EIM – CHO (GALACTOSEMIA)
•
Doença rara do metabolismo da galactose;
•
Alteração se encontra em uma das enzimas responsáveis pela
conversão da galactose em glicose e dependendo da enzima
defeituosa, a doença se manifesta de maneira diferente.
CLASSIFICAÇÃO:
Galactosemia tipo 1, 2 e a tipo 3.
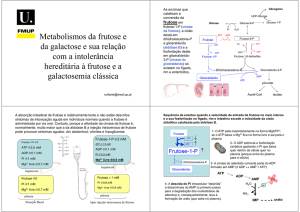
galactose-1-fosfato
uridil transferase
EIM – CHO (GALACTOSEMIA)
A) Galactosemia tipo 1
•
Deficiência da enzima galactose-1-fosfato uridil transferase
(GALT).
•
Acúmulo de galactose-1-fosfato e galactose tecidual.
•
Incidência é de 1: 50.000 na população branca e a idade alvo
é preferencialmente neonatos.
•
Os danos podem se iniciar na fase pré natal a partir da
galactose transplacentária vinda da mãe heterozigota.
•
As características clínicas da doença podem se apresentar de
diversas maneiras diferentes e surgem poucos dias a
semanas após o nascimento:
MANIFESTAÇÕES HEPÁTICAS E GASTROINTESTINAIS:
irritabilidade, letargia, vômitos, dificuldade de alimentação,
baixo ganho de peso, hepatoesplenomegalia, ascite, cirrose
hepática e outras complicações.
SÍNDROME DE FANCONI:
vômitos, desidratação, fraqueza e febre inexplicada, anorexia,
constipação, polidipsia, poliuria e distúrbios do crescimento.
EIM – CHO (GALACTOSEMIA)
A) Galactosemia tipo 1
OUTRAS MANIFESTAÇÕES:
Neurológicas: retardo mental (irreversível), problemas da
fala e coordenação motora.
Endócrinas: disfunção ovariana com amenorréia e
provável aumento de risco de câncer ovariano.
Oculares: catarata, no cristalino a galactose é convertida
pela aldolase redutase em galactitol, um açúcar ao qual o
cristalino é impermeável, e em conseqüência ocorre
hidratação excessiva, redução do glutation no cristalino e
formação de cataratas.
Infecciosas.
EIM – CHO (GALACTOSEMIA)
B) Galactosemia tipo 2
• Deficiência da atividade da galactoquinase (galactokinase)
resultando em danos principalmente nos olhos.
• A incidência é de 1: 40.000 crianças, nas quais desde a
infância podem se formar cataratas.
• Gera acúmulo de galactose no sangue e nos tecidos.
• Características clínicas: catarata, anormalidades do sistema
nervoso
central:
retardo
mental,
neurofibromatose,
deterioração neurológica, epilepsia e pseudotumor cerebral.
C) Galactosemia tipo 3
• Deficiência da atividade da uridil difosfo galactose-4epimerase, resultando em 2 formas da doença, a saber: 1forma inicial; 2- forma grave.
• A incidência é muito rara.
• Gera acúmulo de galactose.
FORMA INICIAL: assintomática (enzima deficiente apenas nas
células do sangue, sendo normal nos outros tecidos).
FORMA GRAVE: sintomática (sintomas clínicos idênticos a forma
da Galactosemia tipo 1).
TRATAMENTO
• Dieta isenta de galactose e lactose.
• Monitoramento constante do nível de galactose e seus
metabólitos no sangue do paciente, deve ser feito para
verificação da eficácia da dieta.
• Monitoramento multidisciplinar.