Percurso dos fármacos no corpo
humano
1
Vias de administração de fármacos
1.
Oral: podem ser formas de dosagem sólidos ou líquidos.
Dentro dos
líquidos temos as gotas e os comprimidos efervescentes. Os sólidos podem ser:
a. Comprimidos: têm a forma de um disco, são obtidos por pressão
mecânica da substância activa e excipientes, que permitem serem de
fácil manuseamento e de engolir. A quantidade de substância activa é
em pequena dose (alguns miligramas). O uso de um desintegrante como
o amido que aumenta de volume em contacto com a água, ou o NaHCO 3
que liberta CO2 em contacto com o ácido gástrico, permite principio
activo fique liberto para ser absorvido.
b. Os comprimidos revestidos: contêm a substância activa no centro e
coberto por uma “concha” que pode servir: (1) de protecção para um
farmaco que seja de fácil decomposição; (2) mascarar o cheiro ou sabor
desagradável; (3) facilita a passagem ou o ser engolido; (4) permitir
código de cor.
c. Comprimido do tipo matriz: a substância activa é embebida numa matriz
inerte a partir do qual se vai libertando por difusão devido a ir-se
molhando.
Em contraste com o que ocorre com as soluções, que permitem uma absorção directa
do farmaco as formas de dosagem sólida requerem no caso dos comprimidos a sua
quebra e no caso das cápsulas a sua abertura (desintegração) antes que o farmaco
seja dissolvido e passe a mucosa do trato gastrointestinal onde é absorvido. Dado que
a desintegração do comprimido e a dissolução do farmaco demora tempo a absorção
ocorre principalmente no intestino. No caso de serem soluções a absorção ocorre
principalmente no estômago.
2.
Rectal ou vaginal: são os supositórios ou ovos (?). A aplicação rectal tem
como objectivo a absorção para a circulação sistémica, enquanto a aplicação
vaginal o efeito é local. O principio activo é incorporado numa gordura sólida
À temperatura ambiente, mas que funde a temperatura corporal (pf próximo
dos 35 ºC)
3.
Ocular: são os colírios.
2
4.
Nasal: são usados os aerossóis; têm uma aplicação que é um nebulizador
que permite a dispersão de um líquido ou partículas de sólido sob a forma de
gás, no trato respiratório.
5.
Cutânea: são pós pastas ou unguentos. São aplicados na pele e
normalmente não contêm substâncias farmacologicamente activas. São usados
como protecção ou cuidado. Podem ser adicionados substâncias activas para
actuar localmente ou raramente têm um efeito sistémico
6.
Parenteral (injectáveis):
a. Intravenosa: o farmaco é introduzido directamente no sangue
b. subcutânea e intramuscular: o farmaco difunde-se do sitio de aplicação
para o sangue
Desde a aplicação até à distribuição no corpo
Com regra geral os farmacos chegam até aos órgãos alvo via sangue. Pelo que entram
no sangue normalmente pela circulação venosa. Existem vários sítios de entrada:
1- Se o farmaco for injectado intravenosamente então é introduzido
directamente no sangue;
2- Se a aplicação do farmaco for por injecção subcutânea ou intramuscular o
farmaco difunde-se desde o sitio de aplicação até ao sangue;
3- Se for de aplicação oral, o fármaco tem que passar a mucosa
gastrointestinal.
A desvantagem desta via de administração é que o
farmaco tem que atravessar o fígado antes de atingir a circulação geral. O
fígado é um órgão de metabolização de substâncias pelo que o farmaco
pode ser alterado e eliminado.
4- A No acaso de administração do farmaco ser rectal uma parte dele pode
“driblar” o fígado e entrar na circulação geral via veia portal que comunica
com a veia cava inferior que conduz ao coração.
5- Se administração do farmaco for sublingual ou por inalação a irrigação da
cavidade bucal drena directamente para a veia cava superior, pelo que
entra na circulação geral sem passar pelo fígado.
Biodisponibilidade: é definida como a fracção de um dado fármaco que atinge a
circulação sem ser alterada e se torna disponível para a sua distribuição sistémica.
