prolongada, permite a administração de volumes moderados de soluções, veículos oleosos (p. ex., enantato e decanoato de
flufenazina) e soluções irritantes, o que já não é possível com injeções subcutâneas.
Por outro lado, o diazepam administrado por via intramuscular resulta em absorção
lenta e errática, com picos de concentração plasmática inferiores aos obtidos
após administração oral, provavelmente devido à cristalização do fármaco no
local da injeção.
A via oral é a mais amplamente utilizada. A absorção se processa em toda a
extensão do trato gastrintestinal, sendo o estômago e o intestino os locais de
maior absorção. A absorção em cada local depende da variação de pH, da irrigação
e de características anatômicas, bem como das propriedades físico-químicas do
fármaco. Um dos fatores que favorece a absorção no intestino é a presença de
microvilosidades altamente irrigadas, que proporcionam grande área de superfície.3
Qualquer fator que acelere o esvaziamento gástrico aumentará a velocidade
de absorção da droga, tanto para drogas absorvidas a partir do estômago, porque o seu contato com a parede mucosa será favorecido, quanto para as absorvidas
a partir do intestino, que o atingirão mais rapidamente. Assim, quando se deseja
a rápida absorção de uma droga não-irritante da mucosa gástrica, esta deve ser
administrada em jejum, enquanto para uma absorção mais lenta recomenda-se
ingeri-la após as refeições.
Para as drogas administradas por via oral, a formulação farmacêutica exerce
grande influência na absorção. As soluções são as mais rapidamente absorvidas,
enquanto cápsulas, comprimidos ou drágeas o são mais lentamente, porque dependem da velocidade de dissolução da forma sólida.
Nem toda a concentração da droga ingerida chega à circulação geral. Para
avaliar o quanto estará disponível no sítio de ação, determina-se a biodisponibilidade da formulação farmacêutica. O termo biodisponibilidade indica a fração
de uma droga ingerida que tem acesso à circulação sangüínea. A biodisponibilidade
pode ser baixa se a absorção for incompleta ou se a droga for metabolizada na
parede do intestino ou do fígado antes de atingir a circulação sistêmica (metabolismo de primeira passagem).1
14
DISTRIBUIÇÃO
A etapa seguinte à absorção é geralmente a distribuição da droga para os diversos
tecidos. A velocidade de distribuição depende do grau de perfusão do órgão. O
equilíbrio de distribuição é atingido mais facilmente nos tecidos que recebem
grande circulação de fluidos (coração, cérebro, fígado) e mais lentamente nos
órgãos pouco irrigados (ossos, unhas, dentes e gorduras).3
A água corpórea, que corresponde a aproximadamente 60% do peso do
indivíduo, distribui-se por dois compartimentos funcionais principais: o líquido
intracelular (40%) e o líquido extracelular (20%, sendo 15% intersticial e 5%
vascular). O volume de distribuição aparente (Vd) de uma droga é o volume de
fluido no qual ela está aparentemente distribuída. Portanto, a distribuição de
uma droga depende do compartimento pelo qual ela se distribui. De um modo
geral, as drogas que se distribuem pelo líquido extracelular e que exercem efeitos
em membranas têm início de ação mais rápido do que aquelas que devem penetrar
na célula para atuar. Dado que são lipossolúveis, a maioria dos psicofármacos
distribui-se pela água corpórea total.2
A distribuição também é regulada pela ligação da droga às proteínas plasmáticas (principalmente albumina), o que torna a droga biologicamente inativa. A
competição entre dois fármacos pela ligação às proteínas plasmáticas leva a um
aumento de suas porções livres, o que pode determinar o aumento do efeito
terapêutico e tóxico ou a ineficácia terapêutica.4
15
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
Figura 1.1
Etapas da droga no organismo após a administração.
A droga pode também se acumular no tecido adiposo. Este constitui, aproximadamente, 15% do peso corpóreo, e seu volume é de cerca de 25% do volume
de água total, representando, portanto, um grande compartimento não-polar
do organismo. Uma molécula não-polar, com alto coeficiente de partição óleo/
água, tende a acumular-se consideravelmente no tecido adiposo, não exercendo
ação farmacológica. O acúmulo em um determinado tecido pode prolongar a
permanência da droga no organismo, como ocorre, por exemplo, após administração endovenosa de tiopental, que se acumula no tecido adiposo e sofre redistribuição.2
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
BARREIRA HEMATENCEFÁLICA
Embora o cérebro represente apenas 2% do peso corpóreo de um indivíduo, ele
recebe aproximadamente 16% do débito cardíaco. Devido ao seu alto suprimento
sangüíneo, esperar-se-ia que as drogas passassem rapidamente da corrente circulatória para o espaço extracelular cerebral, mas isso não ocorre devido à restrição
imposta pela barreira hematencefálica. Embora não verdadeiramente definida
do ponto de vista anatômico, essa barreira caracteriza-se pela justaposição das
células do endotélio dos capilares cerebrais. É provável que o arranjo característico das células pericapilares da glia também contribua para a lenta difusão dos
ácidos e das bases orgânicas para o SNC. Além disso, a vascularização das diversas
áreas cerebrais não é uniforme (p. ex., o córtex é mais vascularizado que a substância branca), e as drogas entram mais rapidamente nas áreas mais vascularizadas.2
Além da barreira hematencefálica, existe uma via indireta de passagem, que
é a barreira hematoliquórica, constituída por células epiteliais do plexo coróide,
que regulam o acesso de drogas ao líquido cerebrospinal.
Os principais fatores que determinam a passagem das drogas pela barreira
hematencefálica são semelhantes aos que interferem na sua passagem através
do endotélio gastrintestinal para o sangue, ou seja, a lipossolubilidade, o grau de
ionização e a ligação a proteínas plasmáticas.
Quanto maior for a lipossolubilidade, mais facilmente a droga penetra no
cérebro. A maioria dos psicofármacos são aminas secundárias ou terciárias que,
sendo lipossolúveis, não encontram dificuldade na passagem para o cérebro.
Para aumentar as concentrações cerebrais de substâncias com baixa lipossolubilidade, como a dopamina e a serotonina, é necessária a administração de seus
precursores, L-dopa e L-triptofano, respectivamente, que atravessam a barreira.
