FerreiraBreve
e cols.
Comunicação
Recorrências de comunicação intera rial
Arq Bras Cardiol
volume 73, (nº 2), 1999
Recorrência de Comunicação Interatrial
em Três Gerações
Celso Ferreira, Leila M. S. Farah, Rui M. Póvoa, Bráulio Luna Fº, Andréa Costa, Celso Ferreira Fº
São Paulo, SP
A partir de um portador de comunicação interatrial
(CIA), do tipo defeito da fossa oval, foi identificada
genealogia com quatro elementos afetados, pertencentes
a três gerações sucessivas. As anomalias foram comprovadas, visualmente, em todos os portadores, e eram análogas
anatomicamente. Não ocorreram outras malformações
em qualquer dos portadores da cardiopatia. A genealogia
foi identificada em 1972, ocasião em que recorreu em
duas gerações, e se concluiu por mecanismo de transmissão autossômico recessivo.
O quinto elemento, identificado 21 anos após, portador de anomalia idêntica aos demais, era filho de casal
não consangüíneo, sendo a progenitora membro da
genealogia anteriormente estudada.
Levando em conta a ausência do fenótipo nos pais e a
raridade do gene da CIA na população geral, foi admitida ocorrência plausível do fenômeno da dissomia uniparental para este núcleo familial, e o mesmo mecanismo
autossômico recessivo de transmissão por este afetado.
O presente trabalho tem o objetivo de divulgar a recorrência familial da CIA, pelo mecanismo genético de
transmissão, tornando evidente a necessidade do estudo
genético-clínico dos demais membros do núcleo familial,
para detectar novos portadores, freqüentemente assintomáticos e, assim, proporcionar tratamento precoce e adequado aos eventuais afetados.
A incidência das cardiopatias congênitas varia com a
metodologia utilizada em diferentes averiguações e foram
realizadas por diferentes autores. No período de 1946 a 1953,
no Presbyterian Medical Center de Nova Iorque, foram
observadas 6.053 crianças incluindo-se também natimortos
e conceptos com peso >500g. Verificou-se nesse estudo, a
presença de 50 cardiopatias congênitas, isto é, 8,3 por mil,
variando essa incidência de 7,7% para os nascidos mortos e
falecidos no 1º mês, a 6 por mil para os que viveram mais de
um mês 1. Entre nós, Saldanha e cols. 2 encontraram a inci-
Universidade Federal do Estado de São Paulo – EPM
Correspondência: Celso Ferreira – Rua Leandro Dupret, 317 – 04025-011 – São
Paulo, SP
Recebido para publicação em 29/10/98
Aceito em 7/4/99
dência de 3,4% de malformações, de modo geral, em nascidos vivos no Hospital das Clinicas da Faculdade de Medicina da USP. Particularmente para as anomalias cardíacas, a incidência foi de 2,37 por mil. Para Insley 3, a proporção relatada foi de 6 para mil nascidos vivos. Campbell 4 acredita que
10% correspondem a CIA, o que resultaria na incidência de
0,6 por mil desta afecção ao nascer.
A distribuição das cardiopatias congênitas varia nos
diferentes grupos etários, assim a prevalência torna-se diversa da incidência ao nascer. Campbell 4, comentando este
aspecto, comparou seus dados aos de MacMahon e cols. 5
e de Carlgren 6 sobre a incidência preferencial em relação ao
sexo, com a prevalência obtida por Keith e cols. 7. Para a
CIA, cuja incidência é de 1:1 ao nascer, chega a 2:1 na idade
adulta, o que nada mais é que sua prevalência.
As anomalias congênitas, inclusive as cardiopatias
congênitas, podem ser decorrentes de alteraç es endógenas, ou seja, genéticas, tanto em anomalias microscópicas
ou cromossômicas, quanto submicroscópicas ou gênicas.
Por outro lado, alterações exógenas representadas por teratógenos externos, como irradiações ou mesmo ação de infecções viróticas no período da embriogênese, podem ocasionar anomalias idênticas (fenocópias e genocópias).
