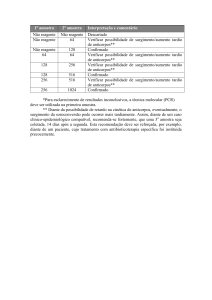
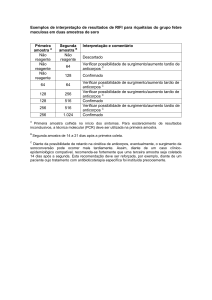
» Rev Bras Neurol, 48 (3): 25-41, 2012
Demências rapidamente progressivas.
Revisão atualizada e etapas diagnósticas
Rapidly progressive dementias. Updated review and diagnostic steps
Eliasz Engelhardt
Resumo
As demências rapidamente progressivas (DRPs) constituem um grupo heterogêneo de condições clínicas (neurodegenerativas, vasculares, infecciosas, imunomediadas, tóxicas, metabólicas, tumorais, psicogênicas) e cirúrgicas. Suas
principais características são início e evolução rápidos, com comprometimento cognitivo, comportamental, psicológico
e motor. Avaliação detalhada é imprescindível, devendo ser seguido protocolo extenso, de modo sistematizado,
compreendendo anamnese minuciosa, exames clínico e neurológico e complementares de sangue, urina, LCR, EEG,
neuroimagem e eventualmente biópsia cerebral. A investigação deve ser rápida, já que podem ser encontradas causas
reversíveis, e uma demora pode levar a morbidade ou mortalidade. As DRPs representam um desafio clínico complexo,
mesmo para os profissionais mais experientes.
Palavras-chave: demência, evolução rápida, demência rapidamente progressiva, diagnóstico
Abstract
Rapidly progressive dementias (RPDs) constitute a heterogeneous group of clinical (neurodegenerative, vascular,
infectious, immunomediated, toxic, metabolic, tumoral, psychogenic) and surgical conditions. Their main characteristic is rapid start and course with cognitive, behavioral, psychological and motor impairment. Detailed evaluation
is essential, and an extensive protocol that must be followed in a systematic way, including a meticulous anamnesis,
clinical and neurological examination, and complementary assessment, including blood, urine, CSF, EEG, neuroimaging exams, and eventually brain biopsy. The investigation must be fast, considering that reversible causes may
be found, and a delay can lead to morbidity ou mortality. The RPDs represent a complex clinical challenge, even for
the most experienced professionals.
Keywords: dementia, rapid course, rapidly progressive dementia, diagnosis
Neurologia Cognitiva e do Comportamento - INDC-CDA/IPUB-Universidade Federal do Rio de Janeiro
Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
jul - ago - set, 2012 »
25
Engelhardt E.
Introdução
As demências rapidamente progressivas (DRPs)
constituem grupo heterogêneo de condições que se
caracterizam por apresentar um ritmo evolutivo rápido.
O termo, apesar de não haver ainda uma definição
plenamente aceita, pode ser utilizado para pacientes
que evoluem para demência em período inferior a 2
anos, sendo mais comuns períodos inferiores a 1 ano,
como meses, semanas ou mesmo dias. Desse modo, as
DRPs progridem de modo muito mais rápido que a
maior parte das demências neurodegenerativas típicas,
que costumam evoluir no curso de diversos anos desde
os primeiros sintomas até o quadro de demência,
como a doença de Alzheimer (DA), degeneração
lobar frontotemporal (DLFT), demência com corpos
de Lewy (DCL), demência associada à doença de
Parkinson (DDP), parkinsonismos atípicos, entre
outras 31, 32, 63.
Considerando que muitas das DRPs são passíveis
de tratamento e reversão do quadro a avaliação do
paciente costuma ser urgente e requer procedimentos
complementares amplos com exames múltiplos
realizados em paralelo. O diagnóstico rápido é
imperioso levando em conta que a demora pode levar
a sequelas irreversíveis ou mesmo à morte. Enquanto
o prognóstico é relativamente bem conhecido nas
demências crônicas, o prognóstico nas DRPs é variável,
na dependência da causa subjacente. Desse modo,
constituem-se em um desafio clínico complexo que
requer avaliação rápida e eficiente, e diagnóstico preciso
32, 42, 46, 63, 65, 82
.
O objetivo deste artigo, além de uma atualização de
artigo previamente publicado20, propor um protocolo
com etapas que possam facilitar o diagnóstico.
Categorias de demências rapidamente
progressivas
As DRPs incluem variadas condições heterogêneas
e podem ser encontradas no grupo etário senil
e pré-senil, incluindo a sua faixa mais jovem,
contando com condições que podem ser classificadas
em neurodegenerativas, vasculares, infecciosas,
imunomediadas, tóxicas-metabólicas, tumorais e
psicogênicas. Estatísticas de centros de referência, que
se dedicam ao estudo das DRPs (e principalmente
da DCJ), mostram que cerca de a das DRPs são
constituídas pela mais frequente doença por prion,
26
» Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
a doença de Creutzfeldt-Jakob (DCJ), sendo que a
fração remanescente compreende numerosas condições
heterogêneas que necessitam de esclarecimento
detalhado33,37,42,63,65,82. Um dos centros de referência
mais importante, o UCSF Memory & Aging Center,
com os estudos mais detalhados sobre o assunto, teve
a oportunidade de avaliar mais que 1.300 casos com
suspeita de doença priônica. Revisão recente desses dados
mostrou que o diagnóstico de doença priônica foi dado
a 48% dos pacientes (32% com as formas esporádicas
provável ou definida, 15% genética e 1% adquirida),
29% dos pacientes tiveram diagnóstico indeterminado
(prontuários com informação insuficiente, rotulados
como sDCJ potencial) e, aspecto muito importante,
foi a demonstração que 23% apresentava outras
condições não-priônicas, muitas das quais tratáveis.
A classificação diagnóstica dessas DRPs não-priônicas
foi neurodegenerativas (26%), autoimunes (15%),
infecciosas (11%), psiquiátricas (11%), miscelâneas
(9%) e 28% se mostraram ainda indeterminadas, sendo
frequentemente leucoencefalopatias ou encefalopatias
de etiologia desconhecida31,34.
Neurodegenerativas
A DCJ, a mais característica das DRPs, e demências
neurodegenerativas clássicas, porém com evolução
atípica, serão vistas a seguir32,33,42.
Doença de Creutzfeldt-Jakob. Será considerada
a forma esporádica (sDCJ), a mais conhecida e
freqüente (85%), com incidência de 0,5 a 1,0 casos/
milhão/ano. Existem também as formas familial
(genética) e adquirida. Os aspectos clínicos da sDCJ
compreendem um quadro de demência associada a
manifestações comportamentais, extrapiramidais,
cerebelares e mioclonia. Ocorre geralmente entre 50 e
70 anos de idade, sem predileção de gênero. O EEG
mostra complexos periódicos de pontas ou ondas
lentas agudas em cerca de dos casos, sua presença
depende da fase da doença, sendo características
quando ocorrem. Esse padrão pode ser encontrado
em outras condições, que podem ser confundentes. A
sensibilidade é de 66% e há relatos que lhe atribuem
uma especificidade de 74%. A RM pode mostrar,
principalmente na sequência em difusão, considerada
a mais sensível para esse fim, hiperintensidade nos
gânglios da base, em particular no caudado (em 60%
dos casos) e/ou corticais (faixas corticais) (em até 80%
dos casos), enquanto o comprometimento talâmico
jul - agos - set, 2012
Demências rapidamente progressivas. Revisão atualizada e etapas diagnósticas
pode ser visto com menor freqüência (14% dos casos).
A sensibilidade da RM em casos de sDCJ provável
comprovados por autópsia variou entre 58 e 70%. Já
a especificidade mostrou-se elevada, entre 81 e 89%.
Eventualmente, a sintomatologia, os exames de EEG
e de RM podem se mostrar focais ou lateralizados,
dificultando o diagnóstico. O LCR mostra elevação da
proteína 14-3-3 em 91% dos casos, com sensibilidade
de 94% e especificidade de 84%. Podem ocorrer
falsos positivos (casos de DA e DCL com evolução
fulminante) e falsos negativos. Outros marcadores que
podem ser utilizados são enolase neurônio-específica
(ENE) e a S100b, com menor especificidade, e a
tau total. Análise genética pode ser realizada com
objetivo de identificar polimorfismos e/ou mutações
no gene do prion celular (PRNP), a presença de
mutações confirmando o diagnóstico de formas
priônicas hereditárias. A biópsia cerebral costuma ser
esclarecedora, com achados histopatológicos clássicos e
principalmente com imunocoloração. Não é realizada
com freqüência, levando em conta a possiblidade
de estabelecer o diagnóstico com os exames acima
mencionados. Devem ser tomados cuidados especiais,
quando realizada11,19,32,41,42,44,55,75,82.
Doenças neurodegenerativas classicamente
crônicas. As doenças demenciantes neurodegenerativas
clássicas costumam ter início insidioso e uma
progressão muito lenta, com duração geralmente
maior que cinco anos (p.ex., DA, DLFT, DCL, DCB,
PSP). Entretanto, podem ser encontradas, de modo
menos freqüente, formas rapidamente progressivas
e inclusive fulminantes, com algumas características
típicas da categoria, e outras atípicas, inclusive a
rapidez da evolução. Desse modo, a avaliação de
uma DRP requer frequentemente considerações
diagnósticas que apresentam sobreposição apenas
marginal com as demências lentamente progressivas.
Excetuando possivelmente a DCL e a DCB, as outras
demências de evolução lenta, como DA e DFT (exceto
a DFT-DNM), raramente se apresentam como DRPs.
Mioclonia e sinais extrapiramidais podem ser vistas na
DCB, DCL e em fases mais adiantadas na DA, de modo
que sua presença não sugere forçosamente o diagnóstico
de DCJ. Essas demências podem se assemelhar a DCJ,
podendo ser algumas vezes enquadradas na categoria
de sDCJ possível. Adicionalmente ao quadro clínico
semelhante, pode ser encontrado EEG sugestivo e
proteína 14-3-3 no LCR. Caso a duração seja maior
que 1 ano, pode se pensar em uma doença não DCJ
como a mais provável. A dificuldade pode surgir diante
de doença neurodegenerativa de evolução rápida ou
de sDCJ de evolução lenta. Outro aspecto que deve
ser considerado é a possiblidade de uma doença
neurodegenerativa (AD, DCL) coexistir com a sDCJ,
como verificado em estudos de autópsia12,32,34,36,42,76,77.