No caso de drogas polares, a velocidade de difusão para o SNC é determinada
pela solubilidade da forma não-iônica.5
Em relação ao grau de ionização, sabe-se que apenas moléculas neutras são
capazes de atravessar barreiras lipídicas e que, portanto, o transporte será tanto
mais rápido quanto maior for a concentração de moléculas. Já a passagem dos
íons é determinada pelo seu tamanho: íons pequenos, como o lítio, são capazes
de atravessar os poros das membranas, enquanto íons maiores dependem da
presença de algum tipo de transporte ativo. Substâncias altamente ionizadas,
tais como as aminas quaternárias, são geralmente incapazes de penetrar no SNC.
16
BIOTRANSFORMAÇÃO
Biotransformação ou metabolismo é o conjunto de alterações químicas que a
droga sofre no organismo sob a ação de enzimas. O principal objetivo da biotransformação é facilitar a eliminação. A maioria das drogas tem alto coeficiente
de partição óleo/água, isto é, são altamente lipossolúveis, o que favorece sua
reabsorção pelos túbulos renais e aumenta seu tempo de permanência no organismo. Assim, drogas lipossolúveis são biotransformadas em compostos mais polares,
passíveis de eliminação. Drogas de baixa lipossolubilidade ou altamente ionizadas,
são excretadas in natura pelos rins (p. ex., barbital e lítio).3
Geralmente, a biotransformação transforma drogas ativas em metabólitos
inativos (p. ex., haloperidol). Entretanto, os metabólitos também podem ser ativos
(p. ex., desipramina, metabólito da imipramina), ou até mais ativos do que o
composto original (p. ex., hidrato de cloral).
A biotransformação ocorre principalmente no fígado, cujas enzimas localizamse na fração mitocôndrica (monoaminoxidase, MAO), na fração microssômica,
responsável pela biotransformação das drogas lipossolúveis (sistema do citocromo
P450, que participa principalmente de reações oxidativas e de algumas redutivas)
e na fração solúvel (p. ex., desidrogenases, amidases, transferases).
As principais reações químicas que as drogas sofrem por ação enzimática são:
a) reações de fase I: oxidação, redução e hidrólise; b) reações de fase II: conjugação, que normalmente produz compostos inativos. Embora grande parte das
reações oxidativas se processe nos microssomas hepáticos, estas também podem
ocorrer em outros locais. Por exemplo, a enzima mitocondrial MAO é responsável
17
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
A ligação a proteínas plasmáticas limita a concentração da droga nos tecidos
e no sítio de ação uma vez que apenas a fração não-ligada tem a capacidade de
atravessar as membranas. Drogas altamente ligadas a proteínas plasmáticas tendem a penetrar no cérebro e no líquido cerebrospinal mais lentamente. A maioria
dos psicofármacos apresenta alta taxa de ligação às proteínas plasmáticas e teciduais, e, portanto, pequenas alterações de sua fração livre, como em estados
carenciais, devidos à desnutrição, por exemplo, ou no envelhecimento, podem
levar à intensificação de seu efeito farmacológico.5
Como o líquido cerebrospinal normalmente não contém proteínas, toda droga
aí presente encontra-se livre. Por outro lado, o cérebro é rico em proteínas, podendo haver intensa ligação protéica. Pequenas modificações na taxa de ligação
podem levar a alterações importantes nas concentrações de droga biologicamente
ativa em seus locais de ação.
Vale ressaltar que recentemente foi identificada uma série de proteínas transportadoras de moléculas expressas nas membranas celulares, que tem conferido
proteção ou toxicidade dependendo de sua localização (células intestinais, renais,
hepáticas, placenta e endotélio cerebral) e do agente envolvido. Um exemplo é
a MDR (multidrug resistant proteins) ou glicoproteína P, cuja superexpressão,
geneticamente definida, na barreira hematencefálica pode ser um dos mecanismos
que explica a resistência ao tratamento antiepiléptico em alguns pacientes.1
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
pela degradação oxidativa de dopamina, noradrenalina, serotonina e tiramina,
existindo em vários órgãos e tecidos, como cérebro, fígado, rins, coração e plaquetas sangüíneas. Já o álcool etílico é oxidado, transformando-se em acetaldeído,
principalmente pela álcool-desidrogenase e, em menor quantidade, pelas oxidases
microssômicas.2
Enquanto as reações de redução e hidrólise não são comuns nos psicofármacos,
as de conjugação são bastante freqüentes. Elas consistem na combinação da
droga ou dos seus metabólitos com moléculas pequenas que existem no organismo, tais como o ácido glicurônico (glicuronidação), o ácido acético (acetilação),
o ácido sulfúrico (sulfatação), os radicais metila (metilação), a glicina, etc. O
objetivo dessas reações é tornar a droga menos lipossolúvel e mais facilmente
excretável pela bile ou pelos rins. A acetilação de drogas como a fenelzina parece
ser geneticamente determinada, sendo que pacientes que a acetilam rapidamente
tendem a apresentar menor resposta terapêutica que os acetiladores lentos.2
Algumas drogas são eficazmente removidas da circulação pelo fígado e biotransformadas antes de atingir a circulação sistêmica. Este metabolismo de primeira
passagem, ou metabolismo pré-sistêmico, exige doses maiores da droga quando
ela é administrada por via oral. Por exemplo, mais de 80% de uma dose oral de
cloropromazina pode ser metabolizada por esse processo, e calcula-se que a
administração por via intramuscular resulte em níveis plasmáticos cinco vezes
maiores que os obtidos com a mesma dose oral dessa substância. Drogas tais
como imipramina, doxepina, levodopa e metilfenidato também sofrem o efeito
da primeira passagem.5
Os avanços recentes em farmacologia molecular permitiram a caracterização,
até o momento, de 267 famílias de citocromo P450, codificadas por mais de
5.000 genes.1 As isoenzimas do citocromo P450 são representadas pela sigla
CYP, seguida de um algarismo que indica a família, uma letra que indica a subfamília
e outro algarismo que indica o gene. Por exemplo, CYP3A2 significa a isoenzima
2 da família 3 e da subfamília A.3,6
Isoformas específicas de CYP são responsáveis pelo metabolismo dos diversos
psicofármacos. Nem todos os indivíduos têm as mesmas enzimas do CYP450, o
que explica a variabilidade individual na taxa de metabolismo das medicações. A
subfamília 3A do citocromo P450 e as isoformas 3A3 e 3A4 (CYP3A3/4) são
particularmente importantes em psicofarmacologia devido ao seu envolvimento
no metabolismo de várias substâncias, como antidepressivos, benzodiazepínicos,
bloqueadores de canais de cálcio e carbamazepina. A participação dos sistemas
enzimáticos no metabolismo das diferentes drogas tem implicações na interação
medicamentosa, como será explicado adiante.5
INDUÇÃO E INIBIÇÃO ENZIMÁTICA
Algumas drogas são capazes de promover estimulação da atividade das enzimas
hepáticas, particularmente pelo aumento da síntese de enzimas. A indução enzimática acelera a biotransformação de muitas drogas, diminuindo a intensidade e
a duração de suas ações. O fenobarbital e a carbamazepina são exemplos de
18
EXCREÇÃO
Os processos básicos mais importantes para a excreção renal de psicofármacos
são a filtração glomerular e a reabsorção tubular.
