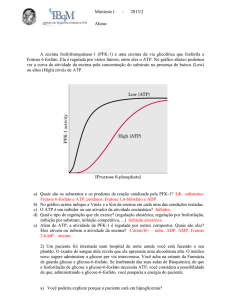
A via dos ácidos urónicos ocorre no fígado e é uma via oxidativa alternativa para
a glicose, no entanto, nesta via não existe produção de ATP. Ela cataliza a conversão da
glicose a ácido glicourónico, a ácido ascórbico e a pentoses.
Inicialmente a glicose 6P é isomerizada a glicose 1P, pela acção da
fosfoglicomutase. A glicose 1P reage com a uridina trifosfato (UTP) para formar glicose
uridina difosfato (UDPGlc), sendo a reacção catalizada pela pirofosforílase da UDPGlc.
Posteriormente, no carbono 6 da UDPGlc ocorre uma oxidação catalizada por uma
enzima NAD-dependente, a desidrogénase da UDPGlc, originando UDP-glicoronato,
que é a forma activa do glicoronato. O UDP-glicoronato participa em reacções que
envolvem a incorporação do ácido glicourónico em proteoglicanos ou nas quais os
substratos são hormonas esteróides, a bilirubina e vários medicamentos. Estes substratos
unem-se ao glicoronato activo, sendo excretados na urina ou na bílis. O glicoronato é
ainda reduzido a gulonato, sendo esta reacção NADH-dependente. Finalmente, o
gulonato é metabolizado a xilulose 5P, sendo este composto um intermediário da via
das pentoses e, como tal, podemos relacionar a via dos ácidos urónicos com esta via,
uma vez que na via dos ácidos urónicos existe a formação de compostos que intervêm
na via das pentoses.
A via dos ácidos urónicos pode dar-se de uma forma deficiente devido a dois
factores: alguns medicamentos e deficiências enzimáticas.
Existe uma doença hereditária rara, na qual consideráveis quantidades de Lxilulose aparecem na urina devido à ausência da enzima necessária à redução deste
composto a xilitol.
A administração de algumas drogas como o barbital ou o clorobutanol
aumentam a taxa de conversão da glicose a glicoronato.
A frutose resulta da hidrólise da sucrose que é um açúcar que se encontra
presente em grandes quantidades em alimentos da nossa dieta. A hidrólise da sucrose é
catalizada pela enzima sucrase.
No fígado a frutose sofre mais rapidamente glicólise do que a glicose, pois a
frutose é capaz de contornar a regulação feita pela fosfofrutocínase. Isto permite que a
frutose sature as vias metabólicas no fígado levando a um aumento da síntese de ácidos
gordos, da esterificação dos mesmos, da produção de VLDL e, consequentemente, a um
aumento da concentração de colesterol LDL.
No fígado, a frutocínase cataliza a fosforilação da frutose a frutose 1P. Esta
enzima não actua sobre a glicose e a sua actividade não é afectada pelo jejum nem pela
insulina, o que pode explicar o porquê da frutose apresentar valores normais no sangue
dos diabéticos. A frutose 1P, pela acção da aldolase, origina gliceraldeído e
dihidroxiacetona. Posteriormente, o gliceraldeído sofre uma fosforilação catalizada pela
triocínase, originando o gliceraldeído 3P. O gliceraldeído 3P e a dihidroxiacetona
podem entrar na via glicolítica e serem degradados ou podem servir de substratos para a
aldolase e, consequentemente entrar na gliconeogénese. Daqui se conclui que da
degradação da frutose vamos obter intermediários da glicólise e da gliconeogénese que
poderão ser importantes no mecanismo de regulação da concentração de glicose no
sangue.
A degradação da frutose no fígado pode provocar problemas de saúde
complicados, tais como, hipertriacilglicerolemia e hiperuricemia que é uma causa do
aparecimento da doença denominada “Gota”.
A falta de frutocínase provoca frutosuria primária e a ausência de aldolase
provoca intolerância hereditária à frutose. A hipoglicemia e o aparecimento de cataratas
nos diabéticos podem estar relacionados com a degradação da frutose no nosso
organismo.
A galactose resulta da hidrólise da lactose que ocorre no intestino, sendo esta
reacção catalizada pela enzima lactase. No fígado, a galactose é prontamente convertida
a glicose.
A galactose é fosforilada a galactose 1P por acção da cínase da galactose,
usando como dador de grupos P o ATP. A galactose 1P reage com a glicose uridina
difosfato (UDPGlc) originando galactose uridina difosfato (UDPGal) e glicose 1P,
sendo a reacção catalizada pela transferase da galactose 1P uridil. Posteriormente, dá-se
a conversão da UDPGal a UDPGlc que é catalizada pela 4-epimerase da UDPGal.
Finalmente, a glicose é libertada da UDPGlc depois da conversão a glicose 1P. Este
composto segue, normalmente, a via da glicogénese originando glicogénio.
A galactose é importante na síntese de lactose na glândula mamária em lactação,
pois nesta ocorre uma condensação entre a glicose e a UDPGal, originando lactose. Esta
reacção é catalizada pela síntase da lactose. A galactose para além de ser importante na
síntese de lactose também é um constituinte importante dos glicolípidos, dos
proteoglicanos e das glicoproteinas.
As galactosemias que se traduzem pela incapacidade de produzir galactose
podem ser causadas por deficiências nas enzimas que participam na via da galactose. As
consequências destas deficiências são variadas. A concentração da galactose no sangue
e nos olhos é reduzida, pela redutase da aldose, a galactitol, que ao acumular-se dá
origem a cataratas. Também, na deficiência da enzima transferase da galactose 1P
uridil, existe acumulação da galactose 1P e há um esvaziamento do fosfato inorgânico
que existe no fígado, provocando a falência deste e uma deterioração mental. Contudo,
se os indivíduos galactosémicos possuírem quantidades adequadas de epimerase, pode
existir na mesma produção de UDPGal a partir da glicose e ocorre um crescimento e
desenvolvimento normais, porém, a dieta destes indivíduos terá que ser livre de
galactose para que possa haver um controlo dos sintomas da galactosemia.
A glicose é o principal precursor das hexoses aminadas, no entanto, também o
glicogénio pode ser um precursor destes compostos. Tanto a glicose como o glicogénio
sofrem uma série de reacções em que intervêm enzimas, tais como, a amidotransferase,
a fosfoglicomutase e epimerases que levam à formação das hexoses aminadas. As
principais hexoses aminadas são o ácido siálico, a glicosamina, a galactosamina e a
manosamina.
As hexoses aminadas são componentes importantes das glicoproteinas, de certos
glicoesfingolípidos e de glicosaminoglicanos.