Os trabalhos que demonstram a recorrência familial de
CIA são relativamente pouco numerosos. Courter e cols. 8,
pioneiros em mencionar a repetição de CIA sem associação
de outra anomalia cardíaca congênita, admitiram em sua
publicação a síndrome de Lutembacher. Embora esta primazia não possa ser confirmada por trabalhos anteriores, o
fato identifica a época como o início desses relatos. Gänsslen e cols. 9 estudaram 68 heredogramas de várias cardiopatias congênitas e descreveram a existência de evidências
de herança recessiva em algumas delas, particularmente,
naquelas com persistência do canal arterial e com CIA.
Courter e cols. 8 apresentaram duas irmãs, cujo diagnóstico
clínico foi CIA, sem associação com outras anomalias cardíacas congênitas. Concomitantemente, diagnosticaram estenose mitral reumática em ambas, razão pela qual foram enquadradas na síndrome de Lutembacher. Carleton e cols. 10,
baseados na apresentação de uma única família, concluíram
por mecanismo de transmissão recessivo; Howitt 11, também baseado na apresentação de uma única genealogia,
concluiu por herança dominante; Nora e cols. 12 concluíram
por mecanismo multifatorial; Zetterqvist e cols. 13 admitiram
211
Ferreira e cols.
Recorrências de comunicação interatrial
herança dominante, apresentando três genealogias e Volti e
cols. 14 concluíram por herança dominante.
Em tese de doutorado, Ferreira 15 realizou estudo genéticoclínico da CIA do tipo fossa oval isolada em oito genealogias e
concluiu por mecanismo de herança do tipo autossômico
recessivo, em função da casuística e metodologia utilizadas.
O presente trabalho relata a ocorrência de um quinto
portador de idêntica anomalia, detectada em 3ª geração consecutiva na mesma genealogia.
Em 1972, ocasião em que se desenvolvia na UNIFESP
estudo genético-clínico da CIA do tipo defeito da fossa
oval isolada, dentre as oito genealogias estudadas, foi destacada a do presente relato.
O propósito (III - 9) (fig. 1), portador de CIA de importante repercussão hemodinâmica, foi internado na enfermaria de cardiologia para se submeter a correção cirúrgica,
efetivamente ocorrida dias após, comprovando-se visualmente a presença e características da anomalia. A partir
desse paciente, foram examinados 24 elementos desse heredograma. Eram submetidos a exames clínicos e de rotina,
onde se incluíam entre outros, eletrocardiograma (ECG) e
radiografia de tórax. Quando havia suspeita de anomalia
cardíaca, o exame era complementado por estudo hemodinâmico. Com esta metodologia, foram detectados mais
três portadores de CIA que, por critérios similares, foram
submetidos à cirurgia para a correção, evidenciando-se
idêntica malformação.
Em 1993, foi encaminhado à UNIFESP, outro portador
de cardiopatia congênita que, estudado clinica e complementarmente por radiologia, eletrocardiografia e ecocardiografia, demonstrou a recorrência de CIA, evidenciando-se
no tratamento cirúrgico tratar-se do mesmo tipo anatômico
das demais anomalias da genealogia. A partir desse paciente, foram estudados pela mesma metodologia os pais e irmãos, tendo sido excluídas novas recorrências.
Para tratamento mais eficaz de qualquer afecção em
medicina é primordial a abordagem da etiologia, freqüentemente, de difícil elucidação para os portadores de anomalias
congênitas. Em conseqüência, conforme Gordon 16, que
comparou as taxas de mortalidade na infância decorrentes
de óbitos por doenças diarréicas, e anomalias congênitas,
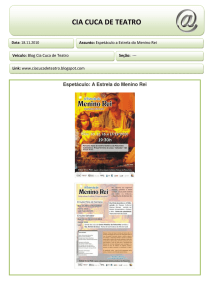
Fig. 1 – Heredograma demonstando a recorrência de CIA do tipo fossa oval em
5 elementos de três gerações.
212
Arq Bras Cardiol
volume 73, (nº 2), 1999
no período de 1910 a 1965, constatou-se a inversão da importância relativa dessas situações clinicas.