3
Quanto maior a eliminação pré-sistémica, menor é a biodisponibilidade de um
farmaco administrado oralmente.
Distribuição no corpo
Barreiras externas
1- Epitélio intestinal: é a 1ª barreira a quando da administração oral do farmaco.
É uma mono camada de células, sendo formada por enterócitos e células
globulares que produzem muco. Na parte luminal estas células são mantidas
juntas por junções constituídas por proteínas trans-membranares. Desta forma
uma bicamada de fosfolípidos continua faz a separação entre o lúmen
intestinal e o espaço intersticial.
É a chamada barreira Barreira intestinal
mucosa-sangue. Os farmacos que ultrapassam esta barreira têm propriedades
físico-químicas que permite atravessar o interior lipofílico da membrana ou
têm um mecanismo de transporte. A superfície de absorção é muito grande
devido ao epitélio em forma de “escova”.
2- Células epiteliais do trato respiratório: são células que têm cílios e igualmente
estão juntas devido à acção das proteínas, formando-se uma bicamada de
fosfolípidos que separa o espaço bronquial e o interstício.
3- Epitelio da mucosa oral: é uma multicamada de células epiteliais não
queratinizadas. Entre as células formam-se bicamadas fosfolípidicas continuas
o que implica que apenas fármacos lipifilicos a consigam atravessar
4- Pele: tem uma multicamada de células epiteliais queratinizadas, com bicamada
de fosfolípidos continuas (tal como acontece no epitelio da mucosa oral).
Barreiras tecido – sangue
Os fármacos são transportados pelo sangue até aos diferentes tecidos do corpo. Para
chegarem ao seu sítio alvo têm que abandonar a circulação sanguínea. A permeação
dos fármacos ocorre a nível capilar, onde a área superficial é elevada e a velocidade
do sangue é baixa. A parede capilar forma a barreira tecido-sangue. Basicamente é
constituída por uma camada endotelial de células, que estão juntas por acção de
4
proteínas, não havendo poros, ou falhas, etc entre elas, de modo a que o fármaco se
possa escapar.
Esta barreira é diferente a nível dos capilares do (1) músculo cardíaco; (2) sistema
nervoso central (SNC); (3) Pâncreas e (4) fígado.
No musculo cardíaco as células endoteliais são caracterizadas pelo por uma intensa
actividade endo e transcitotica, visualizando-se a presença de invaginações e
vesículas. A actividade transcitótica faz o transporte de líquidos ou macromoléculas
do sangue para o interscicio e vice-versa. Os farmacos são assim transportados pelo
que as suas propriedades físico químicas não têm aqui grande importância.
As células epiteliais do pâncreas exibem poros que estão fechados por um diafragma.
Este tal como a membrana podem ser atravessados por moléculas de baixo peso
molecular (ex: insulina). Este tipo de endotelio com “janelas” ocorre também no
intestino e nas glândulas endócrinas.
No fígado existe um livre intercâmbio entre o sangue e o interstício dado que as
células epiteliais dos capilares têm fendas (poros) de grande diâmetro.
No sistema nervoso central as células capilares não têm poros e têm pouco actividade
transcitótica. Os farmacos para atravessarem a barreira hemato-encefalica têm que
se difundir transcelularmente, ie. Penetrar as membranas luminal e basal do
endotelio. Este movimento do fármaco requer pois que ele tenha propriedades físicoquímicas específicas.
Permeação da membrana
A capacidade de penetrar a bicamada lípidica é um pré-requisito para que ocorra
absorção de fármacos, para a sua entrada nas células ou organelos celulares e para
passar a barreira hemato-encefálica. As substâncias podem atravessar a bicamada
lípidica de 3 diferentes maneiras (1) Difusão (2) Transporte (“carrier”) (3) Endocitose
mediado ou não por receptores.
5
A difusão de substâncias lipofílica (desenho são as bolas encarnadas) podem
atravessar a membrana do espaço extracelular, acumular-se na membrana e sair para
o citosol. A direcção e a velocidade de permeação depende da concentração relativa
nas fases fluidas e na membrana.
Os farmacos podem ser transportados pela membrana através de sistemas de
transporte (“carrier”). O pré-requisito necessário é que a substância tenha afinidade
para o carrier. Este mecanismo é sujeito a inibição competitiva.
