Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
Adaptação e
Desregulação
Metabólicas
Jejum
Prioridade no jejum:
manutenção da glicémia para utilização de glicose pelos tecidos
insulino-independentes
fornecimento de substratos energéticos alternativos aos tecidos insulinodependentes;
manutenção da homeostasia energética do sistema nervoso central.
Principais reservas energéticas do organismo
glicogénio hepático (utilização como glicose após glicogenólise
hepática);
triacilglicerol do tecido adiposo (utilização como ácidos gordos livres,
ou após β-oxidação e síntese de corpos cetónicos no fígado);
proteínas (principalmente musculares; utilização dos aminoácidos como
substrato da gliconeogénese hepática).
Jejum imediato (3-10h)
Manutenção da normoglicémia e aumento da glicagina pelos efeitos da
diminuiçao da insulina
Activação da glicogenólise e gliconeogénese hepáticas, devido à baixa
relação insulina/glicagina.
Diminuição do consumo de glicose pelos tecido insulino-dependentes
A glioconeogénese ocorre a partir do piruvato, lactato e aminoácidos,
até então utilizados na lipogénese que é interrompida.
Há um baixo balanço proteico, inicialmente, devido à diminuição de
insulina.
Alguns aminoácidos livres libertados, especialmente de cadeia ramificada
são oxidados no músculo e os seus grupos amina transferidos para o
piruvato que resulta da glicogenólise formando alanina que é levada para
o fígado como substracto.
A libertação de NEFA pelo tecido adiposo é elevada devido à não
supressão da Lipase hormono-dependente pela insulina.
O ciclo de cori e da alanina tornam-se importantes.
Página 1 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
Fome precoce (10-24h)
Substituição progressiva da glicogenólise pela gliconeogénese, enquanto
principal fonte de glicose para a manutenção da normoglicémia.
Os ciclos de Cori e da Alanina têm papéis importantes, mas não fornecem
carbonos para a sintese de glicose. O lactato e a alanina, que atingem o
figado, limitam-se a fornecer Carbonos para aformação de glicose, que
substitui aquela que foi convertida aos mesmos pelos tecidos periféricos.
Na verdade, estes ciclos funcionam como um mecanismo de
transferência de energia, obtida através da -Oxidação hepática, para os
tecidos que não têm capacidade para oxidar ácidos gordos.
Mas, a síntese de glicose é inevitável no jejum, já que há tecidos, como o
cérebro, que oxidam completamente a glicose a CO2 e H2O, não
havendo possibilidade de recuperação do composto utilizado.
O glicerol e os aminoácidos gerados a partir da lipólise (no tecido
adiposo) e proteólise (no músculo), respectivamente, constituem as
principais fontes de carbono para a síntese de glicose.
As proteínas são hidrolisadas no interior das células musculares.
Dos aa resultantes, apenas 2 – alanina e glutamina – são libertados na
corrente sanguínea em grandes quantidades, a partir da qual atingem o
fígado.
Os restantes são metabolizados a piruvato ou a -cetoglutarato, a partir
dos quais se forma alanina e glutamanto, respectivamente.
Os -cetoácidos derivados de aminoácidos de cadeia ramificada, por
transaminação, são, também, libertados no sangue, para captação do
fígado, que sintetiza glicose a partir de valina, corpos cetónicos a partir de
leucina e ambos a partir da isoleucina.
Grande parte da glutamina libertada pelo músculo é parcialmente
oxidada nos enterócitos para produção de energia e percursores para
biossíntese de pirimidinas e purinas, que essas células de divisão rápida
executam. Grupos de carbono e amina remanescentes são libertados, de
volta, na corrente sanguínea, sob a forma de alanina e NH4. Essa via é
chamada de glutaminólise
A gliconeogénese hepática durante o jejum está intimamente ligada à
sintese da ureia
Devido aos baixos níves de insulina, a lipólise é activada no tecido
adiposo, proporcionando o aumento do nível sanguíneo de ácidos
gordos, que passam a ser utilizados por muitos tecidos enquanto
combustivel alternativo à glicose.