Para as cardiopatias, devido a dificuldade da elucidação da etiologia nas anomalias congênitas, associada ao
grande avanço nas prevenções primária e secundária das
cardiopatias adquiridas, cuja etiologia é freqüentemente
bem definida, torna-se cada vez mais aparente sua relativa
importância e faz com que o estudo genético-clínico das cardiopatias congênitas torne-se cada vez mais relevante.
O maior obstáculo à aceitação da hipótese genética para
as cardiopatias congênitas foi levantado por alguns estudos de
gêmeos realizados em uma análise passível de crítica 17-25. Em
gêmeos monozigóticos, idênticos quanto à sua constituição
gênica, seria esperada maior concordância de cardiopatias entre seus pares, do que nos gêmeos dizigóticos, que são apenas
irmãos da mesma idade. Esses estudos, entretanto, não mostraram estas diferenças. No entanto, Nora e cols. 26, que reviram
a literatura pertinente ao assunto, demonstraram que os resultados em gemelares apóiam a hipótese genética para explicar as
cardiopatias congênitas. Assim, conseguiram 88 pares
monozigóticos, 104 dizigóticos e 78 cuja zigozidade não havia
sido diagnosticada. Nesse material, fizeram uma seleção daqueles em que os diagnósticos de zigozidade e de cardiopatia
eram seguros, resultando um número bem menor, ou seja, 36
monozigóticos e 41 dizigóticos, entre os quais incluíram seu
próprio material de investigação. Nesses gêmeos, a concordância foi bem maior que a obtida pelos autores anteriormente
citados, ou seja, 25% para os monozigóticos e 4,9% para os
dizigóticos. Com estes resultados, a influência genética é evidente e a importância do ambiente na determinação das
cardiopatias congênitas, tornou-se incontestável.
Analisando-se a recorrência verificada na presente genealogia, constata-se que ocorreu apenas na linha horizontal, afetando tanto pacientes do sexo feminino quanto do
masculino, além da presença de consangüinidade, o que
apoia o mecanismo de transmissão autossômica recessiva.
Aliás, no estudo genético clínico realizado com as oito genealogias, a recorrência em irmãos com as referidas anomalias do septo atrial foi de 11,5% com desvio padrão de 6,3%,
quando incluídos apenas os pacientes cuja comprovação
do defeito foi completa, e 16,7% com desvio padrão de 6,8%,
quando incluídos também irmãos falecidos, mas com probabilidades de apresentarem CIA 15. Vale ressaltar que esses
resultados não diferem significativamente das proporç es
esperadas para a herança recessiva, isto é, de três normais
para um afetado.
Ademais, deve ser lembrado neste trabalho, que se trata
de complementação de estudo anterior, onde se avaliaram mais
seis genealogias com a mesma metodologia, concluindo-se,
naquela oportunidade, o mencionado mecanismo genético 15.
A recorrência de cinco portadores de CIA em uma genealogia chama a atenção, mesmo a observadores menos
familiarizados com a genética, em levantar tal mecanismo
como fator etiológico. Esta possibilidade torna-se mais evidente, ao se cotejar a incidência desta anomalia na população geral, ou seja, 6:10 000 4 com a expressiva recorrência na
genealogia apresentada. Por outro lado, deve-se notar que
Ferreira e cols.
Recorrências de comunicação intera rial
Arq Bras Cardiol
volume 73, (nº 2), 1999
a anomalia presente, ou seja, defeito da fossa oval, foi idêntica em todos os afetados, o que mais uma vez reforça a etiologia genética.
A importância do estudo genético-clínico para os portadores de cardiopatias congênitas, aqui particularizandose a CIA, é demonstrada pela identificação de novos pacientes evidenciados pela pesquisa sistemática. Assim, o
exemplo da paciente III-7 que havia falecido há anos, e as informações de seus parentes próximos, não eram condizentes com o diagnóstico de cardiopatia congênita, mas sim
com o de cardiopatia hipertensiva. Nesse caso, o registro
hospitalar foi de real valia, pois, além de ser bastante minucioso, apresentava a descrição do exame necroscópico, podendo-se comprovar, não só a existência do defeito cardíaco, como também, suas particularidades anatômicas.