A filtração glomerular permite a eliminação de moléculas não muito grandes
(peso molecular inferior a aproximadamente 20.000) e não-ligadas às proteínas
plasmáticas.
As substâncias lipossolúveis, após sofrerem filtração glomerular, são reabsorvidas por difusão passiva nos túbulos renais. A reabsorção tubular é altamente
influenciada pelo pH urinário. Como ocorre na absorção, as formas não-ionizadas
tendem a ser reabsorvidas pelos túbulos renais. Assim, ácidos fracos são excretados
melhor em urina alcalina, e bases fracas, em urina ácida. Daí as vantagens de
alcalinizar a urina de pacientes com superdosagem de barbitúricos e de usar
cloreto de amônio para acidificar a urina de intoxicados por anfetamina.1
A excreção de drogas na bile e sua conseqüente reabsorção intestinal geram
o chamado ciclo êntero-hepático, que é parcialmente responsável pela longa
permanência de drogas como a cloropromazina no organismo.
19
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
drogas que possuem essa capacidade. A indução da CYP3A4 ocorre geralmente
após alguns dias de tratamento e desaparece cerca de uma semana após a suspensão de seu uso. A conseqüência mais comum da indução enzimática é que várias
drogas podem estimular seu próprio metabolismo, levando assim à ineficácia da
medicação e à necessidade de aumento da dose para obtenção dos efeitos originais
(tolerância). Como essas enzimas são inespecíficas, o uso de um indutor pode
causar aceleração da biotransformação de outras drogas administradas concomitantemente. Em fumantes, por exemplo, pode ser necessário o ajuste da dose
de drogas metabolizadas pela CYP1A2A, que é induzida pela nicotina.2
O fenômeno oposto é a inibição enzimática, que determina o acúmulo das
substâncias degradadas pela enzima inibida. O exemplo mais típico em psiquiatria
é o dos antidepressivos inibidores da MAO. Essas drogas, além de acarretarem
acúmulo dos neurotransmissores, que são seus substratos naturais, potencializam
os efeitos pressórios de aminas simpatomiméticas administradas, como descongestionantes nasais e broncodilatadores, ou ingeridas na alimentação, como é o
caso da tiramina. A inibição da MAO intestinal e hepática leva ao acúmulo de
tiramina, que libera noradrenalina da terminação nervosa, acarretando crises hipertensivas. Esse risco é minimizado pela contra-indicação de drogas de ação indireta
e pela restrição dietética (alimentos ricos em tiramina, como queijos envelhecidos,
arenque defumado, etc.).2 Com os inibidores reversíveis da MAO, como a moclobemida, essas interações são potencialmente muito menos perigosas. Outro exemplo
de inibidor enzimático é o dissulfiram, que inibe a aldeidodesidrogenase, promovendo,
assim, o acúmulo de aldeído acético, responsável pelas manifestações desagradáveis
da ingestão de álcool. O sistema enzimático P450 também é alvo de inibição
enzimática. Por exemplo, antidepressivos como a fluoxetina e a fluvoxamina inibem CYP3A3/4, aumentando a concentração de outros substratos da enzima, o
que pode levar à intoxicação.
Os psicofármacos podem também ser excretados no leite, mas seus efeitos
no lactente são pouco conhecidos. Sabe-se, por exemplo, que o diazepam excretado dessa maneira pode produzir efeitos sedativos na criança.
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
DURAÇÃO DA AÇÃO
Conforme já abordado, o início de ação das drogas depende da via de administração, da formulação farmacêutica, da velocidade de absorção e da passagem para
o cérebro, que, por sua vez, depende, dentre outros fatores, da lipossolubilidade.
Já a duração dos efeitos depende principalmente das meias-vidas de eliminação
(β) da droga ou de seus metabólitos ativos. No caso de doses únicas de drogas
muito lipossolúveis, a duração de ação é geralmente determinada pela meiavida de distribuição. Por exemplo, a meia-vida do diazepam, após atingir o pico
de concentração no sangue, é rápida, e ele é extensivamente distribuído aos
tecidos, o que pode resultar em uma curta duração do efeito após doses únicas.2
Por outro lado, quando a administração é repetida, a meia-vida de eliminação
é que determinará a ocorrência ou não de acúmulo do fármaco e/ou de seus
metabólitos. Drogas de meia-vida de eliminação curta, em geral, são completamente eliminadas antes da administração seguinte. Intervalos de administração
inferiores a aproximadamente quatro vezes a meia-vida de eliminação (tempo
necessário para a eliminação completa) provocarão acúmulo. Drogas de meia-vida
curta são as que usualmente determinam fenômenos rebote, isto é, a expressão
exagerada da condição original (insônia rebote, insônia de fim de noite, ansiedade
diurna) e aquelas cuja síndrome de abstinência se manifesta mais rapidamente.5
Algumas drogas, como os inibidores da MAO, exercem efeitos muito mais
prolongados do que sua meia-vida biológica. A tranilcipromina, por exemplo,
tem uma meia-vida plasmática de apenas algumas horas, mas doses únicas de
10 mg desse composto promovem intensa inibição da atividade da MAO de
plaquetas, que somente retorna aos valores pré-tratamento após mais de uma
semana.