Nesses tecidos a -oxidação inibe a glicólise e a descarboxilação
oxidativa do piruvato a acetil-coA.
No fígado, esta via metabólica, é ainda, responsável pelo fornecimento
da maior parte do ATP necessário para a gliconeogénese.
Página 2 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
Fome intermédia (1-24 dias)
Nesta fase, a lipólise periférica encontra-se fortemente activada, com os
ácidos gordos libertados a constituirem a principal fonte energética para
muitos tecidos.
Contudo, no fígado, muito pouco acetil-CoA gerado pela -oxidação é
oxidado completamente. Ao invés disso, é convertido em corpos
cetónicos, que são libertados para a corrente sanguínea e distribuidos
pelos tecidos periféricos.
Contando que constituem uma fonte de energia para a generalidade
dos tecidos, inclusivé o cérebro (uma vez que consegue atravessar a
barreira hemato-encefálica), os corpos cetónicos diminuem a
necessidade de glicose corporal, promovendo a inibição da
gliconeogénese e supressão da proteólise e a oxidação de aa de cadeia
ramificada no músculo, por diminuição de substratos gliconeogénicos,
com redução da perda muscular.
No entanto, é preciso ter em conta que, os corpos cetónicos são
incapazes de substituir completamente a necessidade de glicose
corporal, pelo que a gliconeogénese continua a ocorrer.
Fome prolongada (mais de 24 dias)
Os corpos cetónicos adquirem uma maior importância enquanto
metabolito energético
Verifica-se a inibição do ciclo da ureia, com a excreção de azoto a
ocorrer sob a forma de NH4, formado directamente no rim, a partir da
glutamina.
O carbono remanescente é utilizado no processo de gliconeogénese
renal
Página 3 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
Regulação hormonal na situação de jejum
Diminuição da insulina
Diminuição de T3 (triiodotironina) redução do metabolismo basal
Aumento da glicagina
Aumento da epinefrina (por activação do SNS)
Aumento da somatotrofina
Aumento de cortisol
Efeitos metabólicos da epinefrina:
no fígado (receptores β2):
activação da glicogenólise e da gliconeogénese;
nos ilhéus de Langerhans:
diminuição da libertação de insulina (células β, receptores α2)
aumento de glicagina (células α, receptores β);
no músculo (receptores β2)
diminuição da captação de glicose
aumento da libertação de alanina e lactato (substratos
gliconeogénicos);
no tecido adiposo (receptores β1 e β2)
activação da lipólise, com aumento dos ácidos gordos livres e do
glicerol.
Sintomas de hipoglicémia:
sintomas neuroglicopénicos
(resultantes de diminuição do
fornecimento de glicose ao
sistema nervoso central)
versus
sintomas neurogénicos
(resultantes da activação do
sistema nervoso simpático e da
libertação de catecolaminas)
Resposta metabólica à realimentação
Jejum nocturno:
Refeição promove a libertação de insulina, com activação da
glicogénese e inibição da glicogenólise e da gliconeogénese;
Melhor adequação de refeição rica em glícidos, para promover a
utilização da glicose e evitar hipoglicémia.
Jejum prolongado:
situação associada a degradação acentuada de proteínas corporais,
incluindo vias enzimáticas de degradação de aminoácidos, com
utilização dos aminoácidos preferencialmente por desaminação, e sem
destoxificação do NH3 pelo ciclo da ureia;
realimentação rica em proteínas origina hiperamoniémia (tóxica para o
sistema nervoso central).
Página 4 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
Exercicio Físico
Exercício físico anaeróbio (intenso e curta duração)
Pouca coorperação inter-orgãos
A contracção leva a compressão dos vasos sanguíneos do músculo,
levando ao isolamento das células do resto do organismo
Obtenção de energia através da creatina-fosfato, numa primeira fase, e
depois através do glicogénio muscular.
