55º Congresso Brasileiro de Genética
Resumos do 55º Congresso Brasileiro de Genética • 30 de agosto a 02 de setembro de 2009
Centro de Convenções do Hotel Monte Real Resort • Águas de Lindóia • SP • Brasil
www.sbg.org.br - ISBN 978-85-89109-06-2
203
Detecção de uma nova mutação no gene COL1A1 em
uma família com Osteogênese Imperfeita Tipo I
Moraes, MVD1; Quirino, GA1; Almada, BVP1; Rebouças, MRGO2; Sipolatti, V2; Nunes, VRR2; Akel, ANJ2;
Perrone, AMS1; Louro, ID1; Paula, F1
Núcleo de Genética Humana e Molecular (NGHM), Departamento de Ciências Biológicas, Universidade Federal do Espírito Santo (UFES)
Hospital Infantil Nossa Senhora da Glória de Vitória – ES (HINSG).
[email protected]; [email protected]
1
2
Palavras-chave: Osteogênese Imperfeita, colágeno tipo I, COL1A1, correlação genótipo: fenótipo, aconselhamento genético.
A Osteogênese Imperfeita (OI) é uma desordem Mendeliana do tecido conjuntivo caracterizada por fragilidade
óssea e suscetibilidade a fraturas. A OI Tipo I é resultante de mutações nos genes COL1A1 ou COL1A2, que
codificam as cadeias de pró-colágeno alfa-1(I) e alfa-2(I) do colágeno Tipo I, respectivamente. A expressão reduzida
de uma das cadeias de pró-colágeno alfa-1(I) ou a expressão de cadeias de pró-colágeno alfa-1(I) defeituosas são
fatores determinantes para a OI Tipo I. Este trabalho teve como objetivo o estudo molecular do gene COL1A1 em
indivíduos de uma mesma família (mãe e filho) com fenótipos clínicos característicos de OI Tipo I. A partir do
gDNA extraído de 5mL de sangue periférico, fragmentos de exons e regiões flanqueadoras de todo o gene COL1A1
do paciente, e fragmentos dos exons 37 e 40 do gene COL1A1 da mãe, foram amplificados por PCR, submetidos à
triagem de mutações por Single Strand Conformation Polymorphisms (SSCP), sequenciamento automatizado dos
fragmentos alterados e análise, de acordo com as seqüências de referência do gene COL1A1 GenBank NG_007400.1
(gDNA) e GenBank NM_000088.3 (cDNA), para fins de correlação genótipo: fenótipo. A análise molecular do gene
COL1A1 no paciente OI tipo I, atendido no Hospital Infantil N. Sra. da Glória, Vitória/ES-Brasil, do sexo masculino,
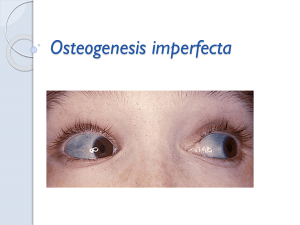
16 anos, 154 cm, 40 kg, IMC/I = P3, P/I < P3, E/I < P3, com histórico de 2 fraturas, escoliose, esclerótica azulada
e comprometimento auditivo, revelou a presença da mutação patogênica inédita c.2750delG (exon 40), além do
polimorfismo não patogênico p.Pro823Ala (exon 37), previamente descrito por Mackay et al. (1993). A análise
molecular de fragmentos dos exons 37 e 40 do gene COL1A1 na mãe, 45 anos, 164 cm e 72 kg, que apresentou
esclerótica azulada, comprometimento auditivo e um histórico de 9 fraturas, revelou a presença das mesmas
alterações, sugerindo um padrão de herança autossômico dominante. O pai (II-3), com 43 anos de idade, e os tios
maternos do paciente (seis homens e duas mulheres), com idade média de 36 anos (DP=10,50), não apresentaram os
sinais ou sintomas típicos da doença. Contudo, os familiares assintomáticos também deverão ser analisados uma
vez que a variabilidade fenotípica intrafamiliar em OI Tipo I é frequente e significativa, o que pode comprometer
o aconselhamento genético familiar e a prevenção do surgimento de novos casos da doença. A deleção de um
aminoácido guanina codificante (c.2750delG) no gene COL1A1 resulta na interrupção precoce da síntese de uma
das cadeias de pró-colágeno alfa-1(I) pela formação de um códon de parada prematuro (p.Gly917AspfsX191), o que
corrobora com os achados literários em OI tipo I associados à haploinsuficiência do gene COL1A1. Assim, estudos
moleculares em Osteogênese Imperfeita visam uma melhor compreensão da etiologia genética e variabilidade
clínica da doença.
Apoio Financeiro: Arcelormittal Tubarão, FAHUCAM, FACITEC e FAPES.