Assim, quando se emprega o termo meia-vida, deve-se especificar meia-vida
farmacológica (tempo necessário para que os efeitos de uma droga sejam reduzidos pela metade); meia-vida plasmática (tempo para que concentrações plasmáticas sejam reduzidas em 50%); e meia-vida biológica (tempo no qual as concentrações corpóreas totais de uma substância são reduzidas em 50%).
FATORES QUE MODIFICAM O EFEITO DAS DROGAS
Vários fatores intrínsecos e extrínsecos podem modificar os processos que ocorrem
desde a administração até a eliminação das drogas, alterando, portanto, seu efeito.
Entre os fatores intrínsecos (i.e., dependentes do organismo), destacam-se os
constitucionais, ou seja, a variabilidade individual, os fatores genéticos e a idiossincrasia, bem como a idade, o peso e a composição corpórea. A variabilidade individual é responsável pela variação na intensidade dos efeitos observados com uma
20
21
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
determinada dose dentro de uma população. Os fatores genéticos são os principais
determinantes da insensibilidade ou da sensibilidade exagerada a uma droga,
decorrentes do aumento ou da diminuição da quantidade de enzimas de biotransformação. Uma resposta inesperada é chamada idiossincrasia e pode ser
devida à ausência ou alteração genética de uma enzima.3
Diferenças genéticas podem afetar a resposta aos fármacos, já que são responsáveis por variações nas atividades de enzimas e transportadores envolvidos na
absorção, na distribuição, no metabolismo e na excreção de drogas. A grande
variabilidade genética (étnico-racial), decorrente do polimorfismo de diferentes
isoformas do CYP, pode alterar a metabolização dos fármacos. Os polimorfismos
podem também interferir no padrão de efeitos colaterais, como, por exemplo, o
aumento de peso observado com antipsicóticos, que apresenta alta correlação
com o dimorfismo na região promotora do receptor de serotonina 5-HT2C. Contribuem para o maior risco de discinesia tardia dos antipsicóticos típicos (como o
haloperidol) os fatores ambientais (p. ex., o tabagismo) e o polimorfismo genético
do receptor de dopamina (D3) e da isoforma CYP1A2 envolvida no seu metabolismo.5
A idade assume um papel importante, principalmente nos extremos, quando
os sistemas enzimáticos responsáveis pela biotransformação não estão completamente desenvolvidos (recém-nascidos) ou quando a capacidade de excreção renal está diminuída (idosos). O peso e a constituição corpórea adquirem importância para indivíduos muito magros ou obesos, casos nos quais a proporção
entre água e gordura é diferente da normal. Por exemplo, o obeso necessita de
uma dose maior de drogas que se acumulam no tecido adiposo e menor de
drogas que se distribuem preferencialmente no compartimento extracelular, pois
seu volume de água é relativamente menor.3
Os fatores condicionais são os ligados a condições especiais do organismo,
tais como estado patológico e psicológico. Entre os estados patológicos, destacamse as condições que podem interferir na biotransformação (p. ex., insuficiência
hepática), na ligação das drogas a proteínas plasmáticas (p. ex., subnutrição), na
distribuição (p. ex., edema e desidratação) e na excreção (p. ex., insuficiência
renal, diarréia, acidose e alcalose).3 O estado emocional, por sua vez, pode modificar ou mesmo inverter os efeitos de uma droga. Por exemplo, a administração de
um benzodiazepínico pode levar a um efeito ansiogênico paradoxal em um indivíduo que está muito ansioso e agitado. Além disso, na gravidez, a presença de
quantidades anormais de hormônios pode interferir no efeito de algumas drogas.
No caso particular dos psicofármacos, o efeito placebo, isto é, o efeito não
atribuível à ação farmacológica, assume uma dimensão maior. Esse efeito inespecífico, presente na administração de qualquer medicamento, resulta, entre outros fatores, da interação médico-paciente e da expectativa do paciente em relação
ao resultado do tratamento em termos de efeitos benéficos e colaterais. Estudos
de neuroimagem em pacientes deprimidos demonstraram que, ao menos em
parte, a remissão dos sintomas depressivos produzida pelo placebo é mediada
pelas mesmas alterações cerebrais que as produzidas por antidepressivos ativos.
Os fatores extrínsecos são os dependentes da droga, tais como suas características físico-químicas e sua formulação farmacêutica, e de suas condições de uso,
ou seja, via de administração, dose, administração aguda ou crônica e interação
com outras drogas.3
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
MECANISMOS DE AÇÃO
Os psicofármacos, em última análise, interferem na neurotransmissão. É crescente
o número de neurotransmissores ou neuromoduladores identificados no SNC
(Tab. 1.1). Os principais neurotransmissores são as catecolaminas – noradrenalina
(NA), adrenalina (A), dopamina (DA) –; as indolaminas – serotonina (5-HT), histamina (H) –; os aminoácidos excitatórios – glutamato e aspartato –; os aminoácidos inibitórios – ácido γ-aminobutírico (GABA) –; e o óxido nítrico (NO). Embora
sua participação efetiva no mecanismo de ação dos psicofármacos não esteja
totalmente estabelecida, aparentemente NA, 5-HT, DA e GABA são, provavelmente, os neurotransmissores mais envolvidos com a ação dessas drogas.1,2
É importante lembrar que os sistemas neurais que regulam a atividade do
SNC formam interações complexas entre si. Os neurotransmissores que exercem
funções excitatórias interagem com os que desempenham funções inibitórias,
modulando as funções nervosas de forma balanceada. Conseqüentemente, qualquer manipulação que afete um ou mais componentes desses sistemas afeta o
equilíbrio e manifesta-se por meio de alterações funcionais. Assim, dificilmente
pode-se pensar de forma simplista e julgar que apenas uma determinada ação
farmacológica é a responsável pelo efeito terapêutico de um psicofármaco.1,2
A alteração provocada por uma droga pode decorrer de uma ou de múltiplas
ações em alguma das etapas ou processos que ocorrem nas terminações nervosas.