A creatina fosfato serve como fonte de Pi para a síntese de ATP, enquanto
a glicogenólise e a glicólise não são estimuladas.
Ocorre glicólise anaeróbia, produzindo lactato
É possivel ocorrer glicólise aeróbia, dependendo da oximioglobina
Exercicio fisico aeróbio (intensidade média e longa duração)
Obtenção de energia através do glicogénio muscular e dos ácidos
gordos.
As reservas de glicogénio podem ser aumentadas por exercicio exaustivo
que esgota o glicogénio seguido de dieta rica em hidratos de carbono
O aumento da entrada de glicose na célula é aumentada como
consequência da translocação do GLUT 4 para a membrana plasmática
O quociente respiratório (CO2 expirado/O2 inspirado) desce durante o
exercício, logo vai haver uma troca entre glicogenólise para lipólise
Aumenta a importância dos ácidos gordos após 2h de exercicio
Aumenta a -oxidação, devido à menor [malonil-CoA] que vai aumentar
a actividade da Carnitina Palmitoil Transferase I.
Aumenta o AMP, devido ao aumento da hidrólise de ATP, aumentando
assim a actividade das cinases activadas por AMP.
Existe um pequeno aumento dos corpos cetónicos no sangue.
Página 5 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
Efeito do treino de endurance:
melhor capacidade cardiovascular;
aumento da massa muscular;
diminuição do tecido adiposo;
aumento da resistência óssea;
alterações musculares estruturais
o maior densidade capilar,
o número e tamanho das mitocôndrias aumentado
o maior concentração de mioglobina
alterações musculares metabólicas
o maior sensibilidade à insulina
o maior expressão do transportador da glicose GLUT4
o aumento da actividade das enzimas
lipoproteína lipase,
enzimas oxidativas mitocondriais – ciclo de Krebs e βoxidação
glicogénio sintase.
Efeito da dieta no exercício físico
Dieta rica em hidratos de carbono após exercício físico promove o
armazenamento muscular de glicogénio (devido ao aumento da
expressão do transportador GLUT4 e à activação da glicogénio sintase).
Cafeína (inibidor da fosfodiesterase) como activador da lipólise periférica
e da β-oxidação muscular, acelerando a capacidade muscular de utilizar
ácidos gordos livres como principal metabolito energético e poupando o
glicogénio muscular.
Página 6 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
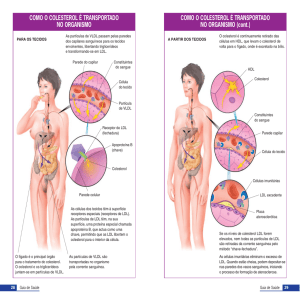
Transporte lipidico
A energia disponível nos ácidos gordos necessita de ser distribuída através do
organismo, a partir do local de absorção, biossíntese ou armazenamento para
os tecidos funcionais que a utilizem.
Formas de transporte de lipidos no sangue
1. Lipoproteinas plasmáticas, nas quais os triacilgliceróis e outros lípidos são
transportados como gotículas de lípidos recobertas por proteínas;
2. Ácidos Gordos Livres ligados à Albumina sérica; (tecido adiposo fígado)
3. Corpos Cetónicos: Ácido Acetoacético e β-Hidroxibutírico (fígado
músculos, coração, cérebro)
Lipoproteínas plasmáticas
Estrutura
Um "core" (núcleo central) contendo lípidos neutros ou apolares (ésteres
do colesterol e triacilgliceróis).
Ao redor há uma camada de proteínas e lípidos antipáticos (formando
uma mono camada lipídica que fazem a interface entre as fases
aquosa e lipidica) composta de colesterol livre (não esterificado) e
fosfolípidos, sobretudo Lecitina (fosfatidilcolina).