Vasculares
Doença cerebrovascular. Lesões cerebrovasculares
causadas por doença arteriosclerótica de artérias de
grande calibre e/ou de pequeno calibre, podem levar
a comprometimento cognitivo vascular e demência
vascular (DV), de modo agudo ou subagudo, com
características de DRP. As lesões teciduais ocorrem sob
a forma de infartos córtico-subcorticais ou subcorticais
em áreas heteromodais ou límbicas-paralímbicas ou
em estruturas subcorticais (tálamo, gânglios da base).
Lesões podem também ocorrer na substância branca
subcortical, sob a forma de leucoaraiose e lacunas,
interrompendo feixes que interconectam estruturas
diversas. As lesões das estruturas cinzentas e da
substância branca devem ter características de localização
e extensão que atendam aos critérios diagnósticos
estabelecidos. Lesões hemorrágicas intracerebrais
também podem ocorrer e ocasionar síndromes
semelhantes, agudas. As regiões e os feixes atingidos
devem ser relevantes para a integração cognitiva e/ou
comportamental, causando síndromes cognitivas por
lesão específica ou por desconexão. As manifestações
clínicas são variadas, cognitivas e neuropsiquiátricas,
na dependência das estruturas atingidas. A disfunção
executiva, o comprometimento da atenção, memória,
linguagem, função visuoespacial, entre outras, podem
estar presentes, em combinações e intensidade variadas.
O diagnóstico de DV é estabelecido por critérios
próprios, onde a neuroimagem tem lugar de destaque,
principalmente a RM do crânio. As sequência básicas,
com ênfase para a sequência em FLAIR, podem mostrar
os diversos tipos de lesão acima descritas21,47,50,53,73.
A angiopatia amilóide cerebral (AAC) é uma
desordem caracterizada por depósito de β-amilóide
nas paredes de artérias e arteríolas leptomeníngeas
e corticais cerebrais. A forma esporádica é a mais
comum e sua incidência encontra-se associada à idade
avançada. Pode ser assintomática, ou causar diversas
condições clínicas em consequência à ruptura de vasos,
levando à micro-hemorragias ou à hemorragia lobar.
Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
jul - agos - set, 2012 »
27
Engelhardt E.
A hemorragia intracerebral lobar relacionada à AAC é
a condição clínica mais bem estudada. Os depósitos
amilóides podem também obliterar a luz vascular
levando a infartos e leucoaraiose. As manifestações
clínicas compreendem comprometimento cognitivo/
demência de instalação rápida (infarto, hemorragia).
A TC de crânio sem contraste pode ser útil no
diagnóstico diferencial entre infarto e hemorragia.
Caso seja uma hemorragia em localização suspeita para
diagnóstico de AAC deve ser realizada RM, incluindo
a sequência gradiente-eco, adequada para detecção de
micro-hemorragias crônicas córtico-subcorticais. Além
disso, a RM nas sequências em FLAIR e gradiente-eco
pode mostra hiperintensidades na substância branca
(leucoaraiose) e micro-hemorragias9,13.
A trombose microangiopática na púrpura
trombocitopênica trombótica pode levar à DRP. A
redução da perfusão cerebral devido a síndromes
de hiperviscosidade com policitemia ou gamopatias
monoclonais, ou ainda trombose de fístulas
arteriovenosas durais, podem evoluir com demência
de instalação rápida, parkinsonismo ou ataxia. Casos
com história familial sugestiva de demência de início
precoce e ictus vasculares permitem considerar a
possibilidade de doenças mitocondriais no diagnóstico
diferencial32,33,74,82.
A s va s c u l i t e s c e n t r a i s , e m b o r a d e c a u s a
imunomediada, podem ser incluídas nesse ponto,
considerando seus efeitos diretos nos vasos arteriais e
na integridade do tecido cerebral.
Vasculites centrais. A vasculite primária do SNC
(vasculite [angiite] cerebral) é uma condição autoimune
rara, não apresenta uma causa subjacente definida e pode
ocorrer associada a certas condições clínicas (infecções,
vasculites sistêmicas), a determinados medicamentos
e a tumores. Acomete geralmente vasos pequenos e
médios, com inflamação e necrose segmentar de vasos
leptomeníngeos e parenquimatosos, podendo levar a
lesões múltiplas. A evolução é progressiva e flutuante
ou escalariforme, com cefaléia, alterações do estado
mental, sinais neurológicos focais, e com a progressão
da doença são observadas manifestações multifocais.
Apresentação similar pode ser vista também nas
vasculites com células gigantes e a poliarterite nodosa.
Infecções bacterianas, fúngicas e virais podem levar a
vasculites semelhantes à primária. Outras vasculites
compreendem a vasculite granulomatosa do SNC,
a poliarterite nodosa e as relacionadas à sarcoidose,
28
» Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
lupus eritematoso, síndrome de Sjögren, doença de
Behçet, síndrome hipereosinófila. Exames laboratoriais
visando avaliação reumatológica incluem VHS, PCR,
complemento (C3), complemento (C4), complemento
total (CH50), anticorpo antinuclear (AAN), fator
reumatóide (FR), anticorpos aos antígenos Ro/SSA
e Lo/SSB, anticorpos anticitoplasma de neutrófilos
(ANCA), padrão perinuclear (p-ANCA) e padrãocitoplasmático (c-ANCA) devem ser realizados. Esses
testes podem mostrar positividade ou ter apenas o
VHS e o PCR elevados. Os exames complementares
podem oferecer esclarecimento. A RM revela alterações
variadas, como lesões isquêmicas múltiplas de idade
diferente, hemorragias, leucoencefalopatia, realce
meníngeo ao contraste. A angio-RM pode ser útil,
porém a angiografia cerebral (AGC) convencional
é mais precisa, podendo mostrar estreitamentos
multifocais e oclusão de artérias de médio e grande
calibre, apesar de alto índice de falsos negativos. O LCR
é geralmente anormal, com leucocitose leve e aumento
de proteínas. A biópsia leptomeníngea-cerebral pode
ser esclarecedora33,56,61,62.
Infecciosas
Causas infecciosas de DRPs compreendem vírus,
bactérias, fungos e parasitas. As infecções do SNC,
meningites e encefalites, geralmente são acompanhadas
por manifestações sistêmicas típicas de infecção, como
febre, leucocitose e eventualmente sinais meníngeos
(em caso de menigoencefalites). A maioria das infecções
que causam alterações do estado mental se manifestam
de modo agudo, algumas podendo se instalar de modo
mais lento. O exame do LCR mostra padrão citológico
e bioquímico que varia com o agente etiológico. A
detecção do agente causal pode ser realizada através de
exames imunológicos10,32,33,82.
Encefalites. Entre as infecções, as encefalites virais
podem-se apresentar como uma DRP. Os vírus Herpes
simplex (VHS-1 e 2), Epstein–Barr, citomegalovirus
(CMV), vírus varicela-zoster (VVZ) e enterovirus,
embora tipicamente se apresentarem sob a forma
de uma encefalite aguda, podem se manifestar com
alterações mais graduais do estado mental e do
comportamento. A detecção dos vírus mais comuns é
possível pela PCR, mais rápida que a cultura, podendo
o teste diferenciar o agente causal, assim como a
detecção de anticorpos IgM e IgG para outros vírus.
A encefalite pelo VHS-1 (EHS-1) é a mais
jul - agos - set, 2012
Demências rapidamente progressivas. Revisão atualizada e etapas diagnósticas
comum no adulto, com altas taxas de morbidade e de
mortalidade. Constitui-se em caso de urgência e deve
ser considerada na avaliação de alterações cognitivas ou
comportamentais de instalação rápida. As manifestações
compreendem amnésia, desinibição ou outros aspectos
psiquiátricos devido ao comprometimento de
estruturas límbicas. Ocasionalmente o quadro pode
ser precedido por alucinações olfatórias ou gustativas.
O EEG é bastante sensível e mostra anormalidade
de modo freqüente, com atividade delta rítmica
intermitente, lentificação temporal não rítmica ou
lentificação frontotemporal, podendo se desenvolver,
com a evolução da doença, um padrão periódico.
Apesar de sua menor especificidade em comparação
à RM e ao LCR, é importante na monitoração de
atividade de crise, quando presente ou suspeitada. A
RM apresenta-se anormal na grande maioria dos casos,
nas sequência em difusão e FLAIR, assim como ao
contraste, com edema da região inferomedial do lobo
temporal, uni- ou bilateral, algumas vezes hemorrágico,
eventualmente associado comprometimento da insula
e do giro do cíngulo. O LCR apresenta pleocitose
linfocitária moderada, leve aumento de proteínas e
glicose normal, podendo ser encontradas hemácias e
leve xantocromia. O VHS-1 pode ser diagnosticado no
LCR pela detecção do seu DNA pela PCR, com alta
sensibilidade e especificidade1,5,54,71.
A encefalite pelo vírus da raiva é uma doença
fulminante desde o início, após um período de
incubação médio de 1-2 meses (3 semanas a raramente
dois anos) pós-exposição ou na aparente ausência
desta. Aparecem inicialmente espasmos faringolaríngeos, seguidos por aspectos comportamentais
e neuropsiquiátricos marcantes, incluindo agitação,
comportamento bizarro, alucinações, excitabilidade
e rápida progressão para o estado de coma. O
exame de sangue pode mostrar hiponatremia. Testes
sorológicos específicos podem mostrar resultados
variáveis. A RM pode mostrar áreas com hipersinal
mal definido em T2 na substância branca subcortical,
hipocampos, núcleos cinzentos profundos e córtex
cerebral, e tronco cerebral, de gravidade variada na
dependência do estágio, com realce ao contraste
em fases mais adiantadas. O LCR pode ser normal.
O diagnóstico pode ser feito através de biópsia de
pele da nuca, com a demonstração do antígeno do
vírus na inervação de folículos pilosos por técnica
com anticorpo fluorescente em cortes congelados. O
teste da impressão corneana ou salivar pode não ser
confiável. Pode haver prevenção com imunoterapia
ativa e passiva e quando aplicada em período
adequado pode ser eficaz. Embora considerada
incurável, já há relatos de raros casos de cura4,38.