Como as ações não são exclusivas, isto é, a maioria dos psicofármacos não possui
uma ação seletiva, interagindo com múltiplos sistemas simultaneamente, aumenta
a possibilidade de efeitos colaterais e a potencialidade das interações medicamentosas.
Em relação ao seu local de ação na terminação nervosa, os mecanismos de
ação de uma droga podem ser pré e pós-sinápticos (Fig. 1.2). Por meio da ação
pré-sináptica, a droga vai interferir na regulação da liberação de um neurotransmissor. Desse modo, uma droga pode aumentar ou diminuir a síntese do neurotransmissor, sua liberação, sua estocagem, sua recaptação e sua biotransformação.
A síntese pode ser aumentada pela maior disponibilidade de substratos ou cofatores, como é o caso da L-dopa, precursora da síntese de dopamina, ou pode
ser diminuída pela administração de inibidores enzimáticos, tais como a p-clorofenilalanina, que bloqueia a síntese de serotonina.
Certas substâncias podem ainda facilitar a liberação de neurotransmissores,
tais como os psicoestimulantes anfetamina e fencanfamina, que aumentam a
liberação de DA e de NA ou, ainda, inibir o armazenamento de neurotransmissores
como, por exemplo, a reserpina. Os antidepressivos de primeira geração, tais
como a tranilcipromina e a fenelzina, inibem irreversivelmente a MAO, e os tricíclicos, tais como a imipramina, a amitriptilina e a clomipramina, inibem a recaptação de NA e de 5-HT, em diferentes proporções. As gerações seguintes de
antidepressivos são compostas por grupos heterogêneos de drogas que inibem
22
Tabela 1.1
NEUROTRANSMISSORES/NEUROMODULADORES DO SNC
Peptídeos hipofisários
Corticotrofina (ACTH)
Hormônio do crescimento (GH)
Lipotrofina
Ocitocina
Vasopressina
Prolactina
Neurocininas/taquicininas
Substância P
Neurocinina A
Neurocinina B
Aminoácidos
Ácido gama-aminobutírico (GABA)
Glicina
Ácido glutâmico (glutamato)
Ácido aspártico (aspartato)
Peptídeos opióides
Dinorfina
β-endorfina
Met-encefalina
Leu-encefalina
Hormônios circulantes
Angiotensina
Calcitonina
Glucagon
Insulina
Leptina
Fator natriurético atrial
Estrógenos
Endrógenos
Progestinas
Hormônios tireoideanos
Gases
Óxido nítrico (NO)
Monóxido de carbono (CO)
Outros peptídeos
Bombesina
Bradicinina
Carnosina
Neuropeptídeo Y
Neurotensina
Galanina
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
Aminas
Serotonina (5-HT)
Dopamina (DA)
Norepinefrina (NE)
Epinefrina
Acetilcolina (Ach)
Histamina
Melatonina
Feniletilamina
Octopamina
Hormônios intestinais
Colecistocinina (CCK)
Gastrina
Motilina
Secretina
Peptídeo vasoativo intestinal
Neurotransmissor lipídico
Amandamida
seletivamente a recaptação de 5-HT (fluoxetina, paroxetina, sertralina), de NA
(reboxetina) e de DA (bupropiona), de NA e 5-HT (venlafaxina) ou que têm
outros mecanismos de ação (p. ex., mirtazapina, nefazodona, trazodona).5
Qualquer alteração na liberação do neurotransmissor poderá interferir também
nos mecanismos inibitórios de controle de sua concentração exercidos pelos autoreceptores pré-sinápticos. Esses receptores também podem ser diretamente esti23
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
Figura 1.2
Principais eventos sinápticos.
mulados ou inibidos por substâncias como, por exemplo, a clonidina e a ioimbina,
respectivamente.
Em resumo, uma droga de ação pré-sináptica pode, por exemplo, promover
um efeito excitatório quando, por meio de um ou mais dos mecanismos mencionados, aumenta a concentração de um neurotransmissor excitatório na fenda
sináptica ou diminui a concentração de um neurotransmissor inibitório.
Da mesma forma, a ação de um psicofármaco pode ocorrer em algum dos
eventos pós-sinápticos. Mais uma vez, o efeito final vai depender da ativação ou
da inibição de vias excitatórias ou inibitórias. A droga pode interferir na própria
ligação do neurotransmissor a seus receptores ou nos mecanismos moleculares
pelos quais o neurotransmissor produz alterações na membrana pós-sináptica
(potencial de membrana ou segundo mensageiro), e pode, ainda, atuar diretamente nos receptores de um ou mais neurotransmissores.7
A ação pós-sináptica direta requer que a substância possua um grau de afinidade relativamente alto pelos receptores. Drogas que mimetizam a ação dos neurotransmissores endógenos nos seus receptores são denominadas agonistas. Os agonistas têm afinidade pelos receptores e a atividade intrínseca (ou eficácia), uma
vez que sua interação determina um efeito. O agonista pode ser total, quando
24
25
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
exerce o efeito máximo, ocupando ou não 100% dos receptores disponíveis (p.
ex., apomorfina, LSD e muscimol, que são exemplos de agonistas nos receptores
de dopamina, serotonina e GABA, respectivamente). Já o agonista parcial, mesmo
ocupando 100% dos receptores, produz efeito sempre menor do que o efeito
máximo exercido pelo agonista total (p. ex., ação do aripiprazol nos receptores
dopaminérgicos).1
As drogas também podem se ligar ao receptor e não desencadear nenhuma
resposta (sem atividade intrínseca), sendo nesse caso denominadas antagonistas.
Seu efeito resulta da inibição da ação do neurotransmissor ou de uma droga
agonista, uma vez que elas impedem o acesso dessas substâncias aos receptores,
como no caso de competir pela ocupação deles. Acredita-se que o efeito antipsicótico dos neurolépticos deva-se à sua ação antagonista nos receptores dopaminérgicos mesolímbicos preferencial por receptores D2 (clorpromazina, haloperidol,
flufenazina, olanzapina, risperidona).