Nessa camada superficial encontram-se as proteínas – apoproteínas.
Quanto menor a partícula, maior a densidade proteica no envoltório
externo e menor o teor lipidico no núcleo.
Classificação
Lipoproteína
Conteúdo
Apoproteína
Origem
Quilomicra
Triacilgliceróis
da dieta
(exógenos)
Colesterol da
dieta
Tracilglicerois
endógenos
AI, AII, AIV,
B48, C, E
Intestino
Quilomicra
remanescente
VLDL
(very low density
lipoproteins)
IDL
(intermediate DL)
LDL
(low DL)
HDL
(high DL)
Colesterol
endógeno
B48, E
B100, C, E
Fígado e
intestino
B100, E
VLDL
B100
IDL
AI, AII, C e E
Fígado e
intestino
Mecanismo de libertação
do conteúdo
Hidrólise pela lipoproteina
lipase
Endocitose mediada por
receptores (fígado)
Hdróliese pela lipoproteina
lipase
Endocitose mediada por
receptores (fígado)
Endocitose mediada por
repectores (fígado e outros
tecidos)
Transferência dos ésteres de
colesterol para outras
lipoproteínas (LDL/IDL)
Página 7 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
Apolipoproteinas
−
−
−
−
Conferem estabilidade estrutural às lipoproteinas
Determinam o destino metabólico das partículas que as contém
São co-factores enzimáticos;
Servem de ligação para interacção com os receptores de lipoproteinas
nos tecidos.
Tipos
AI
AII
AIV
B-48
B-100
CI
CII
CIII
E
Funções
Cofacto (LCAT); estrutura (LDL)
Actividade (lipase hepática); estrutural (HDL)
Controlo da saciedade ?
Estrutural (quilomicra)
Estrutural (VLDL e LDL); ligação ao receptor celular
Cofactor (LCAT) ?
Activador (Lipoproteina Lipase)
Inibidor (Lipoproteinas Lipase) ?
Ligação ao receptor celular
Enzimas envolvidas
Lipoproteína Lipase (LPL) Plasmática
− Está presente na superfície capilar de todo o organismo.
− Hidrolisa os triacilgliceróis presentes na VLDL e nos quilomícrons, gerando
ácidos gordos livres e glicerol a nível tecidual.
− Os ácidos livres difundem para os tecidos subjacentes, mas uma pequena
parte regressa à circulação onde se liga à proteína albumina.
− A insulina estimula a síntese e a actividade da LPL.
− Na diabetes mellitus, a diminuição ou ausência do efeito da insulina,
prejudica a depuração de triacilgliceróis, o que agrava a
hipertriacilgliceridémia.
− Os estrógenios criam uma distribuição diferenciada da Lipoproteína lipase
plasmática, definindo o padrão corporal feminino.
− A sua actividade é dependente da Apo C-II e da insulina.
Triacilglicerol 2 ácidos gordos + 2-Monoacilglicerol
Lipoproteina lipase
Lipase Sinusoidal hepática
− Hidrolisa triacilgliceróis e fosfolipidos da IDL nos sinusóides hepáticos,
convertendo-a em LDL.
− Converte a HDL em HDL3
Lipase Ácida Lisossómica
− Hidrolisa os remanescentes dos quilomícrons, VLDL (IDL) e a HDL 2 captada
pelo fígado.
Página 8 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
LCAT (Lecitina Colesterol Acil Transferase)
− Catalisa a transferência do acil (em geral ácido linoleico, ácido
araquidónico) da lecitina para o colesterol livre – ambos componentes da
monocamada lipidica externa, gerando éster de colesterol e lisolecitina.
− A reacção transforma a molécula polar do colesterol (externo) num éster
apolar.
− Os ésteres de colesterol têm dois destinos:
1. Migram para a região apolar central da HDL ou;
2. São transferidos para VLDL e LDL.
− A Apo A-I é cofactor para a acção da LCAT.