Também o paciente IV-8, produto de casamento consangüíneo, desconhecia a existência de qualquer anomalia cardíaca, era assintomático, e o diagnóstico resultou unicamente
da investigação sistemática referida neste artigo.
A utilização de informações obtidas em registros hospitalares e mesmo de certidões de óbitos podem, por vezes,
merecer criticas, já que nem sempre chegam a um grau de
precisão desejado. As distorções acarretadas pelo diagnóstico não permitem ser aceitas sem rigorosa análise, conduzindo a estimativas, via de regra, menores que as reais. Na
presente genealogia, teve-se a oportunidade de constatar
um exemplo enquadrado em uma dessas situações. O paciente III-13 era assintomático, com vida ativa e desconhecia
qualquer anormalidade cardíaca. O levantamento dos dados
de seu registro hospitalar, feito por ocasião de internação
para pequena cirurgia, fazia referência a resultado normal do
exame do coração. Contudo, ao ser examinado, por ocasião
da avaliação para este estudo, com a atenção voltada para a
detecção de anomalias cardíacas, foi feito o diagnóstico clinico de CIA, comprovado a seguir visualmente pelar cirurgia. Finalmente, deve-se referir a recorrência do paciente V3, encaminhado com o diagnóstico de CIA, comprovado
pela correção cirúrgica.
A identificação do quinto afetado, produto do casamento de jovens não consangüíneos, permitiu por sua vez, a
avaliação dos pais e irmã, tanto clinicamente, quanto por exames complementares, incluindo o ECG, radiografia de tórax e
ecocardiograma, concluindo-se por indivíduos saudáveis.
A nova recorrência chamou a atenção no sentido de
reconsiderar o mecanismo de transmissão autossômico recessivo, anteriormente admitido, já que os progenitores
não eram consangüíneos. Para tal mecanismo, sem que os
pais exibissem o fenotipo da CIA, conforme os conhecimentos clássicos de transmissão recessiva, seria necessário que ambos fossem portadores do gene em heterozigoze, fato que para o heredograma apresentado, seria plausível para a progenitora, porém de muito baixa probabilidade para o pai do afetado, tendo em conta a freqüência do
gene na população geral. A explicação coerente para o fato
baseia-se na hipótese da ocorrência do fenômeno da
dissomia uniparental. Este mecanismo, aplicado a este
heredograma, admitiria que apenas a progenitora do paciente V-3, embora fenotipicamente normal, apresentasse o
gene para CIA em heterozigoze.
A dissomia uniparental é um fenômeno que tem sido
aventado para explicar a ocorrência de doenças autossômicas recessivas em portadores em que apenas um dos progenitores é heterozigoto para o gene em questão 27. Este mecanismo pode resultar da correção de uma trissomia em período inicial do desenvolvimento embrionário, quando aí estariam representados dois cromossomos maternos, carregando o gene da anomalia, e um cromossomo paterno, onde estaria localizado o gene normal e que seria eliminado. Outra
possibilidade seria o restabelecimento muito cedo na gravidez, do estado dissômico a partir de uma monossomia do
cromossomo materno, onde estaria localizado o gene da
anomalia, pela duplicação do cromossomo único, configurando uma isodissomia deste cromossomo.
Independente das teorias que possam justificar a
recorrência da anomalia, é importante frisar que, decorrendo
o tempo e a 4ª geração entrando na idade reprodutiva e levando-se em conta, também, a penetrância e expressividade
observadas no presente heredograma, tornar-se-ia mandatória a retomada da avaliação genético-clínica para identificar novos afetados. É conveniente enfatizar a necessidade
de pesquisa em novos portadores com objetivos não só da
prevenção secundária mais precoce, como eventualmente a
prevenção primária pelo aconselhamento genético nesta e
em todas as situações onde ocorra a CIA aparentemente esporádica.
Referências
1.
2.
3.
4.
5.
6.
7.
Richards MR, Merrit KK, Samuels MH, Langmann AG. Congenital malformations
of cardiovascular system in series of 6.053 infants. Pediatrics 1955; 15: 12-32.
Saldanha PH, Cavalcanti MA, LEMOS ML. Incidência de defeitos congênitos
na população de São Paulo. Rev Paul Med 1963; 63: 211-29.