Um efeito inibitório também pode ser observado com agonistas parciais, isto
é, drogas que têm a capacidade de se ligar aos receptores (afinidade), porém
cuja atividade intrínseca é menor do que a de um agonista forte.
Existe ainda um grupo de substâncias que se ligam a receptores e promovem
efeitos opostos aos dos agonistas. É o caso dos agonistas inversos (β-carbolinas),
que atuam em receptores de benzodiazepínicos e induzem ansiedade, rigidez
muscular, distúrbios de sono e convulsões. Esses efeitos são bloqueados tanto
por agonistas como por antagonistas e constituem, até o momento, a única exceção na farmacologia de drogas que atuam em receptores.1
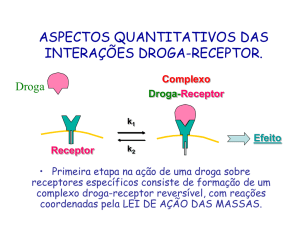
A primeira etapa na ação de uma droga em receptores específicos envolve a
formação de um complexo droga-receptor reversível. Os receptores podem ser
classificados em quatro tipos principais: os canais iônicos, os ligados à proteína G
ou metabotrópicos, os que controlam diretamente sistemas enzimáticos (p. ex.,
receptor de insulina e de fatores de crescimento, ligados à tirosina quinase) e os
intracelulares (p. ex., receptores para hormônios esteroidais e tireoideanos).7
O canal iônico acoplado ao receptor abre-se quando o receptor associado é
ocupado por um agonista. Este é o caso dos benzodiazepínicos cujos receptores
próprios formam um complexo molecular com os receptores GABAA e o ionóforo
de cloro, modulando a freqüência de abertura do canal de cloro. Já os barbitúricos,
por exemplo, modulam a função do canal de cloro pela ligação direta a um sítio
do canal.7
Receptores de dopamina, serotonina e adrenérgicos, por sua vez, pertencem
à família de receptores acoplados às proteínas G (assim denominadas devido à
sua interação com os nucleotídeos guanínicos, GTP e GDP). Nesse caso, a ligação
receptor-agonista desencadeia a ativação de uma proteína G localizada na face
citoplasmática da membrana.
A proteína G liga o receptor a uma via intracelular específica, dependente do
tipo de proteína G presente: estimulatória (Gs), inibitória (Gi/o) ou ativadora do
sistema fosfolipase C (Gq). A proteína G ativada altera a atividade de um elemento efetor (enzima ou canal iônico), que, por sua vez, altera a concentração
do segundo mensageiro intracelular (p. ex., AMPc, diacilglicerol). Para o AMPc,
a enzima efetora é a adenililciclase, uma proteína que converte ATP intracelular
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
em AMPc. A ativação do sistema AMPc pode ser feita a partir de diferentes
sistemas de neurotransmissores e de diferentes tipos de receptores, sendo que as
respostas variam de acordo com o tipo de proteína G ativada pelo receptor. Por
exemplo, os receptores dopaminérgicos D1 e D2 estão acoplados a proteínas G
que, respectivamente, ativam e inibem a adenililciclase. Os receptores β-adrenérgicos e serotoninérgicos 5-HT1 atuam através de uma proteína G estimulatória,
ativando a formação de AMPc.7-10
Outro exemplo de sistema de segundo mensageiro é o fosfoinositídeo. Ele
envolve receptores acoplados a proteínas G que ativam a fosfolipase C, uma
enzima responsável pela formação de diacilglicerol e IP3 (1,4,5-trifosfato de inositol). O IP3 libera cálcio, que, por sua vez, gera uma série de estímulos, entre
eles, a ativação da enzima responsável pela formação do óxido nítrico, a óxido
nítrico sintase. A ativação da guanililciclase solúvel pelo óxido nítrico leva à formação do segundo mensageiro, GMPc. Um exemplo de receptor que atua por
essa via é o 5-HT2 (Fig. 1.3).7-10
Um exemplo de psicofármaco que atua diretamente nos sistemas de segundos
mensageiros é o lítio, que bloqueia a formação do fosfatidilinositol pela inibição
Figura 1.3
Regulação da proteína G e dos segundos mensageiros em sistemas efetores
celulares. (Adaptada de Rang; Dale, 1995.2) GMPc = guanina monosfato cíclico;
AMPc = adenosina monosfato cíclico; IP3 = trifosfato de inositol;
DAG = diacilglicerol; AA = ácido araquidônico; [CA+2] = cálcio intracelular;
PKG = proteína quinase G; PKC = proteína quinase C;
PKA = proteína quinase dependente de AMPc.
26
FENÔMENOS DE NEUROADAPTAÇÃO
Sabe-se que a administração crônica de drogas leva a alterações adaptativas, por
exemplo, nos receptores. Os receptores não só iniciam a regulação de uma função
fisiológica ou bioquímica como também são alvos de controles regulatórios e
homeostáticos. A contínua estimulação de um receptor por um agonista geralmente resulta em um estado de dessensibilização ou subsensibilidade (down-regulation), isto é, uma diminuição do efeito obtido em condições normais. Essa alteração
de resposta pode envolver uma modificação do receptor, sua destruição ou diminuição de síntese ou, ainda, sua realocação na célula. A subsensibilidade é, muitas
vezes, responsável pela tolerância observada após uso crônico.11
O contrário também é freqüentemente observado, ou seja, a hipersensibilidade
do receptor quando exposto cronicamente a uma redução no nível de estimulação.
Em alguns casos, essa hiper-reatividade decorre do aumento de síntese do receptor.
A hipersensibilidade resultante do bloqueio prolongado de receptores dopaminérgicos parece estar envolvida no desenvolvimento da discinesia tardia observada
no tratamento crônico com neurolépticos.11
27
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
da enzima inositol monofosfatase e pela modificação das respostas mediadas
pelo sistema adenililciclase e AMPc.2
Apesar dos inúmeros estudos realizados e dos avanços obtidos, nenhuma
descoberta foi capaz de explicar, de forma satisfatória, o mecanismo responsável
pela ação terapêutica dos psicofármacos a partir do seu efeito agudo sobre os
neurotransmissores ou das alterações na sensibilidade dos receptores produzidas
após administração crônica (ver a seção a seguir, “Fenômenos de neuroadaptação”). Além disso, verificou-se que as alterações metabólicas e funcionais observadas inicialmente nos tratamentos agudos de animais na maioria das vezes não
só não persistem após administração repetida como também são substituídas
por alterações que podem, de fato, ser opostas às observadas agudamente.11
Passou-se, assim, a estudar as alterações moleculares (plasticidade), enfatizando as alterações intracelulares (pós-receptor) promovidas pelos psicofármacos.