Lecitina + Colesterol Lisolecitina + Éster de colesterol
LCAT
ACAT (Acil-CoA Colesterol Acil Transferase)
− Tem como função a formação de ésteres de colesterol
Colesterol + Oleil-CoA Oleato de colesterol + CoASH
ACAT
Proteínas de Transferência
CETP (proteína de transferência de ésteres de colesterol)
− É produzida no fígado e circula em associação com LCAT na HDL.
− Actua na transferência de ésteres de colesterol das HDL por troca dos
triacilgliceróis das VLDL, LDL e em menor escala para os quilomícrons.
− É um activador indirecto da LCAT, uma vez que remove o seu produto de
inibição.
− É de salientar que esta troca só se realiza se existirem triacilgliceróis.
− Se a quantidade de triacilgliceróis ingerida for elevada vão existir
triacilgliceróis suficientes para a troca entre as lipoproteinas e
consequentemente o colesterol vai passar das HDL para as outras
lipoproteinas. Ora tal processo não é benéfico uma vez que o colesterol
transportado pelas HDL destina-se à sua eliminação no figado.
− Por outro lado existe o aumento de partículas LDL densas e pequenas.
− Deficits nesta proteína levam ao aumento da HDL.
PLTP (proteínas de transferência de fosfolipidos): participam na troca de
fosfolípidos e triacilgliceróis dos quilomícrons e das VLDL por ésteres de
colesterol da HDL3.
Página 9 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
1. Transporte de Lípidos Exógenos - Quilomícrons
Após a digestão, o colesterol e os triacilgliceróis são absorvidos e
incorporados no core dos quilomícrons nascentes.
A Apo B-48, Apo A-I e Apo A-IV são geradas no retículo endoplasmático
rugoso do enterócito e combinam-se com o colesterol, fosfolípidos e
triacilgliceróis (gorduras) absorvidos.
Os quilomícrons formados vão para vesículas secretórias, são lançados no
líquido extracelular e captadas pelo sistema linfático (devido ao seu
tamanho).
Somente os compostos hídrossolúveis são absorvidos pelos capilares
sanguíneos mesentéricos e levados ao fígado pelo sistema porta, os
compostos lipossolúveis são absorvidos pelo sistema linfático.
Os quilomicras interagem com a HDL, de quem recebem Apo E
(necessária para que os quilomícrons remanescentes possam ser captados
pelo fígado) e a Apo C-II (essencial para a hidrólise dos triacilgliceróis). As
apo C são devolvidas à HDL.
A Apo C-II activa a LPL, que hidrolisa os triacilgliceróis, os quilomicra
perdem
90%
dos
triacilgliceróis
formando-se
os
"quilomicras
remanescentes".
Os ácidos gordos são captados e utilizados pelos tecidos.
Os "quilomicras remanescentes" pesam cerca de metade relativamente
ao quilomicra, sendo a sua fracção em colesterol e ésteres de colesterol
maior devido à perda dos triacilgliceróis.
Os quilomicras remanescentes são captados pelo fígado (sendo
degradados pela lipase lisossómica hepática).
Somente deste modo o colesterol (e os demais lipídos ingeridos na dieta)
atinge o fígado.
O colesterol captado pelo fígado, podendo seguir por três caminhos:
1. Excretado na bílis como colesterol livre;
2. Convertido a ácidos biliares e lançado na bílis;
3. Secretados para o sangue dentro das VLDL.
Página 10 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
2. Transporte de lípidos endógenos – VLDL e LDL
Excedentes calóricos, consumo de álcool e diabetes mellitus levam à
lipogénese, com a conversão de glicose em ácidos gordos, fosfolípidos e
colesterol e destes em triacilgliceróis (gorduras) e ésteres de colesterol.
Estes lípidos são reunidos com apo B-100, C-II, C-III e E, gerando a VLDL,
que é libertada pelo fígado com destino ao tecido adiposo.