Transcitose (transporte vesicular) forma-se uma vesícula de transporte com a
substância.
Endocitose mediada por um receptor, é um sistema mais complexo em termos gerais
envolve 1- ligação do receptor ao farmaco, 2- ligação do complexo formado com uma
proteína do citosol, 3- Migração do complexo de modo a juntar-se a outros, 4, 5, 6invaginação com formação de uma vesícula que contém o complexo, 7- Formação do
revestimento diferente da vesícula, 8- regresso do receptor à membrana celular, 9transporte do farmaco para o sitio alvo (ex: Golgi, núcleo, lisossoma, etc).
Possíveis modos de distribuição
Depois de entrar no corpo , o fármaco é distribuído pelo sangue (1) e pelos vários
tecidos do corpo. A distribuição pode ficar restrita ao espaço extracelular (plasma e
espaço intersticial (2) ou pode-se extender ao espaço intracelular (3).
Muitas
substâncias ligam-se fortemente a a estruturas tecidulares, pelo que a sua
concentração no plasma desce significativamente mesmo antes de começar a
eliminação (4).
As substâncias que permanecem no espaço vascular (em circulação) ou estão ligadas a
proteínas plasmáticas ou não são capazes de atravessar as barreiras tecido-sangue.
A distribuição das substâncias no corpo depende da capacidade de penetrar as
membranas. As substâncias hidrofilicas como a inulina é usada para determinar o
6
fluido extracelular (caso 2). Algumas substâncias lipidicas atravessam as membranas
pelo que têm uma distribuição uniforme.
Ligação às proteínas plasmáticas
Depois de entrar no sangue os fármacos podem-se ligar às proteínas aí presentes,
formando-se complexos farmaco-proteína. As proteínas ligantes são a albumina (4,6
g/100 mL, tem dois sítios de ligação por molécula), -globulinas, glicoproteinas
acídicas.
As ligações são instantâneas e reversíveis e podem ser do tipo iónico,
dipolo-dipolo, ião dipolo e de van der Waals.
Estas ligações são de grande importância pois delas dependem a concentração de
farmaco livre o qual determina a intensidade do seu efeito terapêutico. O aumento
ou diminuição de farmaco livre também causa efeito na sua biotransformação e
eliminação, dado que apenas a forma livre entra nos hepatocitos para a
metabolização ou sofre filtração glomerular.
O aumento da ligação do farmaco com a proteína provoca um aumento da duração do
efeito.
Eliminação dos fármacos
O Fígado é o orgão responsável pelo metabolismo dos fármacos. As enzimas que
têm estas funções estão localizadas na mitocôndria, nas membranas do reticulo
endoplasmático rugoso (RER) ou liso (REL). As mais importantes estão situadas no
REL.
Se o fármaco for polar então será rapidamente excretado pelos rins. Contudo se
não o for não será excretado e o objectivo do metabolismo é convertê-lo em
compostos mais polares de modo a serem excretados.
As vias metabólicas têm sido divididas em duas grandes categorias:
1- Reacções de fase 1 (biotransformações): inclui oxidação, hidroxilação,
reducção e hidrólise. Estas reacções conduzem à formação de metabolitos com
efeitos terapêuticos (pró-farmacos) ou com efeitos tóxicos.
7
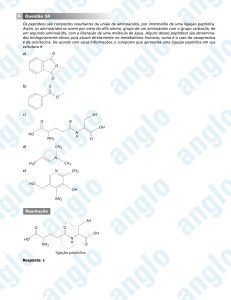
2- Reacções de fase 2 (conjugações): são sínteses enzimáticas onde o grupo
funcional é mascarado pela adição de um novo grupo; grupo acetil 1, sulfato 2,
acido glucuronico 3, glutationa 4 ou certos aminoácidos como a glicina 5, que
aumentam a polaridade da molécula.
HO2C
O
O
R
CH3
R O S OH
OH
HO
O
2
1
O
HS
HN
O
HO
OH
3
CO2H
N
H
O
CO2H
NH2
O
OH
H2N
5
4
Ambas as fases podem ocorrer independentemente ou sequencialmente.