Essas alterações envolvem subunidades das diferentes proteínas G, os segundos
mensageiros (AMPc, GMPc, fosfatidilinositol, diacilglicerol), as proteínas envolvidas na síntese e na liberação de neurotransmissores, os mensageiros gasosos
intra e intercelulares (monóxido de carbono e óxido nítrico), os mecanismos de
fosforilação protéica através do estudo das proteínas quinases dependentes destes
segundos mensageiros (PKA, PKC e PKG) e das proteínas reguladoras de proteínas
fosfatases, além dos fatores de transcrição e de receptores de corticosteróides
que regulam a expressão gênica neural.11,12
Espera-se que a incorporação de técnicas sofisticadas, tais como as técnicas
moleculares, o uso de marcadores genéticos e as técnicas de neuroimagem, auxiliem na elucidação dos mecanismos de ação dos psicofármacos, explicando por
que drogas com ações bioquímicas agudas diferentes têm efeitos clínicos similares
e, conseqüentemente, levando ao desenvolvimento de novas drogas mais eficazes
e específicas.12
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
Os mecanismos de neuroadaptação têm sido relacionados não só com fenômenos de tolerância e de dependência, mas também vêm sendo propostos como
subjacentes ao mecanismo de ação de muitos psicofármacos, além de estarem
possivelmente implicados na fisiopatologia dos próprios transtornos psiquiátricos.
Por exemplo, sabe-se que a administração crônica de muitos psicofármacos
antidepressivos, ainda que não de todos, leva, entre outros resultados, ao desenvolvimento de subsensibilidade de receptores α2 pré-sinápticos e de receptores
β-adrenérgicos e de hipersensibilidade de receptores α1. A diminuição na densidade e na função dos receptores β-adrenérgicos é temporalmente relacionada com
o efeito clínico e é observada após administração prolongada de inibidores da
MAO, tricíclicos, antidepressivos atípicos e eletrochoque. Os mecanismos subjacentes a essa subsensibilidade envolvem mecanismos de fosforilação do próprio
receptor por proteínas quinases, como, por exemplo, a β-ARK (quinase do receptor
β-adrenérgico), e o acoplamento de proteínas associadas, como a β-arrestina.
Outro exemplo dessas alterações adaptativas envolve o sistema serotoninérgico. No tratamento agudo com antidepressivos que bloqueiam a recaptação de
5-HT, os auto-receptores inibitórios 5-HT1A, localizados nos corpos celulares dos
neurônios serotoninérgicos no núcleo da rafe, estão expostos a uma concentração
mais alta de 5-HT em função de seu bloqueio da recaptação. Em conseqüência
disso, há uma diminuição no disparo neuronal e liberação de 5-HT. Já no tratamento prolongado, ocorre uma dessensibilização desses receptores, levando a um
aumento na liberação de serotonina, que se correlaciona temporalmente com a
melhora clínica. Tem sido proposto que esse efeito adaptativo está implicado na
eficácia clínica desses compostos.11
Finalmente, é importante considerar que muitas dessas alterações envolvem
a regulação da expressão gênica neural por meio de fatores de transcrição, tais
como o CREB (proteína de ligação ao elemento de resposta ao AMPc), o Fos e o
Jun, que modulam a atividade de proteínas celulares específicas.11-12
TOLERÂNCIA E DEPENDÊNCIA
O uso continuado de uma droga pode levar a uma diminuição da resposta, levando
à necessidade da administração de doses crescentes para a obtenção do efeito
original. Esse fenômeno é chamado de tolerância. Ela pode ser inata, ou seja,
uma sensibilidade preexistente, ou adquirida – neste caso, pode ser farmacocinética (metabólica) ou farmacodinâmica.2
A tolerância farmacocinética, conforme visto anteriormente, decorre da indução enzimática, com conseqüente aumento da biotransformação e diminuição
da concentração do composto ativo no sangue. Por exemplo, a atividade das
enzimas hepáticas aumenta após um período prolongado de consumo de álcool,
acelerando sua eliminação.1
Já a tolerância farmacodinâmica depende dos fenômenos de neuroadaptação:
ocorre por diminuição do número dos receptores ou da sensibilidade neuronal.
28
29
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
Por exemplo, os efeitos anoréxicos das anfetaminas, os efeitos analgésicos dos
opióides e os efeitos sedativos dos benzodiazepínicos e do etanol podem diminuir
de intensidade após administração crônica.
Fatores ambientais também são capazes de desencadear um tipo de tolerância,
conhecida como tolerância comportamental. Para um indivíduo habituado a
consumir determinada droga em um mesmo ambiente, esta pode apresentar
efeitos mais intensos quando administrada em um ambiente diferente. Além disso,
vários psicofármacos apresentam tolerância cruzada com outras drogas da mesma
classe farmacológica (opióides) ou entre compostos de efeitos semelhantes
(barbitúricos e etanol).
O fenômeno oposto à tolerância é a sensibilização, também conhecida como
tolerância reversa, que determina um aumento da resposta inicial após administração crônica. Ocorre, por exemplo, com psicoestimulantes (cocaína, anfetamina),
opióides e etanol. É possível que a sensiblização tenha um papel na manutenção
da dependência, tornando os dependentes mais suscetíveis a recaídas, mesmo
após longos períodos de descontinuação. A sensibilização é uma forma de plasticidade neuronal e está associada com mudanças neuroadaptativas no circuito
da recompensa, decorrentes do uso crônico de drogas de abuso, sendo a dopamina
o principal substrato neuronal envolvido nesse fenômeno.13
A dependência física consiste no estado de adaptação induzido pelo uso contínuo de uma droga e manifesta-se pelo aparecimento de síndrome de abstinência,
isto é, um conjunto de sintomas que ocorrem quando da retirada absoluta ou
relativa de uma substância psicoativa consumida de modo prolongado. A gravidade da síndrome de abstinência é variável e específica para cada droga ou conjunto de drogas.