Encefalopatia por HIV. O vírus da imudeficiência
humana (HIV ) acomete o SNC causando a
encefalopatia por HIV ouencefalopatia por AIDS. O
comprometimento cognitivo compreende deficiência
nas áreas da atenção, aprendizado, memória,
velocidade de processamento, além de manifestações
comportamentais (alteração de personalidade, apatia,
instabilidade do humor, alucinações). Quadro de
demência costuma ocorrer geralmente em fases mais
avançadas da doença e em percentual mais baixo em
fases iniciais. A progressão é variável, alguns casos
evoluindo de forma rápida, em 3 a 6 meses, ou
outras o quadro pode durar de 1 a 2 anos, entrando
no diagnóstico diferencial de DRPs. Entretanto,
sua incidência tem diminuído com a introdução de
medicação anti-retroviral. O diagnóstico é baseado em
teste de HIV no soro, baseado na técnica de detecção
de anticorpos contra o vírus. Existem outras técnicas
(detecção de antígenos, cultura viral e amplificação do
genoma do vírus), utilizadas em situações específicas.
A contagem de células CD4 no sangue é uma medida
de imunocompetência celular e tem implicações
prognósticas na evolução da infecção, sendo mais útil
no acompanhamento de pacientes infectados. Outros
exames podem ajudar na avaliação, como a RM, que
pode mostrar lesões hiperintensas e eventualmente
atrofia cerebral. O EEG pode mostrar lentificação
generalizada. O LCR pode mostrar citologia com
contagem normal e leve aumento das proteínas. A
presença de bandas policlonais e um aumento da
IgG indica produção intratecal de imunoglobulinas.
Além do comprometimento direto do SNC pelo
vírus, podem ocorrer diversas infecções oportunistas
subagudas ou crônicas (fungos [criptococose], parasitas
[toxoplasmose], bactérias [tuberculose])10,4a,13a,17.
Poliomavirus. Esta família conta com dois
subgrupos principais – os virus BK (iniciais de um
paciente) e JC (John Cunningham), com genoma
semelhante. São muitos comuns na população geral e
costumam permanecer latentes e assintomáticos, vindo
a se manifestar em condições de baixa da imunidade,
como na imunodeficiência adquirida (AIDS – 50
a 80% dos casos) e em determinados tratamentos
Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
jul - agos - set, 2012 »
29
Engelhardt E.
(transplante de órgãos, neoplasias, LES). O vírus JC
pode causar a leucoencefalopatia multifocal progressiva
(LMP), quadro desmielinizante subagudo pouco
comum, com comprometimento cognitivo, ataxia,
crises. A evolução é progressiva, com sobrevida de 1-4
meses, raramento mais que 1 ano. A RM mostra lesões
da substância branca subcortical, incluído a interface
córtico-subcortical, inicialmente múltiplas e pequenas,
eventualmente coalescem para formar lesões maiores,
sendo mais frequentes em localização parieto-occipital e
frontal. O LCR pode ser normal ou com leve aumento
de células e proteínas. A PCR permite identificar o virus
JC, que tem alta sensibilidade e especificidade na LMP
(relatos de 95-100%). A biópsia é o critério padrão
de diagnóstico, porém, com clínica e neuroimagem
características, e PCR positivo no LCR, têm sido
evitada7,18.
Panencefalite esclerosante subaguda. É doença
decorrente de infecção persistente do vírus do
sarampo imuno-resistente (mutante) que pode ocorrer
raramente em adultos jovens. A evolução da PESS
é relativamente rápida com sobrevida média de um
ano e meio, podendo ser fulminante, com óbito em 3
meses. O quadro compreende demência progressiva,
crises focais ou generalizadas, mioclonia, ataxia,
espasticidade.
O EEG apresenta caracteristicamente pontas
de alta amplitude ou complexos periódicos de
descargas de ondas delta polifásicas de alta voltagem,
sincrônicas e bilateralmente simétricas (complexos
de Rademecker) (frequentemente correlacionadas a
mioclonia clínica) ou outros complexos periódicos.
Pode se apresentar normal ou evidenciar apenas
lentificação do traçado no início e em fases tardias
mostrar-se desorganizado com atividade lentificada e
de alta amplitude. À medida que a PESS progride, a
atividade de fundo apresenta-se suprimida, resultando
em um padrão de descarga-supressão. A RM tem um
papel limitado e pode mostrar no início lesões mal
definidas na substância branca subcortical occipital
e frontal, com hipersinal em T2, evoluindo para
desmielinização subcortical extensa em fases mais
tardias. O LCR revela frequentemente leve pleocitose
e aumento das proteínas, gamaglobulina elevada (com
banda oligoclonal) e títulos elevados de anticorpos
ao virus do sarampo (estes também no soro). A PCR
permite detectar o RNA do vírus30,54,57,71.
30
» Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
Criptococose e outros fungos. A meningite por
criptococose pode apresentar-se com evolução subaguda
e comprometimento cognitivo. Outros fungos
(cândida, aspergilos, mucormicose, histoplasmose)
também podem causar quadros semelhantes. Embora
mais comum em pacientes com imundepressão, podem
também ocorrer em indivíduos imunocompetentes.
A RM mostra, na criptococose, espessamento
das meninges da base e eventualmente nódulos
intracerebrais com realce ao contraste, além do
aparecimento de hidrocefalia. Podem ocorrer infartos
isquêmicos em território de artérias grande e/ou
pequenas. Raramente observam-se leões intracerebrais
maiores (criptococomas). O LCR mostra pleocitose
com predominância mononuclear, aumento leve a
moderado de proteínas e glicose diminuída. O exame
com tinta nanquim geralmente mostra-se positiva,
assim como a cultura (no sangue menos). Os títulos
para antígeno capsular do fungo encontram-se elevados
(também no soro)8,49,51.
Tuberculose. A meningite tuberculosa ou outras
espécies de micobactérias podem cursar de modo
subagudo e comprometimento cognitivo. Pode
apresentar-se também em casos com imunodepressão.
A RM mostra espessamento das meninges da base
com realce ao contraste e eventualmente granulomas
e hidrocefalia. Podem ocorrer ictus vasculares com
infartos isquêmicos em território de artérias grande e/
ou pequenas. A angio-RM pode mostrar estreitamentos
vasculares compatíveis com arterite. O LCR mostra
geralmente pleocitose linfocitária, elevação das
proteínas e glicose baixa. Podem ser vistos BAAR no
esfregaço. A cultura demora no mínimo duas semanas.
Mais da metade dos casos não são confirmados
por microbiologia. A PCR pode detectar ácidos
nucléicos de micobactérias com boas sensibilidade e
especificidade39,51.
Ne u r o s s í f i l i s . Po d e s e apre s e nt ar c om
comprometimento cognitivo e demência, desordens
de linguagem, transtornos de comportamento, crises.
A avaliação para essa infecção é sempre necessária,
apesar de disfunção cognitiva seja geralmente uma
complicação tardia. Entretanto pode apresentar-se de
modo mais agressivo em casos com imunodepressão.
A RM não é específica e frequente pode não haver
alterações. Pode haver atrofia temporal medial, atrofia
jul - agos - set, 2012
Demências rapidamente progressivas. Revisão atualizada e etapas diagnósticas
cortical ou áreas de desmielinização. Eventualmente
há evidência de lesão vascular isquêmica e raramente
goma. O LCR pode mostrar pleocitose linfocitária
moderada, leve aumento de proteínas, glicose
normal, com freqüente reatividade positiva ao teste
do VDRL e ao TPHA (também no soro)6,69,83.
Imunomediadas
Os tipos dessa categoria são numerosos,
compreendendo a condições imunomediadas
paraneoplásicas (associadas a antígenos-anticorpos
específicos) e não-paraneoplásicas. Geralmente
acometem estruturas límbicas e neocorticais, podendo
também atingir regiões subcorticais. Devem ser
diagnosticados prontamente, pois podem evoluir de
modo rápido e são passíveis de tratamento eficaz.
Entre as formas imunomediadas nãoparaneoplásicas podem ser mencionadas as doenças
desmielinizantes, a encefalopatia de Hashimoto, as
encefalopatias ligadas ao lupus eritematoso sistêmico,
à síndrome de Sjögren. Além disso, ocorrem demências
sem antígenos-anticorpos específicos, porém com
evidência de inflamação, como a doença de Behçet,
sarcoidose, vasculite primária do SNC32,33,62,79.
Algumas das encefalopatias imunomediadas nãoparaneoplásicas encontram-se a seguir.
Doenças desmielinizantes. As condições
desmielinizantes imunomediadas compreendem,
entre outras, a esclerose múltipla e a encefalomielite
disseminada aguda.
A esclerose múltipla (EM) é a doença
desmielinizante mais frequente em adultos jovens.
Estudos de prevalência indicam taxas elevadas de
pacientes que apresentam comprometimento cognitivo,
inclusive em fases precoces da doença. Entretanto,
demência é menos comum, podendo ser observada
principalmente nos estágios finais da doença. As funções
cognitivas que são mais frequentemente acometidas
compreendem atenção, função executiva, velocidade de
processamento, memória. O declínio cognitivo nesses
pacientes vem sendo correlacionado com alterações
cerebrais macro e microscópicas, demonstráveis por
neuroimagem estrutural. Estudos recentes mostram
que a disfunção cognitiva na EM resulta em grande
parte por desconexão de estruturas relacionadas
com processamento cognitivo, por ruptura de feixes
de substância branca córtico-corticais e córticosubcorticais25,32,35,79. Foi descrito comprometimento
cognitivo/demência com evolução de alguns meses
da doença, portanto sob forma de DRP, porém de
modo raro24,28. Os dados clínicos, associados a exames
complementares permitem o diagnóstico da doença,
compreendendo neuroimagem e exame de LCR, estudo
de potenciais evocados, entre outros. O diagnóstico
diferencial pode ser feito com doenças granulomatosas
e vasculíticas cerebrais, que podem se manifestar sem
manifestações sistêmicas ou serológicas informativas.
A RM é fundamental, com a sequência em FLAIR
para demonstrar as lesões na substância branca, que se
mostram hiperintensas, constituindo-se em marca de
desmielinização. Lesões mais novas poderão mostrar
realce após infusão de agente paramagnético. Outras
sequências podem ser utilizadas para detalhar variados
aspectos da doença (DTI, ERM). O LCR pode mostrar
evidência de inflamação crônica, com leve aumento
de linfócitos e de proteínas, e síntese intratecal de
imunoglobulinas. As bandas oligoclonais de IgG são
sintetizadas por linfócitos B próximos das áreas de
desmielinização – duas ou mais bandas oligoclonais
de IgG encontradas no LCR, não observadas no soro
correspondente, comprovam a síntese local. Estudo no
nosso meio demonstrou bandas oligoclonais em 85%
de pacientes com EM definida. Entretanto, pode ser
encontrada até 95% dos casos utilizando focalização
isoelétrica com imunotransferência de IgG. Não
representa um marcador da doença, considerando não
ser específica da EM, podendo ser encontrada em outras
desordens neurológicas centrais14,58,59,70.
