O nitrogênio molecular ( N2)
abundante na atmosfera
• Para ser utilizado pelos animais, ele
precisa ser fixado
• reduzido de N2 para NH3 (amônia)
•
(microorganismos, plantas, descargas elétricas)
Fixação de nitrogênio
• Somente algumas poucas bactérias de solo ou que
vivem associadas a raízes de plantas podem
converter N2 a NH3 -> Fixação de nitrogênio
• N2 + 8e-+ 8H+ + 16 ATP +16 H2OÆ 2NH3 + 16ADP +
16Pi + H2
As rotas de biossíntese de aminoácidos e
nucleotídeos necessitam de nitrogênio na forma
solúvel
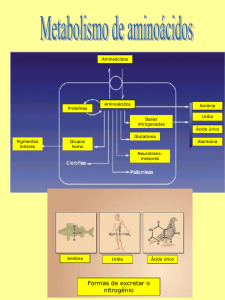
Amônia, aminoácidos e nucleotídeos são utilizados
de forma econômica pela maioria dos organismos
• Reciclagem e Reutilização
Reciclagem
• Aminoácidos em excesso não são
armazenados.
• Indivíduo saudável, com dieta adequada,
elimina nitrogênio correspondente a cerca
de 100g/proteína/dia.
• 400 g de proteínas devem ser renovadas/dia.
• Portanto 100 g devem ser repostos pela
alimentação
Proteínas da dieta
Proteínas endógenas
Aminoácidos
Grupo amino
uréia
Compostos nitrogenados
não-proteicos
A degradação das proteínas ingeridas
ocorre no trato gastrointestinal
A digestão de proteínas pode ser
dividida em fases
• Gástrica
• Pancreática
• Intestinal
Gástrica
• HCl do suco gástrico – mata microorganismos e
desnatura proteínas.
• A mucosa gástrica é recoberta por uma camada de
muco, que a protege da agressão do suco gástrico,
bastante corrosivo
• A denaturação torna as proteínas mais susceptíveis à
hidrólise de proteases gástricas (família da pepsina).
• Pepsinas são enzimas que são ativas em pH ácido.
• Pepsina ativa é gerada a partir da pró-enzima
pepsinogênio (remoção de 46 aminiácidos do NH2
terminal), em pH abaixo de 2.
• Pepsina corta proteínas em peptídeos menores e
aminoácidos livres
Fase Pancreática e Intestinal
• O suco pancreático é rico em pró-enzimas
que só são ativadas ao chegarem ao lúmen
do intestino delgado
Intestino Delgado
À medida que o conteúdo ácido do estômago chega ao
intestino estimula a secreção de secretina.
A secretina estimula o pâncreas a secretar bicarbonato
de sódio. O pH aumenta para 7.
A entrada dos peptídeos na parte superior do intestino
libera o hormônio colecistoquina.
A colecistoquina estimula a secreção de enzimas
pancreáticas (tripsina, quimiotripsina,
carboxipeptidase).
Uma protease produzida pelo duodeno
(enteropepdidase) ativa o tripsinogênio a tripsina.
Tripsina ativa as outras pró-enzimas.
Os aa livres entram nos capilares sanguíneos das
vilosidades e são transportados até o fígado.
Estômago:
A entrada das proteínas
no estômago estimula a
mucosa gástrica a secretar
gastrina ->estimula
secreção de
HCl e pepsinogênio.
As proteínas
denaturam
Em pH baixo->
ligações
acessíveis a
hidrólise
enzimática.
Pepsinogênio
é convertido em
pepsina.
A pepsina
hidrolisa as proteínas
Em peptídeos
O grupo amino e o esqueleto de carbono seguem vias
separadas mas conectadas
• A maioria dos amino ácidos é metabolizada no fígado.
Leucina, isoleucina e valina são oxidados
principalmente como combustível no músculo, tecido
adiposo, rim e cérebro). Esses tecidos têm uma
aminotransferase, ausente no fígado, que age nesses
aminoácidos ramificados.
O nitrogênio é abundante na atmosfera mas muito inerte
para ser usado na maioria dos processos bioquímicos.
• A amônia gerada é reciclada é usada nas sínteses .
• O excesso de amônia é excretado.
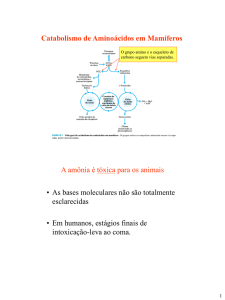
A amônia é tóxica para os animais
• As bases moleculares não são totalmente
esclarecidas
• Em humanos, estágios finais de intoxicação-leva
ao coma.