Principais vias oxidativas
A conversão metabólica mais comum é a enzimática.
As estruturas mais
susceptíveis de oxidação são grupos N-metil, aneis aromaticos poisições terminais da
cadeia alquilica e as posições menos impedidas dos anéis não aromáticos. Tabela 1
mostra as reacções oxidativas como exemplo.
Tabela 1. Vias oxidativas catalisadas by CYP450
Reacções
Exemplo1
Reacções
oxidativas
N- Dealquilação
R N CH3
H
O- Dealquilação
ROCH3
Hidroxilação
R CH2CH3
RNH2 + CH2O
Codeína
ROH + CH2O
Ibuprofeno
R CHCH3
alifática
OH
Hidroxilação
R
R
aromática
Morfina
HO
R
Fenobarbital
O
8
N- Oxidação
R NH2
R1
R N OH
H
R1
NH
R2
S- Oxidação
R1
R1
S
Cimetidine
S O
R2
CH3
R
N OH
R2
R2
Desaminação
Acetaminofeno
OH
R
NH2
Anfetamina
O
CH3
+ NH2
R
NH2
CH3
1 As estruturas dos compostos estão no apêndice;
O sistema enzimático envolvido na biotransformação de fármacos chamado
citocromo P450 (abreviadamente CYP450).
Exemplos de fármacos metabolizados
Acetaminofeno = N-acetil-p-aminofenol = paracetamol
É um analgésico e antipirético muito comum. È normalmente encarado como um
fármaco seguro quando administrado no intervalo terapêutico. Contudo uma dose
exagerada ou depois de administração prolongada e repetida pode causar
hepatotoxicidade.
O
HN
CH3
NHCOCH3
OC2H5
OH
6
NHCOCH3
7
8
Acetanilida, 7 e a fenacetina 8, são os outros membros deste conjunto de
fármacos, mas o paracetamol ganhou popularidade depois de ser reconhecido como a
substância mais activa.
9
Metabolismo
Primeiramente, o acetaminofeno (6, figura 1) é metabolizado no fígado por
conjugação (fase 2 do metabolismo), com ácido glucurónico (cerca de 60%), ácido
sulfúrico (cerca de 35 %), ou cisteína (cerca de 3%), formando-se metabolitos não
tóxicos.
Uma pequena parte do paracetamol por acção do CYP 450, ocorre a N-
hidroxilação formando-se o N-acetil-benzoquinoneime (NAPQI, 9, passo 1, Figura 1)
que é um intermediário muito reactivo.
Este metabolito normalmente reage com
grupos sulfidril da glutationa (passo 2, Figura 1).
Este passo é considerado de
desintoxicação dado que o NAPQI livre exerce a sua toxicidade ligando-se a
macromoléculas, como proteínas, lípidos e DNA.
Os compostos conjugados são
excretados pela bílis ou pela urina.
10
sulfato
glucuronido
major
NHCOCH3
major
NHCOCH3
major
NHCOCH3
major
7
minor
OH
6
minor
NH2
OC2H5
OC2H5
8
minor
NH2
HO
N
COCH3
HO
OH
Anemia hemolitica
COCH3
N
Anemia hemolitica
OC2H5
Passo 1
hepatóxico
COCH3
N
O
9
N-Acetilbenzoquinoneima, NAPQI
(metabolito tóxico)
proteínas hepáticas
N-acetilcisteina
Passo 2 glutationa
NHCOCH3
NHCOCH3
NHCOCH3
NHCOCH3
proteínas hepáticas
S CH2CH CO2H
OH
OH
glutationa
necrose hepatica e falha renal
OH
excrecção renal como ácido mercapturico
Figura 1. Metabolismo do paracetamol
11
Toxicidade
O excesso de consume de paracetamol pode causar necrose hepática. Isto resulta
da activação das enzimas para formar a forma reactiva NAPQI em grande quantidade
o que causa o esgotamento da glutationa hepática. Daqui resulta que a conjugação
passa a ser feita com proteínas com grupos SH (tiois) das células do fígado. Está
igualmente provado que ocorre perturbação da homeostase do cálcio.