A encefalomielite disseminada aguda (ADEM) é
uma doença desmielinizante imunomediada que pode
ocorrer após infecção viral ou bacteriana em até 75%
dos casos, pode ser pós-vacinal ou aparentemente sem
causa prévia. Embora mais frequente na faixa infantojuvenil, pode ocorrer de modo mais raro em adultos
jovens e idosos. A ADEM tem um início agudo e
evolução monofásica, embora tenham sido descritos
casos recorrentes ou multifásicos, com sintomas focais
ou multifocais que aparecem em 1-3 semanas após a
infecção ou vacinação. Inicialmente pode ocorrer febre,
cefaléia mal estar, e as manifestações centrais podem
se instalar de modo agudo ou subagudo, com quadro
de encefalopatia manifestada por estado confusional,
transtornos psiquiátricos (irritabilidade, depressão,
alterações de personalidade, psicose), alteração do
estado de consciência (sonolência, torpor, coma),
crises focais ou generalizadas, sinais focais motores,
Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
jul - agos - set, 2012 »
31
Engelhardt E.
podendo haver ataxia ou movimentos anormais.
Os exames paraclínicos não são específicos. O EEG
geralmente é anormal, com lentificação generalizada
de leve a acentuada, algumas vezes com lentificação
focal e descargas irritativas. A RM é essencial para o
diagnóstico, mostrando lesões focais ou multifocais
assimétricas em T2 e FLAIR na substância branca
subcortical, tálamo, gânglios da base, tronco cerebral,
medula, que podem mostrar realce ao contraste, que
tendem a diminuir e até desaparecer, ou permanecer
sem alteração e sem aparecimento de novas lesões
(ausência de disseminação temporal, diagnóstico
diferencial com EM). O LCR pode ser normal ou
apresentar pleocitose mononuclear leve e pequeno
aumento de proteínas. A gamaglobulina geralmente
não se encontra elevada e bandas oligoclonais podem
ser observadas em até metade dos casos, podendo ser
transitórias (em contraste à EM). Pode haver aumento
da proteína básica da mielina, achado não específico3
1,39,41a,51a,54a,67a,71a
.
Encefalopatia de Hashimoto. Além do termo
encefalopatia de Hashimoto (EH), outros são também
utilizados para designar essa condição autoimune rara,
possivelmente subdiagnosticada – meningoencefalite
inflamatória autoimune não-vasculítica e encefalopatia
responsiva a esteróide. Esta, associada à tireoidite
autoimune (SREAT) é a frequentmente referida como
EH. O nome reflete a presença de autoimunidade
à tireoide, frequentemente sem evidência clínica o
laboratorial de disfunção da glândula. Os anticorpos
anti-tireoperoxidase (a-TPA) ou anti-tireoglobulina
(a-TG) podem estar presentes no soro. O título dos
anticorpos, geralmente elevado, não se correlaciona
com a gravidade ou com qualquer dos aspectos clínicos.
Parece duvidosa uma relação causal direta entre os
anticorpos anti-tireoideanos e a encefalopatia. Pode
ocorrer comprometimento cognitivo, manifestações
psiquiátricas, tremores, mioclonia, crises. Foram
identificadas duas formas de apresentação, uma com
evolução surto-remissão e episódios semelhantes
a ictus vascular e outra de instalação insidiosa, em
indivíduos mais idosos. O diagnóstico é confirmado
pelo achado de acentuado aumento dos níveis de
anticorpos a-TPA e/ou a-TG no soro. Apresenta
aspectos que podem se assemelhar a outras DRPs,
particularmente a sDCJ, a vasculites centrais e a
encefalite paraneoplásica, tornando-se importante
32
» Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
considerar sempre no diagnóstico diferencial de
uma DRP. O EEG é inespecífico, podendo ser
encontrado, geralmente, padrão de ondas lentas,
focal ou com descargas periódicas como vistas na
sDCJ. A RM mostra-se variável, podendo ser normal,
com atrofia generalizada ou com hiperintensidades
na substância branca subcortical. O LCR pode
apresentar uma pleocitose linfocitária e leve elevação
da taxa de proteínas. Anticorpos anti-tireoideanos
também podem ser encontrados, mas sua presença
não parece se correlacionar com aspectos clínicos da
doença3,23,31,32,33,62,66,79.
Sarcoidose. Essa condição pode imitar diversas
condições neurológicas, com manifestações clínicas e de
neuroimagem variáveis. As manifestações neurológicas
podem ser periféricas e centrais, estas podendo ser
resultantes também de vasculite de vasos intracranianos.
Pode ocorrer comprometimento cognitivo e um
quadro de DRP. A RM pode ser normal ou evidenciar
granulomas com realce ao contraste, hiperintensidades
na substância branca, espessamento da leptomeninge
da base, hiperintensidades nas sequência em difusão
(parecidas com sDCJ). Manifestações sistêmicas e
imagem torácica podem ajudar no diagnóstico. O LCR
pode ser normal, mas é comum pleocitose e aumento
de proteínas. A biópsia do tecido afetado pode ser
necessária para o diagnóstico32,33,61,62.
Fo r a m i d e n t i f i c a d a s d i v e r s a s s í n d r o m e s
imunomediadas não-paraneoplásicas, que se
assemelham à encefalopatia límbica (EL) (ver abaix).
Algumas dessas síndromes tiveram identificados os
anticorpos e algumas vezes seus antígenos. Entre essas
síndromes, duas de EL não-neoplásica são devidas a
anticorpos anti-VGKC e a anticorpos antineurópilo,
além da síndrome por anticorpos anti-GAD, que pode
causar algumas vezes leve comprometimento cognitivo.
Quadros com características de EL sem os marcadores
já reconhecidos, paraneoplásicos ou autoimunes,
revelaram anticorpos (soro ou LCR) que ligam a
receptores sinápticos, AMPA-R ou GABAB-R31,62,79.
Entre as formas imunomediadas paraneoplásicas
são apresentadas a seguir algumas das síndormes mais
importantes.
As síndromes paraneoplásicas correspondem a
um grupo de condições que resultam da produção de
anticorpos antineuronais (onconeuronais), que podem
ser encontrados no soro e/ou no LCR, resultando
jul - agos - set, 2012
Demências rapidamente progressivas. Revisão atualizada e etapas diagnósticas
em sintomas neurológicos focais. Esses anticorpos,
expressos pela neoplasia, reagem com proteínas
neuronais, podendo preceder a detecção do tumor
subjacente com grande frequência. A variedade de
manifestações e a evolução rápida das encefalopatias
paraneoplásicas tornam o diagnóstico muitas vezes
difícil. O resultado do processo diagnóstico revela
um quadro autoimune e a síndrome encontrada
clinicamente pode sugerir o tipo de pesquisa em
relação aos anticorpos e, em muitos casos, a neoplasia
maligna subjacente32,33,62,79. Fazem parte desse grupo
a encefalopatia límbica paraneoplásica (ELP), como
principal representante de quadro demencial, além de
outras síndromes centrais.
Encefalopatia límbica. Doenças cerebrais
autoimunes associadas a anticorpos antineuronais
que causam EL são relativamente comuns, sendo
que cerca de 60% são de causa paraneoplásica. Os
sintomas da forma paraneoplásica (ELP) constituem
uma síndrome com comprometimento da memória
de curto prazo, amnésia retrógrada, crises focais (em
⅔ dos casos, geralmente crises parciais complexas
temporais) e sintomas psiquiátricos (depressão, psicose
ou mudança de personalidade. Podem ocorrer crises
parciais complexas curtas, que podem ser sutis e não
reconhecidas, com apresentação comportamental e
cognitiva que pode imitar uma DRP. Os sintomas
podem se desenvolver em dias ou semanas. A ELP
encontra-se associada à neoplasia maligna (pulmonar
[carcinoma de células pequenas], testicular, timoma,
mamária, teratoma ovariano, linfoma de Hodgkin),
sendo os anticorpos mais comuns encontrados o antiHu (ANNA-1), anti-Ma2, CV2 (anti–CMRP-5), Yo
(PCA-1) e provavelmente o antineurópilo. Os sintomas
da ELP com frequência precedem o diagnóstico da
neoplasia, podendo imitar outras complicações da
mesma ou as decorrentes do tratamento, o que pode
ser confundente. O EEG encontra-se alterado na
maior parte dos pacientes, com freqüente atividade
epileptiforme temporal, assim como lentificação difusa.
A RM pode mostrar-se normal em fases iniciais ou
com edema uni- ou bilateral em estruturas temporais
mediais, com hiperintensidade na sequência com
FLAIR. Após a resolução do edema ficam alterações
variadas, como hiperintensidade em hipocampo(s)
com dimensões normais, ou mais frequentemente,
atrofia das estruturas temporais mediais (inclusive
hipocampos) com hipersinal. Também podem ocorrer
alterações na amígdala, tálamo, hipotálamo, tronco
cerebral. Há semelhanças desses achados com fases
iniciais da encefalite herpética. Em fases mais tardias
da doença frequentemente se desenvolve atrofia
hipocampal marcante, associado a comprometimento
cognitivo permanente e epilepsia. O LCR, com
características inflamatórias (pleocitose mononuclear,
aumento das proteínas, bandas oligoclonais, síntese
intratecal de proteínas) na maioria dos casos é
sugestivo de ELP. A ELP pode ser confirmada quando
anticorpos onconeuronais são encontrados no LCR
(e/ou no soro), o que nem sempre ocorre. Quando
presentes, esses anticorpos facilitam o diagnóstico da
encefalopatia e frequentemente permitem a detecção
da neoplasia associada. O diagnóstico diferencial
inclui encefalopatias autoimunes não-paraneoplásicas,
encefalites virais, DCJ, vasculites29,33,60,62,64,78,82.
Outras encefalopatias. A encefalopatia por
anticorpos anti-receptor, como anti-receptor NMDA,
pode ocorrer em mulheres com teratoma ovariano,
homens com tumores de células germinativas
testiculares, ou não associado a neoplasia. A síndrome
característica é constituída por um quadro gripal
prodrômico seguido por sintomas psiquiátricos
(ansiedade, depressão) ou psicose (comportamento
bizarro, delírios, alucinações), muitas vezes levando a
psiquiatra com suspeita de esquizofrenia. Geralmente
ocorre discinesia orofacial ou de membros, seguido
por catatonia, mutismo, hipertonia, permanecendo
os olhos abertos, progressão eventual para coma e
insuficiência respiratória. Podem ocorrer crises e
instabilidade autonômica. A RM pode ser normal, mas
¼ pode ter achados de EL, como hiperintensidades
temporais mediais, estruturas subcorticais e raramento
hiperintensidades corticais. O EEG mostra lentificação
delta-teta difusa. O LCR apresenta aumento de
linfócitos e aumento de proteínas, o diagnóstico
podendo ser confirmado pela presença de anticorpos
para NMDA-R (também no soro)33,40,79.