Insley J. The heretability of congenital heart disease. Br Med J 1987; 294: 662-3.
Campbell M. Causes of malformations of the heart. Br Med J 1965; 2: 895-904.
MacMahon B, McKeown T, Record RG. The incidence and life expectation of
children with congenital heart disease. Br Heart J 1953; 15: 121-9.
Carlgren LE. The incidence of congenital heart diseases in children born in Gothenburg 1941 - 1950. Br Heart J 1959; 21: 40-50.
Keith JD, Rowe RD, Vlad . Heart Disease in Infancy and Childhood. New York:
MacMillan, 1958: 129-32.
8.
9.
10.
11.
12.
13.
Courter SR, Felson B, Mc Guire J. Familial interauricular septal defect with mitral stenosis (Lutembacher’s syndrome). Am J Med Sci 1848; 216: 501-8.
Gänsslen M, Lambrecht K, Werner M. Die kongenitalen Missbildungen des
Herzen. Handbuch der Erbbiologie des Menschen. Bd. 4. Berlin: J. Springer,
1940: 198-217. Apud Lamy M, Grouchy J, Schwisgut O 18.
Carleton RA, Abelmann WH, Hancok EW. Familial occurrence of congenital
heart disease. N Engl J Med. 1958; 259: 1237-45.
Howitt G. Atrial septal defect in three generations. Br Heart J 1961; 23: 494-6.
Nora J.J. Multifatorial inheritance hypotesis for the etiology of congenital heart
diseases. Circulation 1968; 38: 604-17.
Zetterqvist P, Turesson I, Johansson BW, Laurell S, Ohlsson NM. Dominant mode of inheritance in atrial septal defect. Clin Genet 1971; 2: 78-86.
213
Arq Bras Cardiol
volume 73, (nº 2), 1999
Ferreira e cols.
Recorrências de comunicação interatrial
14. Li Volti S, Distefano G, Garozzo R, Romeo MG, Sciacca P, Mollica F. Autosomal
dominant atrial septal defect of ostium secundum type. Ann Génét 1991; 34:
14-18.
15. Ferreira C. Estudo genético-clínico da comunicação interatrial do tipo defeito da
fossa oval. - Tese de doutoramento – UNIFESP,1972.
16. Gordon H. Gennetic counseling. Considerations for talking to parents and prospective parents. JAMA 1971; 217: 1215-25.
17. Lamy M, Grouchy J, Schwisgut O. Genetic and nongenetic factors in
etiology of congenital heart disease: study of 1.188 cases. Am J Hum Genet
1957; 9: 17-41.
18. Drogamaci I, Green H. Factors in etiology of congenital heart anomalies. J Pediat
1947; 30: 295-301.
19. Mc Aleese J.J. Survey of congenital heart disease in children’s hospital with special reference to surgical treatment. Am J Surg 1952; 83: 755-60.
20. Holman E, Gerbode F, Purdy A. Patent ductus: review of 75 cases with surgical
214
21.
22.
23.
24.
25.
26.
27.
treatment including aneurysm of ductus and one pulmonary artery. J Thorac Surg
1953; 7: 111-42.
Anderson RC. Causative factors underlying congenital heart malformations:
patent ductus arteriosus. Pediatrics 1954; 14: 143-51.
Uchida TA, Rowe RD. Discordant heart anomalies in twins. Am J Hum Genet
1947; 9: 17-41.
Ross LJ. Congenital cardiovascular anomalies in twins. Circulation 1959; 20:
327–42.
CampbellM.Twinsandcongenitalheartdisease.ActaGenetMed(Roma)1961;10:443-56.
Fuhrmann WH. Genetische únd peristatische Ursachen angeborener Angiokardiopathien. Ergebn Inn Med Kinderheilk 1962; 18: 47-115.
Nora JJ, Gilliland JC, Sommerville RJ, McNamara DG. Congenital heart disease
in twins. N Engl J Med 1967; 277: 568-71.
Engel E. Uniparental dissomy revised: the first twelve years. Am G Med Genet
1993; 46: 670.