Resumo do que foi dito durante a apresentação
Introdução teórica
O glicogénio é um polissacarídeo de reserva nos animais e em muitos fungos, sendo
constituído por monómeros de glicose (20.000 a 30.000 moléculas) unidos entre si por ligações
glicosídicas α-1,4 e α-1,6 nas ramificações. Encontra-se essencialmente armazenado no fígado
e nos músculos, onde o seu metabolismo é mais acentuado, quer através de reacções de
síntese, quer de degradação do glicogénio, com o objectivo de manter os níveis de glicose no
sangue dentro dos valores normais. No fígado, esta regulação ocorre para enfrentar as
necessidades de todo o organismo, enquanto que no músculo ocorre para enfrentar as
necessidades glicémicas do próprio músculo (em situações de emergência).
E como se processa, então, este metabolismo?
A síntese de glicogénio começa com a conversão de glicose a glicose-6-fosfato (G6P),
pela acção de uma hexocinase. De seguida, uma fosfoglicomutase isomeriza a G6P em glicose1-fosfato, que por sua vez é convertida em uridina-difosfoglicose (UDP-glicose). Esta tem um
elevado potencial de transferência de fosfato, o que lhe permite adicionar glicose à
extremidade 4’ de uma cadeia de glicogénio, numa reacção catalisada pela glicogénio
sintetase.
Por outro lado, em situações de jejum, quando a concentração de glicose no sangue
diminui, o glicogénio armazenado é, então, degradado, convertendo-se em glicose, num
processo conhecido como glicogenólise.
A G6P tem 3 destinos possíveis:
- pode dar início à glicólise
- pode ser processada pela via das pentoses fosfato (produzindo NADPH e derivados de ribose)
- pode ser convertida em glicose livre (neoglicogénese e glicogenólise) para libertação na
corrente sanguínea (o que ocorre principalmente no fígado), através da G6Pase.
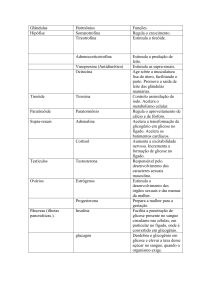
E é através de mensageiros químicos que a glicogenólise é estimulada: a partir da
ligação das hormonas glucagina (no fígado) e adrenalina (no músculo) aos respectivos
receptores membranares, a adenilciclase é estimulada a produzir AMPc que, por sua vez, vai
ser responsável por activar a fosforilase do glicogénio, que é a principal enzima de degradação
do glicogénio, decompondo-o em glicose-1-fosfato. Como devem estar recordados, estas são
hormonas que estimulam a glicogenólise no fígado e músculos, ao contrário da insulina, que
estimula as células do fígado e dos músculos a armazenarem glicose na forma de glicogénio
(estas acções opostas permitem a regulação do metabolismo do glicogénio). De seguida, após
a isomerização da G1P a G6P, por acção da fosfoglucomutase, no fígado, vai ser produzida
glicose através da enzima G6Pase, para ser exportada, pela veia porta, para todo o organismo;
enquanto que no músculo, como não há glicose-6-fosfatase, a G6P (que não pode sair da
célula, devido ao facto da membrana plasmática ser impermeável a ela) não vai ser convertida
em glicose, mas sim metabolizada pela glicólise para a produção de ATP no músculo.
Também a neoglicogénese (ou gliconeogénese – reacção inversa da glicólise, como se
devem recordar) tem como passo final a transformação da G6P em glicose, por acção da
G6Pase.
Quando o glicogénio não consegue ser degradado, devido à deficiência de algumas
das enzimas envolvidas, acumula-se no órgão, e a não libertação de glicose para a circulação
acarreta uma série de consequências para a saúde.
Parte Clínica
As glicogenoses são doenças decorrentes de erros metabólicos hereditários que
resultam em anormalidades da concentração ou da estrutura do glicogénio em qualquer
tecido do organismo. São denominadas de doenças do armazenamento do glicogénio.
Existem mais de 10 diferentes tipos de glicogenoses, dependendo do defeito
enzimático encontrado, pois há defeitos enzimáticos específicos para cada tipo. E dependendo
da distribuição da enzima específica nos tecidos ou órgãos, o armazenamento de glicogénio,
nestas patologias, pode ser limitado a poucos tecidos ou ser mais difundido, havendo
manifestações clínicas variadas (ver slides da apresentação).
Glicogenose tipo I (doença de van Gierke, autossómica recessiva, corresponde a cerca
de 25% das glicogenoses)
Caracteriza-se por uma deficiência do complexo da G6Pase, que é responsável pela
hidrólise da G6P nos órgãos que a possuem (fígado, rim, intestino…), o que faz com que na
glicogenose tipo I não se forme a glicose a partir da G6P, que se acumula, assim, nas células. A
G6Pase é um complexo funcional do RE, constituído por uma unidade catalítica com o centro
activo no lúmen do RE e por transportadores que permitem, quer a entrada do substrato, G6P,
quer a saída dos produtos da reacção, glicose e Pi. E, dependendo do tipo de defeito na
G6Pase, distinguem-se essencialmente 2 subtipos de glicogenose tipo I (ver slides – acredita-se
que o tipo I b seja mais grave…).
Diagnóstico
O gene da G6Pase humana localiza-se no braço longo do cromossoma 17. Assim,
através da análise de DNA (PCR e RFLP) passou a ser possível fazer o diagnóstico desta doença
de uma maneira rápida, precisa e não invasiva e abandonar metodologias anteriores que
exigiam biopsia hepática e o estudo da actividade enzimática. Actualmente, só se fazem
estudos enzimáticos em casos raros, em que o diagnóstico genético não é possível.
Tratamento
Não há cura para a GSD I (Glicogen Storage Disease type I). O controlo glicémico
constitui a base do tratamento:
- Dieta: amido de milho não cozinhado permite manter os níveis de glicose
normalizados.
- Transplante hepático ou renal (quando o tratamento dietético falha)
- Fármacos: alopurinol – hiperuricemia; bicarbonato – acidémia
- Terapêutica génica usando adenovírus parece ser promissora…