Tumorais
Processos expansivos não neoplásicos (p.ex.,
hematoma subdural, uni- ou bilateral), assim como
as neoplasias malignas primárias ou secundárias
(metástases) podem causar DRPs. São identificados
através de neuroimagem adequada e não serão
Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
jul - agos - set, 2012 »
33
Engelhardt E.
considerados no momento. Os tumores focalizados
serão o linfoma primário do SNC e o linfoma
intravascular (linfoma angiotrópico).
Linfomas. O linfoma primário, difusamente
infiltrativo, pode causar quadro de demência subaguda.
Geralmente é do tipo de células B não-Hodgkin e com
frequência ocorre em um estado de imunossupressão.
A RM do crânio mostra sinal isointenso ou levemente
hiperintenso em T2, frequentemente envolvendo os
gânglios da base, a substância branca periventricular
e/ou o corpo caloso, com realce variável ao contraste.
O LCR mostra linfocitose, aumento de proteínas e
frequentemente baixa de glicose. A citologia específica
é frequentemente negativa e o diagnóstico geralmente
requer biópsia cerebral. Quando o linfoma primário
ocorre sob a forma de linfomatose cerebral a RM
mostra anormalidades difusas de sinal na substância
branca, sem realce ao contraste ou efeito de massa,
como visto em um processo infiltrativo difuso
sem quebra da barreira hematoencefálica. Pode
ser necessária biópsia cerebral para esclarecimento
diagnóstico.
O linfoma intravascular é raro, apresentando-se
com sintomas semelhante a ictus vascular e também com
quadro de demência subaguda. Deve ser investigado
o comprometimento da pele ou de órgãos viscerais. A
RM do crânio mostra regiões hiperintensas em T2, com
realce variável ao contraste e algumas vezes edema. A
biópsia cerebral é necessária para o diagnóstico, pois o
estudo angiográfico pode se assemelhar ao da vasculite
central32,82.
Tóxicas-Metabólicas
Numerosas causas metabólicas e tóxicas podem
causar DRPs.
Tóxicas. Intoxicação por metais (chumbo,
arsênico, mercúrio, alumínio, lítio) pode causar
declínio cognitivo rápido. A radioterapia (RT) e a
quimioterpia (QT) em casos oncológicos podem
levar a períodos de sobrevivência que permitem o
aparecimento e manifestação das lesões cerebrais.
Estas podem aumentar de intensidade de modo
progressivo e constituir quadro de encefalopatia com
comprometimento cognitivo expressivo. A RM mostra,
nesses casos, hiperintensidades difusas na substância
34
» Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
branca subcortical, podendo ser mais circunscrita
quando a RT é localizada15,31,32,82.
Metabólicas. Distúrbios eletrolíticos,
hipovitaminoses e outras condições podem ser causa
de DRP aguda ou subaguda. A investigação deve
incluir a dosagem de eletrólitos (NA, K, Ca, P, Mg),
vitamina B1, B12, determinação das funções renal
e hepática. Deficiência de vitaminas (p.ex., tiamina
[B1], niacina, vitamina E), que ocorre geralmente
associada ao alcoolismo ou desnutrição (encefalopatia
de Wernicke, psicose de Korsakoff ) ou em condições
mal-absortivas pode causar manifestações demenciais.
A porfiria pode causar psicose e dor abdominal
inexplicada. Doenças metabólicas hereditárias, mais
habituais na infância, podem se apresentar no adulto
(p.ex., doenças de Kufs, Wilson, HallervordenSpatz, a xantomatose cerebrotendínea, leucodistrofia
metacromática e adrenoleucodistrofia, entre outras),
além de mitocondriopatias, causando quadros de
diagnóstico difícil22,31,32,37,48,82.
Psicogênicas
Condições psicológicas e neuropsiquiátricas
podem eventualmente imitar uma DRP. A demência
relacionada à depressão (“pseudodemência” depressiva)
pode ocorrer de modo rápido em casos com história
prévia de depressão maior. Deve ser ressaltado que
muitos casos com “pseudodemência” podem ter
demência subjacente. Geralmente pode ser observada
a presença de depressão grave acompanhada por
disfunção cognitiva, especialmente em situação de teste,
o que pode ser atribuída a pouco empenho. Transtornos
psiquiátricos não depressivos, como desordens de
personalidade, manifestações conversivas, psicose e
simulação podem estar associados a DRPs. Aspectos
psiquiátricos podem ser sintoma precoce de muitas
doenças neurodegenerativas, como DCJ, DCL, DFT,
entre outras. Uma avaliação completa é muitas vezes
fundamental para excluir condições “orgânicas” que
necessitem de abordagens terapêuticas específicas.32,33.
Etapas diagnósticas
As descrições acima mostram as categorias e a
heterogeneidade das principais causas de DRPs,
reunidas em diversos tópicos, e a frequente semelhança
de suas manifestações clínicas e paraclínicas. O
diagnóstico não é fácil e requer conhecimentos
jul - agos - set, 2012
Demências rapidamente progressivas. Revisão atualizada e etapas diagnósticas
apropriados e um protocolo que conta com itens
sofisticados, estes nem sempre disponíveis em todos os
centros. O diagnóstico de uma DRP pode ser obtido
com maior segurança ao seguir de modo sistematizado
etapas de um protocolo, que compreende várias etapas,
visando distinguir os subtipos potencialmente passíveis
de reversão ou melhora, dos subtipos degenerativos,
irreversíveis (Tabela) 33, 37, 63, 82.
Tabela. Classificação das DRPs e suas principais causas (constantes do texto).
Neurodegenerativas
Tumorais
doenças priônicas
[esporádica, familial, adquirida]
demências clássicas - evolução atípica
[DCL, DCB, DA, DFT, DFT-DNM]
não-neoplásicas
hematoma subdural
[uni- ou bilateral]
neoplasias malignas
primárias
metástases
linfomas primários
células B não-Hodgkin
linfoma intravascular
Vasculares
doença cerebrovascular
angiopatia amilóide cerebral
trombose microangiopática
vasculites centrais primárias
vasculites secundárias
Infecciosas
encefalites
encefalite pelo VHS-1
AIDS
poliomavirus.
panencefalite esclerosante
subaguda
raiva
meningoencefalites
fungos
tuberculose
neurossífilis
Imunomediadas
não-paraneoplásicas
doenças desmielinizantes
esclerose múltipla
encefalomielite disseminada
aguda [ADEM]
encefalopatia de Hashimoto
sarcoidose
encefalopatias tipo límbica
anticorpos anti-VGKC
anticorpos antineurópilo
anticorpos anti-receptores
sinápticos [AMPA, GABAB]
paraneoplásicas
encefalopatia límbica paraneoplásica
encefalopatia anti-receptor NMDA
Tóxicas-metabólica s
intoxicação por metais
[As, Bi, Hg, Li, Pb]
medicamentos
radioterapia
quimioterpia
desquilíbrio eletrólito
[NA, K, Ca, P, Mg]
disfunção paratireoideana
hipovitaminoses
[B1, B12]
disfunção hepatica
disfunção renal
doenças metabólicas hereditárias
[metabólicas, mitocondriais]
Psicogênicas
depressão
(“pseudodemência” depressiva)
transtornos psiquiátricos
desordens de personalidade
manifestações conversivas
psicose
simulação
Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
jul - agos - set, 2012 »
35
Engelhardt E.
Assim, diante de um paciente com suspeita de DRP,
as etapas abaixo são sugeridas visando o esclarecimento
do diagnóstico, e a avaliação de qualquer apresentação
de DRP exige dados concordantes obtidos a partir da
anamnese, exames clínico e neurológico, assim como
de exames paraclínicos (sangue, neuroimagem, EEG,
LCR). A escolha dos exames e a ordem da realização
podem variar na dependência da apresentação clínica
e da suspeita diagnóstica. Deve ser lembrada, a cada
momento, a existência de resultados falsos positivos e
falsos negativos em diversas modalidades de exames,
devendo ser tomadas as devidas cautelas na sua
interpretação.
Etapa nº 2
Delirium e outras condições sistêmicas
Etapa nº 1
Anamnese detalhada
A anamnese deve ser abrangente, com foco na
linha temporal, natureza dos sintomas e potenciais
fatores associados que possam contribuir para o
quadro. É importante avaliar aspectos pré-mórbidos.
Informações de acompanhante confiável podem ser
críticas para evitar inconsistências relativas à linha
temporal e à evolução, importantes na distinção de
demências atípicas das típicas. Qualquer paciente que
apresenta deterioração cognitiva em período inferior
a 2 anos é suspeito de ter DRP. Além disso, a evolução
e a qualidade dos sintomas podem oferecer indicações
quanto a doença subjacente. Considerando que os
pacientes com DRP apresentam disfunção cognitiva
e comportamental, a informação sobre a modalidade
cognitiva comprometida (memória, linguagem) ou o
tipo de comportamento apresentado, pode ajudar no
diagnóstico. Como ocorre disfunção motora de modo
frequente, que pode estar relacionada à doença da via
motora piramidal, gânglios da base ou cerebelo, desse
modo devem ser feito perguntas no sentido de esclarecer
a atividade motora do paciente. Apurar sintomas
sistêmicos e fatores que potencialmente possam
contribuir ao quadro é necessário no caso de causas
reversíveis. É preciso inquirir sobre exposição a tóxicos,
assim como sobre uso de álcool e drogas recreacionais.
Medicamentos de ação central, especialmente de acordo
com os critérios de Beers (considerados inadequados
aos idosos) podem causar alterações mentais agudas e
subagudas (p.ex., anticolinérgicos, anti-histamínicos,
antidepressivos tricíclicos, analgésicos narcóticos,
miorrelaxantes, barbitúricos, entre outros). Devem
ser consideradas neoplasias não diagnosticadas na
36
» Revista Brasileira de Neurologia »
suspeita de possível condição paraneoplásica, com
perguntas sobre tabagismo e perda de peso. Diversas
neoplasias (câncer pulmonar de células pequenas,
timoma, neoplasias mamária, ovariana, testicular)
foram associadas a anticorpos paraneoplásicos de ação
central. Levando em conta que distúrbios do sono
podem contribuir ao comprometimento cognitivo,
deve ser esclarecida a presença de apnéia do sono
e desordens relacionadas. A história familial pode
indicar a possibilidade de causas hereditárias, como
DCJ familial (genética), encefalopatia mitocondrial,
leucodistrofias26,31,32,33,46,63.