• Mudanças de pH celular e diminuição de
intermediários de ciclo de Krebs
• Excesso de NH3 leva a alcalinização de fluidos
celulares
• Um deslocamento do equilíbrio na reação da
glutamina sintetase pode depletar alfacetoglutarato no cérebro reduzindo ATP
A remoção do grupo amino
O papel do glutamato e da
glutamina
• O glutamato e a glutamina desempenham papel
crucial no metabolismo do nitrogênio.
• No citossol de hepatócitos, amino grupos da
maioria de amino ácidos são transferidos para o
alfa-cetoglutarato formando glutamato.
• O excesso de amônia gerado na maioria dos outros
tecidos é convertida no grupo amino da glutamina.
O grupo amino da maioria dos aa é coletado como glutamato
Reação catalisada por aminotrasferases (transaminases)
Reservatório temporário de
grupos amino
Aminotransferases = transaminases
As células têm várias aminotransferases
uma para cada amino ácido
O nome da enzima está relacionado com o
amino ácido doador do amino grupo para o
alfa-cetoglutarato, por exemplo:
Alanina aminotransferase; aspartato
aminotransferase
As reações das aminotransferases são
reversíveis
As aminotransferases têm como coenzima o PLP (derivado da
vitamina B6)
Os amino grupos são coletados no
fígado na forma de glutamato
Em uma segunda etapa, os amino grupos originam aspartato e/ou amônia
Nos hepatócitos, glutamato é
transportado do citossol para a
mitocôndria onde sofre desaminação
oxidativa ou transaminação
O glutamato pode ser desaminado e o grupo amino liberado como
amônia (NH4+) em pH fisiológico
Glutamato desidrogenase
mitocondrial
A glutamato desidrogenase utiliza tanto NAD+ como NADP+
O glutamato também tem outro destino. Transaminação formando
aspartato (o segundo depositário de grupo amino dos
aminoácidos).
aspartato aminotransferase
Alguns aminoácidos são
desaminados por reações especiais
• Glicina, histidina, lisina, metionina,
prolina, serina e treonina
Não participam de reações de transaminação
Ao longo da via de degradação o grupo amino é liberado
Como NH4+ ou forma glutamato . Mesmos produtos dos
Outros aminoácidos
• A ação combinada da aminotransferase e
glutamato desidrogenase resulta na
convergência do grupo amino da maioria
dos aa em 2 compostos:
• NH4+ e aspartato
Glutamina e Alanina são
transportadores de amônia para o
Fígado
Como a amônia é tóxica e a sua conversão em
uréia ocorre no fígado, o NH4+ produzido em
outros tecidos é incorporado em compostos não
tóxicos que atravessam membranas com
facilidade:
Alanina no músculo
Glutamina na maioria dos tecidos extra-hepáticos
Fígado
No rim existe a enzima
glutaminase que permite a
excreção de NH4+
A alanina transporta amônia do
músculo para o fígado
• Ciclo alanina-glicose
• O gasto energético da gliconeogênese é
imposto ao fígado e não ao músculo que
precisa de todo ATP para a contração
muscular.
Amino ácidos
Dieta
Glutamina
Músculo e outros tecidos
Alanina
Músculo
Átonos de nitrogênio da uréia
amônia
aspartato
O ciclo da uréia e o TCA foi descoberto por Sir Hans Krebs e colaboradores
Hans Krebs
The Nobel Prize in Physiology or Medicine 1953
O Ciclo da uréia
• O excesso de amônia é excretado como
uréia pelos organismos ureotélicos.
• A produção de uréia ocorre no fígado.
• A uréia produzida passa para a corrente
sanguínea e vai para o rim onde é excretada
pela urina.
Formação de carbamoil fosfato
Carbamoil fosfato sintetase I
NH4+ formado na mitocôndria
hepática + CO2 produzido pela
respiração
Amônia (primeiro nitrogênio da
uréia) entra no ciclo após
condensação com bicarbonato para
formar carbamoil fosfato
Carbamoil fosfato sintetase I
Carbamoil fosfato sintetase I
Custo de 2 ATPs
Formação da citrulina na matriz
mitocondrial
• Ornitina transcarbamoilase
Ornitina + carbamoil fosfato
citrulina
Citrulina é transportada para for a da mitocôndria , onde as outras
Reações do ciclo da uréia acontece,
Rim e eliminada
Pela urina
Soma das reações
Aspartato + NH4+ + HCO3- + 3ATP + H2O
Uréia + fumarato + 2ADP + 2Pi + AMP + PPi + 4H+
oxaloacetato
Consumo de 4 ligações fosfato ricas em energia
O destino do fumarato
• O fumarato pode ser convertido a
oxaloacetato, por reações analógas ao
Krebs, só que as enzimas envolvidas são
citossólicas.