O tratamento desta intoxicação é a administração de compostos de modo a com
enxofre como N-acetilcisteina 10 ou metionina 11,
Disulfiram 12 é o inibidor mais potente da formação do NAPQI no fígado, ao
bloquear CYP450. 4-Methil-pirazole 13 que pode ser administrado intravenosamente
é igualmente utilizado para este fim.
O
NH2
S
HS
OH
HN
OH
O
O
11
10
(C2H5)N
S
S
S
CH3
N(C2H5)
N
N
H
S
12
13
Anfetamina
Anfetamina, racemica -phenilisopropilamine 14, é um estimulante poderoso do
sistema nervosa central
12
NH2
CH3
14
O seu mecanismo de acção envolve 4 efeitos nos nervos: 1- inibição dos
mecanismos de recapturação de várias aminas como a noradrenalina e dopamina; 2aumento da libertação das catecolaminAs; 3- estimulação dos receptores adrenergicos; 4- inibidor da monoamina oxidase (MOA) em concentração elevada.
Terapêuticamente, anfetamina tem sido usada na narcolepsia, que é um
problema caracterizado por episodeos de sono acompanhado por cataplexia (perda de
tonus muscular frequentemente causada por emoção) e alucinações.
Metabolism
A Figura 2 o metabolismo da anfetaminas. Há 3 vias oxidativas principaisno
metabolismo da anfetamina: 1-envolve a hidroxilação do grupo metileno do benzilico
com a formação da noradrenalina; 2- hidroxilação do anel aromática seguido pela
oxidação do grupo alquilo com p-hidroxinordrenalina como produto da reacção; 3- Noxidação do grupo amina seguido de desaminação catalisada pela MAO (monoamina
oxidase) uma cetona é formada. Esta cetona vai originar o ácido hipúrico no final do
metabolismo.
A hidroxilação do anel aromático ou dos seus substituintes é a principal via
metabólica das anfetaminas.
13
CH3
NH2
14
CH3
CH3
OH
HO
O
CH3
NH2
Fenilacetone
16
NH2
p-Hidroxianfetamina
15
Noradrenalina
COOH
OH
CH3
HO
Ácido benzoico
NH2
17
p-Hidroxinoradrenalina
O
N
H
COOH
Ácido hipúrico
Figura 2. Metabolitos da anfetamina.
Toxicidade
A toxicidade da anfetamina provém da acção do SNC por aumento da quantidade
dos neurotransmissores noradrenalina e dopamina. Isto causa vários efeitos: aumento
da pressão da velocidade de batimentos cardíacos e da respiração; aumento do estado
de alerta, euforia, boa disposição e aumento da performance, etc. Os produtos da
degração da anfetamina causam toxicidade. Portanto, o p-hidroxinoradrenalina 17
pode funcionar como noradrenalina.
A excreção da anfetamine é aumentada acidificando a urina porque a anfetamina
como base que é totalmente ionisada em condições
reabsorção tubular.
ácidas o que evita a sua
O uso de cloreto de amónio é usado para o tratamento da
toxicidade pela anfetamina.
14
Farmacocinética
(def.)
Estuda
a absorção,
metabolização,
distribuição
e eliminação
dos
medicamentos no corpo. Apenas uma pequena porção do fármaco administrado
chega até ao alvo.
A velocidade de absorção depende da via de administração.
Se a via de
administração for intravenosa o tempo necessário para atingir a máxima concentração
no plasma é mínimo. De seguida o fármaco começa a ser eliminada via rins ou via
fígado (pela bílis). A área debaixo das curvas no gráfico concentração do fármaco no
sangue versus tempo é independente da via de administração.
15
Apêndice 1
Estrutura
Nome
R
R = R’ = H, Morfina
R = Me, R’ = H, Codeína
O
N CH3
H
H
R'O
Me
N
HN
N
C S C C N
H2
H2 H2 H
H
CN
N Me
H
Cimedine
NH2
Anfetamina
CH3
O
HN
CH3
Acetaminofeno
OH
CH3
Ibuprofeno
COOH
H
O
N
C2H5
O
Fenobarbital
N
H
O
16