Volume 48 »
Nº 3 »
Deve ser considerada presença de quadro de
delirium devido a processo infeccioso, metabólico
ou tóxico (inclusive medicamentos), independente
ou sobreposto a um eventual quadro demenciante
subjacente (neurodegenerativo., p.ex.), conforme
informações obtidas na anamnese, ressaltando revisão
detalhada de medicamentos com ação central que
possam contribuir para mudança abrupta do estado
mental. Exames paraclínicos (ver abaixo) devem
ser obtidos com ênfase e prioridade na detecção
de condições sistêmicas, como infecção pulmonar
(raios-x de tórax) e urinária (exame de urina com
cultura), desequilíbrio hidroeletrolítico e disfunção
renal e hapática, além de outros exames, conforme
necessário26,27,31,33,46,48,63,81.
É importante lembrar que causas comuns são
comuns, de modo que muitos casos de DRP em
idosos provavelmente resultam de infecções (urinária,
respiratória) causando delirium. Entretanto, quando
causas mais simples são exluídas, torna-se necessário
considerar diversas categorias de etiologias possíveis e
excluir cada uma de modo sistemático 31, 33.
Dessa forma, uma vez esclarecida essa etapa
(delirium vs não-delirium) a investigação já iniciada
pode prosseguir, levando em conta os dados já obtido e
a obtenção de exames adicionais, conforme necessário.
A etapa seguinte ajuda a difenciar as variadas categorias
e etiologias de DRPs relacionadas na Tabela.
Etapa nº 3
Exame neurológico, neuropsicológico e
neuropsiquiátrico
O exame neurológico pode oferecer dados importantes
jul - agos - set, 2012
Demências rapidamente progressivas. Revisão atualizada e etapas diagnósticas
através da evidenciação de comprometimento motor
e oculomotor, movimentos anormais (tremor, coréia,
mioclonia, asterixis), alterações de tônus, exame
de FO. Podem ser encontrados sinais de liberação
frontal (preensão, nasopalpebral, labial, sucção,
palmomentoniano). A avaliação neuropsicológica
(testagem de função executiva, memória, linguagem,
função visuoespacial) pode mostrar padrões de
comprometimento cognitivo sugestivo de tipos de
demência. É necessária avaliação comportamental
(depressão, ansiedade, agitação, apatia, delírios,
alucinações) também capaz de trazer dados relevantes
52, 63, 72
.
Etapa nº 4
Exames paraclínicos
Exame de sangue. Hemograma e bioquímica são
necessários para definir encefalopatias com causas
reversíveis. Distúrbios eletrolíticos [inclusive Na, Ca,
P e Mg] e das taxas de glicose (hipo- ou hiperglicemia)
podem causar alteração aguda do estado mental.
Leucocitose pode indicar processo infeccioso sistêmico.
Exame de função tireoideana (inclusive anticorpos
a-TPA e a-TG), hepática (inclusive amônia) e renal,
e eventualmente paratireoideana, vitamina B12,
homocisteína,ácido metilmalônico, VDRL, marcadores
inflamatórios (VHS, PCR) e painel reumatológico
(complementos C3, C4 e CH50, AAN, FR, ANCA
[p, c], anticorpos anti-Ro/SSA e anti-Lo/SSB), são
necessários para encontrar demências reversíveis.
Em casos específicos devem ser realizados testes para
vírus (VHS-1, HIV, VVZ). Diante de suspeita de
condições paraneoplásicas uma pesquisa de anticorpos
onconeuronais (antineuronais) no soro torna-se
necessária, compreendendo o anti-Hu (ANNA-1),
anti-Ma2, CV2 (anti–CMRP-5), Yo (PCA-1), antiNMDA-R. Pensando em condições não-neoplásicas
devem ser consideraod os anticorpos anti-VGKC e
antineurópilo, anticorpos para GAD e para receptores
AMPA-R e GABAB-R. Essa pesquisa pode também ser
realizada no LCR. O exame de urina, compreendendo
cultura e a existência de fatores que possam contribuir ao
quadro demencial deve ser realizado sistematicamente.
Outros itens são solicitados de acordo com o quadro
clínico1,31,32,33,46,62,63,64,72.
Exame de LCR. É uma das principais etapas.
Oferece informações iniciais quanto a possível natureza
inflamatória da doença e permite obter biomarcadores
de lesão neuronal ou de doença infecciosa. A pressão
inicial deve ser verificada, assim como o aspecto
físico. São realizados exame citológico (incluindo
leucócitos e hamácias) total e diferencial, pesquisa
de células neoplásicas, dosagem das proteínas e da
glicose, eletroforese e IgG (obter um índice de IgG
determinar aumento de sua síntese intratecal) e
bandas oligoclonais, encontrado de modo frequente
na EM e também em outras doenças imunomediadas
ou inflamatórias. O exame bacterioscópico é realizado
com material corado para flora gram-positiva e gramnegativa, BAAR e fungos, acompanhado por estudo
imunológico para bactérias, fungos e vírus e teste
VDRL. Deve ser semeado material para as culturas
específicas. A pesquisa da proteína 14-3-3 e da tau
total, biomarcadores de lesão neuronal, é fundamental.
A presença de proteína 14-3-3 é indicativa de
destruição neuronal significativa e relativamente
rápida. O aumento de sua concentração ocorre na
sDCJ e também em algumas outras DRPs (falsos
positivos, como em casos de DA e DCL com evolução
fulminante) e falsos negativos, devendo-se ter cautela
na interpretação. Outros marcadores que podem ser
utilizados são enolase neurônio-específica (ENE) e
a S100b, com menor especificidade, e a tau total. A
dosagem de β-amilóide, tau e P-tau deve ser solicitada
na suspeita de DA. Havendo suspeita de doença
paraneoplásica devem ser pesquisados anticorpos
onconeuronais1,16,31,32,33,45,59,62,63,64,72,79.
Neuroimagem. Deve ser realizada sempre. A
TC pode ser útil em uma triagem inicial, sendo um
procedimento rápido e importante em situações
agudas (infarto vs hemorragia intracraniana). A RM
do crânio, sem e com contraste, deve ser realizada
utilizando diversas sequências (difusão, gradienteeco, FLAIR) . O padrão das anormalidades na RM
pode ser específico e indicar o diagnóstico, e mesmo
quando inespecífico, pode ajudar no diagnóstico
diferencial e no encaminhamento para outros
exames. Permite evidenciar processos vasculares,
infecciosos, autoimunes, neurodegenerativos. Além
de achados como tumores primários ou secundários,
pode ser encontrada imagem de hematoma subdural
uni- ou bilateral. Achados como infartos (regiões
subcorticais e corticais), hiperintensidades (substância
branca), hemorragias ou micor-hemorragias (AAC),
anormalidades nas regiões mediais dos lobos temporais
(EL, EHS-1), alterações compatíveis com sarcoidose
Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
jul - agos - set, 2012 »
37
Engelhardt E.
(espessamento e realce ao contraste das leptomeninges)
são outros aspectos relevantes para o diagnóstico
específico. A RM (com sequência em difusão) é
imprescindível na distinção de uma DPR priônica de
uma não-priônica. A sDCJ se caracteriza pela presença
de anormalidades corticais (em até 80% ), nos gânglios
da base (em particular no caudado), (em 60%), tálamo
(14%) . Entretanto o diagnóstico deve ser feito com
cautela, já que a concordância entre profissionais
mostrou-se em torno de 64% dos casos, mostrando que
a definição nem sempre é fácil. A sensibilidade da RM
em casos de sDCJ provável comprovados por autópsia
variou entre 58 e 70%, de acordo com o observador. Já
a especificidade mostrou-se elevada, entre 81 e 89%. A
avaliação de padrões de atrofia pode distinguir diversas
demências neurodegenerativas. A EL pode mostrar na
RM, inicialmente, edema uni- ou bilateral em estruturas
temporais mediais, com hiperintensidade na sequência
com FLAIR. O uso de contraste frequentemente não
mostra realce das alterações encontradas. É necessária
a diferenciação com a EHS, na qual a RM mostra
geralmente sinais de edema e efeito de massa, sendo
algumas vezes hemorrágico, e ocorre realce ao contraste.
Deve ser realizada angio-RM e eventualmente AGC
convencional, para o diagnóstico de vasculites. Diante
de suspeita de doença inflamatória ou paraneoplásica
devem ser obtidas imagens de tórax, abdome e pelve
com TC ou RM, conforme necessário33,41,43,44,63,68,72,75,78.
Eletroencefalograma. É um recurso diagnóstico
importante. As alterações encontradas podem ser
variadas, que podem ajudar no diagnóstico diferencial,
com presença de lentificação inespecífica, elementos
irritativos, entre outros. A definição da presença ou
não de crises clínicas ou subclínicas, é importante
em pacientes que apresentam comprometimento
cognitivo subagudo relacionado a estado epiléptico
não-convulsivo (epilepsia focal ou crises parciais
complexas), ou de um estado pós-crítico.
É essencial na sDCJ, onde complexos periódicos de
pontas ou ondas lentas agudas de 0,5 a 2,0 Hz estão
presentes em cerca de 2/3 (dependendo da fase) dos
pacientes com sDCJ (em numero menor em casos
jovens), com sensibilidade de 66% e relatos que lhe
atribuem uma especificidade de 74%. As descargas
periódicas sincrônicas parecem ser a marca da sDCJ,
entretanto, outras apresentações atípicas foram
relatadas sem tais grafoelementos. O EEG é importante
na EL e EH, estas comumente associadas a crises. Pode
38
» Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
apresentar anormalidade em encefalopatias (p.ex.,
hepática com ondas trifásicas). O exame é bastante
sensível e mostra anormalidades e 80% dos casos de
EHS. Considerando o contexto clínico, a ocorrência
de lentificação focal temporal ou difusa e a presença de
complexos periódicos e descargas irritativas lateralizadas
periódicas é fortemente sugestiva de EHS33,54,63,71,72.
Concluídas essas etapas e não havendo uma
definição diagnóstica é considerada a possibilidade de
biópsia meníngea-cerebral.