As VLDL interagem com as HDL recebendo mais apo E e apo C-II. Esta
activa a LPL, que hidrolisa triacilglicerois, reduzindo a VLDL de tamanho e
tornando-se mais densa à medida que vai perdendo triacilglicerois.
No final desse processo, restam VLDL remanescentes ou IDL
Parte das IDL são captados pelo fígado e degradados, o restante é
convertido em LDL.
No processo, IDL perde triacilgliceróis, Apo E e Apo C-II, o que representa
um aumento na concentração de colesterol.
A apo B-100 é a única apoproteína que permanece na LDL.
A LDL contém a maior parte do colesterol circulante, 75 % do qual na
forma de ésteres de colesterol.
A LDL é captada virtualmente por todas as células do organismo, mas os
seus principais órgãos-alvo são as glândulas supra-renais (com maior
concentração de receptores), as gónadas e o fígado, através do receptor
para as LDL.
O receptor para a LDL é uma glicoproteína omnipresente no nosso
organismo que reconhece a Apo B-100.
Captação da LDL:
− Receptores de LDL dispostos na região membranar revestida por
clatrina
− Endocitose dos receptores de LDL – vesícula revestida
− Vesículas revestidas perdem o revestimento – despolimerização da
clatrina
− Receptores desligam as LDL
− Fusão das vesículas desrevestidas com lisossomas
− Receptores das LDL são recicladas para as membranas
− Degradação das proteínas LDL; hidrólise dos ésteres de colesterol.
Regulação da endocitose do complexo receptor-LDL
Colesterol intracelular suprime a actividade de hidroximetilglutarilCoA redutase (feedback negativo)
Colesterol intracelular estimula a actividade do acilo-CoAcolesterol aciltransferase.
Colesterol inibe a síntese dos receptores das LDL.
A meia vida plasmática da LDL é determinada pela actividade do
receptor de LDL. Níveis elevados de LDL implicam baixa actividade do seu
receptor e aterosclerose.
A captação de colesterol mediada por receptores de LDL não gera foam
células (precursoras da aterosclerose). Somente a LDL oxidada
(lipoperoxidação e oxidação da apo B-100) é aterogénica.
Níveis elevados de LDL-colesterol aumentam a hipótese da LDL oxidada esta é a relação entre hipercolesterolémia e aterogénese.
Página 11 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
3. Transporte de lípidos endógenos – HDL
Funções da HDL:
Fornece Apo CII e E às VLDL e quilomicra
Remove colesterol em excesso das células para ser eliminado no
figado.
A HDL nasce no fígado de fosfolípidos com apo A-I (sintetizada no fígado
e intestino), apo A-II, apo A-IV e apo C.
HDL “nascente” ou “discoide” (HDL1) são heterogéneas em tamanho, não
contém colesterol
Quando HDL1 passa a conter estes lípidos, o seu interior arredonda a HDL,
dando-lhe um aspecto esférico, formando HDL3.
A HDL3 é rica em ésteres de colesterol graças à LCAT, que retira colesterol
livre da superfície da HDL – libertando espaço para a captação de mais
moléculas de colesterol dos tecidos.
A HDL3 torna-se maior e menos densa formando HDL2
Esta é captada pelo fígado, podendo libertar os esteres de colesterol,
tornando-se menos densa, reformando HDL3 de novo
As HDL3 podem trocar ésteres de colesterol por triacilgliceróis das VLDL,
com o auxílio das "proteínas de transferência".
Página 12 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
Remoção do Colesterol das Células
Mecanismos:
1. Difusão, em que o colesterol difunde da membrana plasmática para a HDL;
2. Transporte mediado por Apoproteínas, em que a HDL interage com
receptores de membrana para apoproteínas e retira o colesterol da
membrana.
A difusão do colesterol da célula para a HDL só ocorre quando existe um
gradiente de concentração entre a superfície celular e HDL receptora do
colesterol.