O conceito de dependência psíquica está ligado ao impulso psíquico que leva
à administração da droga, geralmente para o indivíduo obter prazer com seu
uso. O conceito dicotômico vem sendo abandonado, e a tendência atual é considerar a dependência como um fenômeno único. No DSM-IV14, a dependência
é considerada como um conjunto de sintomas cognitivos, comportamentais e
fisiológicos que indicam que o indivíduo continua a usar a substância, apesar dos
problemas significativos decorrentes desse uso.
Considera-se que a dependência esteja associada à ativação de sistemas de
recompensa relacionados ao reforço positivo e negativo. De acordo com os princípios de reforço, a auto-administração de uma droga se mantém pelas suas conseqüências positivas, isto é, o prazer que ela proporciona, bem como por evitar as
conseqüências negativas decorrentes de sua retirada, isto é, a síndrome de abstinência. As evidências experimentais sugerem que o sistema dopaminérgico mesolímbico e o hipotálamo lateral, via receptores dopaminérgicos e opióides, estejam
envolvidos nos mecanismos de recompensa. Drogas que mantêm a auto-administração, tais como, anfetaminas, barbitúricos, cocaína, etanol, heroína e nicotina,
promovem ativação do sistema dopaminérgico mesolímbico, aumentando a liberação de dopamina no nucleus acumbens.1
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
INTERAÇÕES MEDICAMENTOSAS
São bastante freqüentes as combinações de drogas para se obter a potenciação
ou a diminuição da latência do efeito terapêutico, ou, ainda, para atingir um
número maior de sintomas. Entretanto, muitas vezes essas interações não são
benéficas, ou são mesmo potencialmente perigosas, devido a efeitos aditivos ou
ao antagonismo entre os agentes. Por exemplo, em geral existe efeito aditivo
entre os depressores do SNC, e combinações como as de barbitúricos e álcool
podem ser letais.3
As interações basicamente ocorrem quando as drogas envolvidas atuam no
mesmo sítio de ação (interação farmacodinâmica), podendo ocorrer sinergismo,
potenciação ou antagonismo, ou quando elas se processam fora dos locais de
ação, acarretando alterações na concentração de droga ao nível de seus receptores.
Estas últimas podem ser farmacocinéticas, decorrentes de alteração na absorção,
distribuição, biotransformação ou eliminação. Por exemplo, drogas que retardam
o esvaziamento gástrico e a motilidade intestinal, como as que possuem efeitos
anticolinérgicos, vão interferir na velocidade de absorção de outras drogas. A
competição por ligação protéica e as alterações no metabolismo (ver biotransformação) por conseqüência da indução enzimática (barbitúricos), com diminuição da concentração plasmática da outra droga, e da inibição do metabolismo
(inibidores da MAO), são alguns dos exemplos de como as drogas podem interagir
quando administradas simultaneamente.3
No caso de drogas com faixa terapêutica estreita, as interações medicamentosas devem ser feitas com especial cautela por poderem desencadear efeitos
tóxicos caso sua faixa terapêutica seja ultrapassada.
O mais prudente é, sempre que possível, prescrever apenas um medicamento.
É preferível aproveitar um efeito colateral sedativo para tratar a insônia associada
a quadros psiquiátricos a prescrever um hipnótico para esse fim. Entretanto, muitas
vezes a monoterapia pode não ser suficiente para a melhora adequada do paciente.
Mais ainda, considerando a cronicidade da maioria dos transtornos psiquiátricos,
é comum que ocorram quadros clínicos associados que necessitem de tratamentos
específicos. Quando necessária, a administração concomitante de diferentes drogas deve ser cuidadosa para evitar as interações indesejáveis.2
REFERÊNCIAS
1. Brunton L, Lazo J, Parker K. Goodman & Gilman’s the pharmacological basis of therapeutics.
11th ed. New York: McGraw-Hill; 2006.
2. Rang HP, Dale MM, Ritter FM, Moore PK. Pharmacology. 5th ed. New York: Elsevier;
2004.
3. Oga S. Fundamentos de toxicologia. 3rd ed. São Paulo: Atheneu; 2007.
4. Gorenstein C, Marcourakis T. Princípios gerais. In: Cordas TA, Barreto OCO, editores.
Interações medicamentosas. São Paulo: Lemos; 1998. p. 17-33.
30
5. Davis KL, Charney D, Coyle JT, Nemeroff C. Neuropsychopharmacology: the fifth
generation of progress. Philadelphia: Lippincott; 2002.
6. Malhotra AK, Murphy GM JR, Kennedy JL. Pharmacogenetics of psychotropic drug
response. Am J Psychiatry. 2004 May;161(5):780-96.
7. Cooper JR, Bloom FE, Roth RH. The Biochemical basis of neuropharmacology. 8th ed.
New York: Oxford University; 2003.
8. Greengard P. The neurobiology of slow synaptic transmission. Science. 2001 Nov
2;294(5544):1024-30.
9. Nestler EJ, Hyman SE, Malenka RC. Molecular Pharmacology: a foundation for clinical
neuroscience. New York: MacGraw-Hill; 2001.
10. Popoli M, Brunello N, Perez J, Racagni G. Second messenger-regulated protein kinases in
the brain: their functional rtole and the action of antidepressant drugs. J Neurochem. 2000
Jan;74(1):21-33.
11. Hyman SE, Nestler EJ. Initiation and adaptation: a paradigm for understanding
psychotropic drug action. Am J Psychiatry. 1996 Feb;153(2):151-62.
13. Wise RA. The role of reward pathways in the development of drug dependence.
Pharmacol Ther. 1987;35(1-2):227-63.
14. American Psychiatry Association. Diagnostic and statistical manual of mental disorders
(DSM-IV). 4th ed. Washington: American Psychiatry Press, 1994.
31
PRINCÍPIOS GERAIS DA AÇÃO DE PSICOFÁRMACOS
12. Stahl SM. Essential psychopharmacology: neuroscientific basis and practical applications.
2nd ed. Cambridge: University Press; 2000.