Etapa nº 5
Biópsia meníngea-cerebral
Determinados casos, nos quais o diagnóstico não
pode ser definido após exaustiva pesquisa conforme
as etapas propostas, podem requerer a indicação
de uma biópsia meníngea-cerebral para esclarecer
uma determinada etiologia, se neurodegenerativa,
neoplásica, inflamatória ou infecciosa. A decisão, no
caso de uma DRP, pode ser uma questão premente,
considerando a oportunidade de diagnosticar uma
condição tratável e potencialmente reversível. A
biópsia ainda vem sendo vista como um último
recurso, e muitas vezes não chega a ser cumprida.
Este aspecto pode ser atribuído à taxa que permanece
com diagnóstico indefinido e à possibilidade de
complicações (morbidade, mortalidade). A decisão
de indicar uma biópsia cerebral depende da análise
de risco-benefício, onde a probabilidade de obter
um diagnóstico e possivelmente alterar o manejo
do caso deve ser ponderado em relação aos riscos
do procedimento e as chances de uma terapêutica
empírica. A sensibilidade diagnóstica em casos de DRPs
oscila entre 20-65% e em alguns serviços de referência
acima desses valores. A biópsia para diagnóstico da
sDCJ praticamente deixou de ser feita com os critérios
clínicos e complementares (EEG, RM com sequência
em difusão, LCR com pesquisa da proteína 14-3-3)
utilizados, eliminando os numerosos riscos associados
à mesma (morbidade, mortalidade, contaminação do
material utilizado e seu descarte)43,63,67,80.
Etapa n° 6
Diagnóstico
Esgotadas as etapas acima a expectativa é a de
chegar ao diagnóstico e, em decorrência, ao tratamento
adequado. Tal expectativa, porém, é muitas vezes
frustrada, pois, mesmo nos grandes centros, fica
jul - agos - set, 2012
Demências rapidamente progressivas. Revisão atualizada e etapas diagnósticas
um percentual de quadros com diagnóstico não
determinado.
Conclusão
As DRPs se constituem em um grupo heterogêneo
de condições que evoluem de modo rápido com
comprometimento cognitivo, neuropsiquiátrico
e motor. Apesar de relativamente infrequentes
representam um dos maiores desafios clínicos, mesmo
para os profissionais mais experientes. A avaliação
diagnóstica de qualquer paciente com suspeita de
apresentar uma DRP é tipicamente mais abrangente
Referências
1. Anderson WE. Herpes Simplex Encephalitis: Differential
Diagnoses & Workup. [serial on the Internet] 2009
[cited 2010 Mai] Available from: URL: http://emedicine.
medscape.com/article/1165183-diagnosis
2. APA. Diagnostic and statistical manual of mental disorders:
DSM-IV. 4ª ed. Washington, D.C.: American Psychiatric
Association, 1994.
3. Aquino RT, Mutarelli EG. Hashimoto’s encephalopathy. Arq
Neuropsiquiatr 2009; 67(3A):724-725.
4. Baevsky RH, Bartfield JM. Human rabies: A review. Am J
Emerg Med 1993;11(3):279-286.
4a. Ballone GJ. Aspectos Neuropsiquiátricos da Infecção por
HIV. In. PsiqWeb, Internet, disponível em www.psiqweb.
med.br, revisto em 2005 [acessado em 2012]
5. Baringer JR. Herpes Simplex Infections of the Nervous
System. Neurol Clin 2008; 26: 657-674.
6. Barreto ML, M. Elvira Balcells M, Ana Fernández S et al.
Neurosífilis en pacientes portadores y no portadores de VIH:
Descripción y comparación de dos cohortes históricas. Rev
Chil Infect 2009;26(6):540-547.
7. Berger JR, Major EO. Progressive multifocal
leukoencephalopathy. Semin Neurol 1999;19(2):193-200.
8. Bicanic T, Harrison TS. Cryptococcal meningitis. Br Med
Bull 2005;72(1):99-118.
9. Biffi A, Greenberg SM. Cerebral Amyloid Angiopathy: A
Systematic Review. J Clin Neurol 2011;7:1-9.
10. Big C, Reineck LA, Aronoff DM. Viral Infections of the
Central Nervous System: A Case-Based Review. Clin Med
Res, 7(4):142-146, 2009.
11. Cambier DM, Kantarci K, Worrell GA,Westmoreland BF,
Aksamit AJ. Lateralized and focal clinical, EEG, and FLAIR
MRI abnormalities in Creutzfeldt–Jakob disease. Clin
Neurophysiol 2003; 114:1724-1728.
12. Boeve BF. Rapidly Progressive Dementias:
Neurodegenerative Disorders. CD da 64ª Reunião Annual
da American Academy of Neurology, New Orleans, 2012.
13. Chao CP, Kotsenas AL, Broderick DF. Cerebral Amyloid
Angiopathy: CT and MR Imaging Findings. RadioGraphics
em comparação às condições neurodegenerativas
crônicas. Deve seguir um protocolo detalhado de
modo sistematizado, visando o diagnóstico de modo
rápido e acertado, objetivando estratégias terapêuticas
adequadas.
A questão mais importante com a qual o clínico
se depara é a de determinar se uma DRP tem uma
possibilidade de ser uma doença passível de ser revertida
ou amenizada ou é uma condições com tratamento
disponível limitado ou inexistente, mas cuja definição
pode contar com vantagens clínicas, epidemiológicas
e em relação à família.
2006; 26:1517-1531.
13a. Christo PP. Alterações Cognitivas na Infecção pelo HIV e
AIDS. Rev Assoc Med Bras 2010; 56(2):242-247.
14. Compston A, Coles A. Multiple sclerosis. Lancet 2002; 359:
1221-1231.
15. Damin AE, Morillo LS, Perroco TR et al. Dementia
post-radiotherapy: improvement with acetylcholinesterase
inhibitor. A case report. Dement Neuropsychol 2009;
3(1):68-72.
16. Deisenhammer F, Bartos A, Egg R et al. Guidelines on
routine cerebrospinal fluid analysis. Report from an EFNS
task force. Eur J Neurol 2006;13:913-922.
17. Dore GJ, Cooper DA. AIDS: Clinical Manifestations.
Encyclopedia of Life Sciences, Nature Publishing Group,
pg 1-8, 2001. [cited 2010 Mai] Available from: URL:
http://web.udl.es/usuaris/e4650869/docencia/segoncicle/
genclin98/recursos_classe_(pdf(/revisionsPDF/AIDSClinical-.pdf
18. Dworkin MS. A review of progressive multifocal
leukoencephalopathy in persons with and without
AIDS. Curr Clin Top Infect Dis 2002;22:181-195.
19. Eduardo MBP, Katsuya EM, Bassit NP (coordenadores/
autores). Vigilância da Doença de Creutzfeldt-Jakob e outras
Doenças Priônicas. Normas e Instruções. São Paulo, 2008.
http://www.cve.saude.sp.gov.br
20. Engelhardt E. Demências rapidamente progressivas. Uma
revisão breve. Rev Bras Neurol 2010; 46 (2):5-15.
21. Engelhardt E, Moreira DM. Demência Vascular. In:
Neurologia Cognitiva e do Comportamento. Teixeira AL e
Caramelli P, Ed. Rio de Janeiro: Revinter, 2012, pg 297-310.
22. Engelhardt E, Laks J, Cavalcanti JLS et al. Demências. In:
Neurologia para o Clínico. Gomes MM, Cavalcanti JLS Ed.
Rio de Janeiro: Editora UFRJ, 2008, pg 437-480.
23. Fatourechi V. Hashimoto’s encephalopathy: myth or reality?
An endocrinologist’s perspective. Best Pract Res Clin
Endocrinol Metab 2005; 19(1):53-66.
24. Feinstein A. The clinical neuropsychiatry of multiple
sclerosis. Cambridge: Cambidge University Press, 2ª Ed.
2007.
25. Ferreira MLB. Cognitive deficits in multiple sclerosis: a
Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
jul - agos - set, 2012 »
39
Engelhardt E.
systematic review. Arq. Neuropsiquiatr 2010;68(4):632-641.
26. Fick DM, Cooper JW, Wade WE et al. Updating the Beers
Criteria for Potentially Inappropriate Medication Use in
Older AdultsResults of a US Consensus Panel of Experts.
Arch Intern Med 2003;163(22):2716-2724.
27. Fong TG, Samir R. Tulebaev SR, Sharon K. Inouye SK.
Delirium in elderly adults: diagnosis, prevention and
treatment. Nat Rev Neurol 2009;5:210-220.
28. Fontaine B, Seilhean D, Tourbah A et al. Dementia in two
histologically confirmed cases of multiple sclerosis: one
case with isolated dementia and one case associated with
psychiatric symptoms. J Neurol Neurosurg Psychiatry 1994;
57(3): 353-359.
29. Foster AR, Caplan JP. Paraneoplastic Limbic Encephalitis.
Psychosomatics 2009;50:108-113.
30. Garg RK. Subacute sclerosing panencephalitis. Postgrad Med
J 2002;78:63–70.
31. Geschwind MD. Rapidly Progressive Dementia (RPDs):
Jakob-Creutzfeldt Disease and Related Disorders. CD da 64ª
Reunião Annual da American Academy of Neurology, New
Orleans, 2012.
32. Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL.
Rapidly Progressive Dementia. Ann Neurol 2008; 64(1):97108.
33. Geschwind MD, Haman A, Miller BL. Rapidly Progressive
Dementia. Neurol Clin 2007; 25:783–807.
34. Geschwind MD, Haman A, Viskontas IV. Prion disorders
and other rapidly progressive dementias. In: The Behavioral
Neurology of Dementia. Miller BL, Boeve BF E. Cambridge:
Cambridge University Press, 2009, pg 345-366.
35. Guimarães J, Sá MJ. Cognitive dysfunction in Multiple
Sclerosis. Frontiers in Neurology 2012;3(Article74):1-8.
36. Haraguchi T, Terada S, Ishizu H et al. Coexistence
of Creutzfeldt-Jakob disease, Lewy body disease, and
Alzheimer’s disease pathology: an autopsy case showing
typical clinical features of Creutzfeldt-Jakob disease.
Neuropathology 2009;29(4):454-459.
37. Heinemann U, Gawinecka J, Schmidt C, Zerr I. Differential
Diagnosis of Rapid Progressive Dementia. Eur Neurol Rev
2010;5(2):21-28.
38. Hemachudha T, Laothamatas J, Rupprecht CE. Human
rabies: a disease of complex neuropathogenetic mechanisms
and diagnostic challenges. Lancet Neurology 2002;1(2):101109.
39. Heringer RR, Fernandes LEBC, Gonçalves RR, PuccioniSohler M. Localização da lesão e achados do líquido
cefalorraqueano na meningite tuberculosa: diferenças
nos compartimentos lombar, cisternal e ventricular. Arq
Neuropsiquiatr 2005; 63(2B):543-547.