• O Oxaloacetato por transaminação forma
aspartato.
A bicicleta do Krebs
Fígado, rim e coração
3 ATPs reduz para 1 ATP
A síntese da uréia
Esquema geral da síntese da uréia
Balanço energético do processo
Glicose
CICLO
DA
URÉIA
CICLO
DE
KREBS
Toxicidade da Amônia
genéticas
• ↓ atividade do Ciclo da Uréia
• Hiperamonemia
ATP
NH3
NAD(P)H
α-cetoglutarato
+ NH3
contribuir
• Encefalopatia Hepática
PODE
• Lesões severas no fígado ou deficiências
Glutamina
Glutamato
NAD(P)+
ADP
GABA
Regulação do ciclo da uréia
• O fluxo de nitrogênio varia com a dieta.
• Todas as enzimas do ciclo da uréia e a a
carbomoilfosfato sintase são sintetizadas em
velocidade maior em animais com dietas
ricas em proteínas.
• Existe também regulação alostérica da
carbomoilfosfato sintase.
Regulação do ciclo da uréia
•O fluxo de nitrogênio
varia com a dieta.
•Todas as enzimas do
ciclo da uréia e a
carbomoilfosfato sintase
são sintetizadas em maior
quantidade em animais
com dieta rica em
proteínas.
•Existe também regulação
alostérica (+) da
carbomoilfosfato sintase
pelo N-acetil-glutamato
Quando
aumenta
degradação de
aminoácidos
A carbomoilfosfato sintase é estimulada por N-acetilglutamato
Quando há maior
Degradação de aa
Degradação da cadeia carbônica
• Piruvato (glicogênicos)
• Intermediários do ciclo de Krebs
(glicogênicos)
• Acetil-CoA (cetogênicos)
Fenilcetonúria: Acumula fenilalanina que pode originar fenilpiruvato
O fenilpiruvato pode competir com o piruvato pela piruvato translocase
Fenilcetonúria
Defeito hereditário frequente no metabolismo
de aminoácido causada por aumento de
fenilalanina hidroxilase
Acúmulo de Phe
O fenilpiruvato pode competir com o piruvato pela piruvato translocase
Encontrados
Aumentados na urina
Compete com o piruvato
pela piruvato translocase
Diminui ATP
Deficiência da fenilalanina hidroxilase = fenilcetonúria
Defeito hereditário mais
frequente do metabolismo
de aminoácidos é a
fenilcetonúria. Ocorre
acúmulo de fenialanina, que
gera fenilpiruvato em reação
de transaminação com αcetoglutarato.
Deficiência da fenilalanina hidroxilase = fenilcetonúria
Na fenilcetonúria ocorre
acúmulo de fenialanina, que
gera fenilpiruvato em reação de
transaminação com αcetoglutarato.
Fenilpiruvato compete com
piruvato pela piruvato
translocase, restringindo a
entrada de piruvato na
mitocôndria e portanto a
produção de ATP a partir de
glicose.
Indivíduos afetados
apresentam retardamento
mental, pigmentação deficiente
da pele e cabelo.
Mais de 100 doenças
hereditárias devido a
deficiências no metabolismo
de aminoácidos já foram
descritas
Aspartame
N-L-alfa-aspartil-L-fenilalanina 1-metilester
Deficiência produz albinismo
melanina
O organismo só sintetiza 11 dos 20
aa constituintes das proteínas
Essenciais
Não Essenciais
Fenilalanina
Histidina
Isoleucina
Leucina
Lisina
Metionina
Treonina
Triptofano
Valina
Alanina
Arginina
Asparagina
Aspartato
Cisteína
Glutamato
Glutamina
Glicina
Prolina
Serina
Tirosina
A partir de
essenciais
tirosina
Cisteína
Glutamato e glutamina são doadores
de nitrogênio para a síntese dos
aminoácidos
Transaminases de Importância
em Diagnóstico
TGP (ALT)Transaminase
Glutâmico-Pirúvica
TGO (AST)- Transaminase
Glutâmico-oxalacético
Transaminases
Distribuição tecidual
• Enzimas intracelulares
•Presentes em vários tecidos
•↑ nos níveis séricos indica lesão tecidual
- infarto do miocárdio
- doenças hepáticas