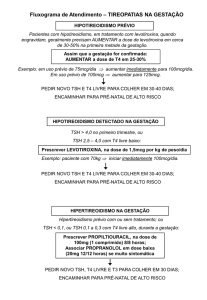
Informati
Informativo Digital - Nº5 – Maio/2014
HIPOTIREOIDISMO CONGÊNITO
O hipotireoidismo congênito (HC) é o distúrbio endócrino congênito
mais frequente. No Brasil, a prevalência do HC varia de 1:2595 a
1:4795. A prevalência é variável entre os grupos étnicos. Crianças com
Síndrome de Down têm um risco 35 vezes maior para apresentar o HC
que a população geral.
CAUSAS
A causa mais frequente de HC permanente resulta de defeitos na
formação glandular durante a embriogênese, denominados disgenesias
tireoidianas (ectopia – localização fora do leito tireoidiano, agenesia –
ausência da glândula e hipoplasia – glândula pouco desenvolvida), e
representam 85% dos casos.
Outras etiologias de HC permanente são os defeitos da produção
hormonal, denominados disormonogênese, e representam cerca de
15% dos casos. São defeitos autossômicos recessivos e incluem
mutações que codificam o transportador de iodo-sódio (NIS), a
tireoperoxidase (TPO), a geração de peróxido de hidrogênio, a
tireoglobulina e iodotirosina deiodinase.
Causas incomuns incluem defeitos no transporte do hormônio
tireoidiano (HT), a resistência à ação do HT (Síndrome de Resistência
ao Hormônio Tireoidiano), a resistência ao TSH e o hipotireoidismo
central.
O hipotireoidismo central decorre da deficiência isolada de TSH ou,
mais comumente, do hipopituitarismo resultando na deficiência de
diversos hormônios da adeno-hipófise. A resistência ao hormônio
liberador de tireotrofina (TRH).
A resistência ao TSH é definida quando as concentrações séricas de
TSH são elevadas na ausência de bócio. Indivíduos afetados têm
glândula normal ou hipoplásica e valores séricos de T4 e T3 normais ou
baixos.
Defeitos no transporte de HT causados por mutações no gene MTC-8,
fazem com que o transporte de T3 fique prejudicado, levando ao
retardo mental. A síndrome é caracterizada por concentrações séricas
elevadas de T3, baixas de T4 e elevadas de TSH.
O HC PODE SER TRANSITÓRIO
O hipotireoidismo neonatal pode ser permanente ou transitório.
Recomenda-se reavaliação após os 3 anos de idade com a suspensão
da levotiroxina (LT4) nas crianças em tratamento, que não apresentem
etiologia estabelecida.
TRIAGEM NEONATAL
O objetivo principal é evitar sequelas, principalmente o retardo mental
secundário ao hipotireoidismo, o que pode ser conseguido com início
da terapêutica adequada nas duas primeiras semanas de vida. A
sensibilidade dos testes de triagem neonatal para HC é elevada, o que
a torna eficaz para rastrear a doença, sendo recomendada para o
rastreamento do HC.
A idade recomendada para coleta do RN para triagem neonatal
é após 48 horas de vida até o quarto dia ou antes que o RN
deixe o hospital nas altas precoces e sempre antes de
transfusões sanguíneas.
Existem diferentes estratégias para a triagem do HC. No Brasil,
a triagem neonatal para HC é realizada pela dosagem de TSH
em papel-filtro, seguida de T4 total e/ou livre no soro, quando
necessária. Esta estratégia é eficaz e adotada em outros
países.
Os testes de triagem não são diagnósticos e resultados alterados
devem ser confirmados por métodos quantitativos de rotina para as
dosagens séricas de TSH, T4 total ou livre.
IDENTIFICAÇÃO DA ETIOLOGIA DO HC
Exames complementares são utilizados para estudar a etiologia do
HC, mas nunca devem postergar o início do tratamento. A
ultrassonografia cervical (US) seria o principal exame inicial, uma vez
localizada a glândula, seriam descartadas a agenesia e a ectopia. O
mapeamento com 99mTc está indicado quando a US não detecta a
glândula ectópica. A cintilografia pode ser realizada com pertecnetato
de tecnésio (99mTc) ou iodo (123I), em vez do 131I, devido à menor
radiação. A dosagem da tireoglobulina apresenta sobreposição entre
as diferentes causas de HC. Se não é visualizado tecido tireoidiano
ao US e há tireoglobulina e T4 mensuráveis, algum tecido funcional
está presente. A dosagens de anticorpos antitireoidianos (TPO,
TRAb) podem ser úteis para justificar a presença de TSH elevado em
filhos de mães com tireoidite de Hashimoto (hipotireoidismo
transitório), assim como em filhos de mães com doença de Graves. A
iodúria poderá confirmar a falta ou excesso de ido em casos
suspeitos.
TRATAMENTO
O início do tratamento deve ser o mais precoce possível,
preferencialmente nas duas primeiras semanas de vida. A idade de
início do tratamento, a dose de LT4 e o monitoramento do tratamento
são essenciais para o desenvolvimento cerebral do paciente com HC.
É recomendada a dose de 10-15mcg/kg/dia. A meta do tratamento é
assegurar à criança o crescimento adequado e o desenvolvimento
psicomotor mais próximo do possível do seu potencial genético.
Cuidado deve se ter para evitar tratamento excessivo por período
prolongado, pois pode levar a craniossinostose e alterações no
temperamento da criança.
Assessoria Científica
Lab Rede
Bibliografia
Maciel LMZ et al. Hipotireoidismo congênito: recomendações do Departamento de
Tireoide da Sociedade Brasileira de Endocrinologia e Metabologia. Arq Bras
Endocrinol Metab. 2013;57(3):184-92