40. Herrero-Velázquez S, Guerrero-Peral AL,Gámez-Leyva
G, Fernández-Buey MN, Conde A, Rodríguez M, RojoMartínez E, Pascual J, Fernández-Herranz MR, DalmauObrador J. Encefalitis por anticuerpos contra el receptor
NMDA. Descripción de una paciente sin tumor asociado y
revisión de la bibliografia. Rev Neurol 2010; 50 (11): 66140
» Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
666.
41. Izco FM, Larrarte LA. Neuroimagen em el diagnostíco de
las encefalopatias espongiformes transmisibles humanas.
Neurologia 2006; 21(8):428-436.
41a. Höllinger P, Sturzenegger M, Mathis J, Schroth G, Hess
CW. Acute disseminated encephalomyelitis in adults: a
reappraisal of clinical, CSF, EEG, and MRI findings. J
Neurol 2002;249(3):320-329.
42. Josephs KA, Ahlskog JE, Parisi JE, Boeve BF, Crum
BA, Giannini C, Petersen RC. Rapidly Progressive
Neurodegenerative Dementias. Arch Neurol
2009;66(2):201-207.
43. Josephson SA, Papanastassiou AM, Berger MS et al. The
diagnostic utility of brain biopsy procedures in patients with
rapidly deteriorating neurological conditions or dementia. J
Neurosurg 2007;106:72-75.
44. Kallenberg K, Schulz-Schaeffer WJ, Jastrow U et al.
Creutzfeldt-Jakob disease: comparative analysis of MR
imaging sequences. AJNR 2006;27(7):1459-1462.
45. Karcher DS, McPherson RA. Cerebrospinal, Synovial, Serous
Body Fluids, and Alternative Specimens. In: McPherson:
Henry’s Clinical Diagnosis and Management by Laboratory
Methods, 22nd ed, McPherson RA, Pincus RM ed.
Philadelphia: Saunders, 2011, pg 480-508.
46. Kelley BJ, Boeve BF, Josephs KA. Rapidly Progressive
Young-Onset Dementia. Cogn Behav Neurol
2009;22(1):22-27.
47. King O, Song X, Rockwood K. Cognitive Impairment of
Acute Onser in the consortium to Investigate Vascular
Impairment of cognition (CIVIC) Study: Occurrence,
correlates, and Outcomes. Am J Geriatr Psychiatry
2006;14:893-896.
48. Kunze K. Metabolic encephalopathies. J Neurol
2002;249:1150-1159.
49. Lan SH, Chang WN, Lu CH, Lui CC, Chang HW.
Cerebral infarction in chronic meningitis: a comparison of
tuberculous meningitis and cryptococcal meningitis. Q J
Med 2001; 94: 247-253.
50. Lanna MEO, Alves CEO, Sudo FK, Alves G, Valente L,
Moreira DM, Cavalcanti JLS, Engelhardt E. Cognitive
disconnective syndrome by single strategic strokes in
vascular dementia. J Neurol Sci 2012 Aug 28. [Epub ahead
of print]
51. Lindenberg ASC, Chang MR, Paniago AMM et al. Clinical
and epidemiological features of 123 cases of cryptococcosis
in Mato Grosso do Sul, Brazil. Rev Inst Med Trop S Paulo
2008;50(2):75-78.
51a. Menge T, Hemmer B, Nessler S, Wiendl H, Neuhaus
O, Hartung HP, Kieseier BC, Stüve O. Acute
disseminated encephalomyelitis: an update. Arch Neurol
2005;62(11):1673-1680.
52. Miller BL. Basal Clinical Approaches. In: The Behavioral
Neurology of Dementia. Miller BL, Boeve BF Eds.
Cambridge: Cambridge University Press, 2009, pg 1-6.
53. Moreira DM, Oliveira Jr AC, Nacif MS, Engelhardt E. A
jul - agos - set, 2012
Demências rapidamente progressivas. Revisão atualizada e etapas diagnósticas
artéria de Percheron e infartos talâmicos bilaterais. Rev Bras
Neurol 2008;44(1):35.
54. Neiman ES, Hanna PA. EEG in Dementia and
Encephalopathy. Medscape, 2012. emedicine.medscape.
com/article/1138235 [acessado em 02-10-2012]
54a. Noorbakhsh F, Johnson RT, Emery D, Power C. Acute
disseminated encephalomyelitis: clinical and pathogenesis
features. Neurol Clin 2008;26(3):759-780.
55. Pereira E. Diffusion-weighted sequence on MRI for the
diagnosis of Creutzfeldt-Jakob disease. Arq Neuropsiquiatr
2002; 60 (4):906-908.
56. Pomper MG, Miller TJ, Stone JH, Tidmore WC, Hellmann
DB. CNS Vasculitis in Autoimmune Disease: MR Imaging
Findings and Correlation with Angiography. AJNR
1999;20:75-85.
57. Prashanth LK, Taly AB, Ravi V, Sinha S, Arunodaya GR.
Adult onset subacute sclerosing panencephalitis: clinical
profile of 39 patients from a tertiary care centre. J Neurol
Neurosurg Psychiatry 2006;77:630-633.
58. Puccioni-Sohler M. Cerebrospinal fluid oligoclonal IgG
bands in multiple sclerosis: what does it mean? Arq.
Neuropsiquiatr 2012;70(8):569-570.
59. Puccioni-Sohler M, Passeri F, Oliveira C, Brandão CO,
Papaiz-Alvarenga R. Multiple sclerosis in Brazil. Analysis of
cerebrospinal fluid by standard methods. Arq Neuropsiquiatr
1999;57:927-931.
60. Ramos-Rivas M, Rojas-Velasco G, Acuña-Hidalgo R et al.
Encefalitis límbica paraneoplásica: una entidad de difícil
diagnóstico. Rev Neurol 2009;48(6):311-316.
61. Rehman HU. Primary angiitis of the central nervous system.
J R Soc Méd 2000;93(11):586–588.
62. Rosenbloom MH, Smith S, Akdal G, Geschwind MD.
Immunologically Mediated Dementias. Curr Neurol
Neurosci Rep 2009;9(5):359-367.
63. Rosenbloom MH, Atri A. The Evaluation of Rapidly
Progressive Dementia. Neurologist 2011;17(2):67-74.
64. Rosenfeld MR, Dalmau J. Paraneoplastic limbic encephalitis
associated with small-cell lung cancer. Commun Oncol
2007;4:491-494.
65. Sala I, Marquié M, Sánchez-Saudinó MB et al. Rapidly
Progressive Dementia Experience in a Tertiary Care Medical
Center. Alzheimer Dis Assoc Disord 2012;26:267-271
66. Schiess N, Pardo CA. Hashimoto’s encephalopathy. Ann N
Y Acad Sci 2008;1142:254-265.
67. Schuette AJ, Taub JS, Hadjipanayis CG, Olson JJ. Open
biopsy in patients with acute progressive neurologic decline
and absence of mass lesion. Neurology 2010;75:419-424.
67a. Schwartz S, Mohr A, Knauth M, Wildemann B, StorchHagenlocher B. Acute disseminated encephalomyelitis:
a follow-up study of 40 adult patients. Neurology
2001;56(10):1313-1318.
68. Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD.
Neurodegenerative diseases target large-scale human brain
Networks. Neuron 2009;62(1): 42-52.
69. Sethi S, Das A, Kakkar N, Banga SS, Prabhakar S, Sharma
M. Neurosyphilis in a tertiary care hospital in north India.
Indian J Med Res 2005;122:249-253.
70. Sicotte NL. Neuroimaging in Multiple Sclerosis:
Neurotherapeutic Implications. Neurotherapeutics 2011;
8(1):54-62.
71. Smith SJM. EEG in Neurological Conditions other than
Epilepsy: When does it Help, what does it add? J Neurol
Neurosurg Psychiatry 2005;76(Suppl II):ii8- ii12.
71a. Sonneville R, Klein I, de Broucker T, Wolff M. Postinfectious encephalitis in adults: diagnosis and management.
J Infect 2009;58(5):321-328.
72. Sorbi S, Hort J, Erkinjuntti T et al. on behalf of the EFNS
Scientist Panel on Dementia and Cognitive Neurology.
EFNS-ENS Guidelines on the diagnosis and management
of disorders associated with dementia. Eur J Neurol
2012;19:1159-1179.
73. Szirmai I, Vastagh I,Szombathelyi E, Kamondi A. Strategic
infarcts of the thalamus in vascular dementia. J Neurol Sci
2002; 203- 204: 91-97.
74. Takada LT, Camiz P, Grinberg LT, Leite CC. Noninflammatory cerebral amyloid angiopathy as a cause
of rapidly progressive dementia: A case study. Dement
Neuropsychol 2009;3(4):352-357.
75. Tschampa HJ, Kallenberg K, Urbach H et al. MRI in the
diagnosis of sporadic Creutzfeldt-Jakob disease: a study on
inter-observer agreement. Brain 2005;128(9):2026-2033.
76. Tschampa HJ, Neumann N, Zerr I et al. Patients with
Alzheimer’s disease and dementia with Lewy bodies
mistaken for Creutzfeldt-Jakob disease J Neurol Neurosurg
Psychiatry 2001;71:33-39.
77. Tsuchiya K, Yagishita S, Ikeda K et al. Coexistence of CJD
and Alzheimer’s disease: an autopsy case showing typical
clinical features of CJD. Neuropathology 2004; 24(1):46-55.
78. Urbach H, Soeder BM, Jeub M et al. Serial MRI of limbic
encephalitis. Neuroradiology 2006;48(6):380-386.
79. Vernino S. Autoimmune Encephalopathies. CD da 64ª
Reunião Annual da American Academy of Neurology, New
Orleans, 2012.
80. Warren JD, Schott JM, Fox NC et al. Brain biopsy in
dementia. Brain 2005;128:2016-2025.
81. Wilson JX, Young GB. Sepsis-Associated Encephalopathy:
Evolving Concepts. Can J Neurol Sci 2003; 30: 98-105.
82. Woodruff BK. Evaluation of Rapidly Progressive Dementia.
Seminars Neurol 2007;27(4):363-375.
83. Yu Y, Wei M, Huang Y et al. Clinical presentation and
imaging of general paresis due to neurosyphylis in patients
negative for human immudodeficiency virus. J Clin Neurosci
2010;17(3):308-310.
Revista Brasileira de Neurologia »
Volume 48 »
Nº 3 »
jul - agos - set, 2012 »
41