Quando a HDL está saturada de colesterol, a difusão continua somente se
houver LCAT (lecitina colesterol acil-transferase), que modifica as
propriedades da HDL, aumentando a sua capacidade de remover o
colesterol das células.
A LCAT esterifica o colesterol presente na superfície da lipoproteína, sendo
o éster deslocado da periferia para o núcleo da HDL.
Desse modo, os fosfolípidos (lecitina) ficam livres na periferia para
incorporar mais colesterol livre.
A retirada de colesterol das células é também dependente da Apo A-I,
que se dissocia da HDL e se aproxima da superfície celular, retomando à
HDL após a captação do colesterol.
A Apo A-I não apenas remove colesterol e fosfolípidos (lecitina)
directamente da membrana plasmática, mas também estimula a
mobilização do stock de colesterol que está disponível para a
esterificação mediada pela ACAT (Acil Colesterol Acil-Transferase), uma
enzima localizada no retículo endoplasmático.
Isto evita o acúmulo intracelular de colesterol, mesmo nos macrófagos e
células espumosas.
Assim, a HDL rapidamente depleta os stocks celulares de colesterol.
A Apo A-I, A-II, Apo A-IV e Apo E fazem o mesmo efeito, porém têm
metade da potência da Apo A-I.
A interacção HDL-célula mediada pela Apo A-I mobiliza o colesterol tanto
da membrana quanto do stock intracelular.
A CEHT (colesteril-ester hidrolase neutra) hidrolisa os ésteres de colesterol
mas esse colesterol não fica disponível para re-esterificação pela ACAT,
sendo então facilmente removido da célula.
O colesterol circula continuamente entre o retículo endoplasmático e a
membrana plasmática, passando antes pelo complexo de Golgi.
As células acumulam mais (ou menos) colesterol de acordo com a sua
necessidade de sintetizar membranas. Quando a membrana fica saturada
com colesterol o excesso é dirigido ao retículo endoplasmático, sendo
guardado sob a forma de ésteres de colesterol.
Página 13 de 14
Bioquimica Fisiológica
Adaptação e Desregulação Metabólica
Mecânica da captação do colesterol
A Apo A-I dissocia-se da superfície da HDL circulante, migrando para o
interstício celular, onde se liga ao colesterol livre.
A redução na concentração local de colesterol permite que este se
difunda da membrana plasmática para o interstício, a favor do seu
gradiente de concentração.
A Apo A-I recém sintetizada é secretada pelo fígado como
apolipoproteína pobre (ou desprovida) em lípidos.
Parte desta Apo A-I interage com receptores do próprio fígado, sendo
convertida em HDL1, mas algumas moléculas circulam para tecidos extrahepáticos, acumulando-se no líquido intersticial.
Através de interacções reversíveis com receptores de membrana, essa
Apo A-I remove o colesterol livre e fosfolípidos, convertendo-se
primeiramente em Préβ -HDL, e posteriormente em HDL1.
A esta altura dos acontecimentos, começam-se a formar as hélices, de
modo que a
HDL1 vai perdendo a sua capacidade de interagir com os receptores de
superfície celular.
Estas partículas entram, a seguir, na cascata de amadurecimento.
A LCAT passa a esterificar o colesterol livre, construindo o núcleo lipidico
dos ésteres do colesterol da HDL3.
Alguns destes ésteres são então transferidos para as VLDL e LDL por acção
da CETP (proteína de transferência de ésteres do colesterol). Outros são
transferidos para o fígado e tecidos que sintetizam hormonas esteróides.
A remoção destes núcleos lipídicos deforma a partícula, o que permite a
dissociação das Apo A-I localizadas na periferia, gerando novos
precursores para a via de remoção do colesterol.
Deste modo, a Apo A-I é usada múltiplas vezes para mobilizar lípidos.
Página 14 de 14