FETUS - CENTRO DE ESTUDOS ESPECIALIZADO EM
MEDICINA FETAL
JOSÉ LEANDRO FATURI
UMA REVISÃO SOBRE MALFORMAÇÕES CARDÍACAS:
ANOMALIA DE EBSTEIN,
TRUNCUS ARTERIOSUS COMMUNIS, ECTOPIA CORDIS E
TUMORES CARDÍACOS.
São Paulo
2011
2
JOSÉ LEANDRO FATURI
UM ROTEINO PARA MALFORMAÇÕES CARDÍACAS:
ANOMALIA DE EBSTEIN,
TRUNCUS ARTERIOSUS COMMUNIS, ECTOPIA CORDIS E
TUMORES CARDÍACOS.
Trabalho de Conclusão de Curso
de pós-graduação em medicina fetal
“lacto sensus” da FETUS – Centro de
Estudos Especializados em Medicina
Fetal.
São Paulo
2011
3
JOSÉ LEANDRO FATURI
UM ROTEINO PARA MALFORMAÇÕES CARDÍACAS:
ANOMALIA DE EBSTEIN,
TRUNCUS ARTERIOSUS COMMUNIS, ECTOPIA CORDIS E
TUMORES CARDÍACOS.
LOCAL: São Paulo - SP
DATA: 18 a 20 de janeiro de 2011
Membros componentes da banca examinadora:
Professora Maria Virgínia Machado
Professor Rogério dos Reis Guidoni
4
DEDICATÓRIA
Dedico este trabalho à minha esposa e
família por se constituírem diferentemente
enquanto pessoas, admiráveis em essência,
estímulos que me impulsionaram a buscar
vida nova a cada dia, meus agradecimentos
por terem aceito se privar de minha
companhia pelos estudos, concedendo a mim
a oportunidade de me realizar ainda mais.
5
EPÍGRAFE
“Quando se está aprendendo, o professor atua
apenas como uma agulha; o aluno é a linha. Como seu
mentor, posso ajudá-lo, apontando-lhe a direção correta.
Mas, como a agulha da linha, devo me separar de você
no fim, porque a força, a fibra e a capacidade de juntar
todas as partes devem ser suas”.
(SECRETAN, Lance H. K. Os Passos do tigre)
6
RESUMO
Este trabalho tem como objetivo fazer uma revisão de literatura sobre as principais
particularidades (definição, incidência, etiopatogenia, classificação, anomalias
associadas, diagnóstico pré-natal, diagnóstico diferencial, conduta pré-natal, conduta
obstétrica, assistência neonatal, tratamento pré e pós-natal) das seguintes
patologias cardíacas: Anomalia de Ebstein, Truncus Arteriosus Communis, Ectopia
Cordis e Tumores Cardíacos.
7
ABSTRACT
This article aims to review the literature on the principal features (definition,
incidence, etiology, classification, associated anomalies, prenatal diagnosis,
differential diagnosis, conduct pre-natal, obstetric, neonatal care, treatment and postnative) of the following heart diseases: Ebstein's anomaly, truncus arteriosus
communis, Ectopia Cordis and Cardiac Tumors.
8
SUMÁRIO
SUMÁRIO.............................................................................................................................................................. 8
LISTA DE TABELAS ......................................................................................................................................... 10
LISTA DE GRÁFICOS ...................................................................................................................................... 11
LISTA DE FIGURAS ......................................................................................................................................... 12
LISTA DE ABREVIATURAS E SIGLAS ........................................................................................................ 13
1
INTRODUÇÃO .......................................................................................................................................... 14
2
DESENVOLVIMENTO ............................................................................................................................ 16
2.1.1
ANOMALIA DE EBSTEIN ............................................................................................................. 16
2.1.2
DEFINIÇÃO .................................................................................................................................. 16
2.1.3
INCIDÊNCIA ................................................................................................................................. 17
2.1.4
ETIOPATOGENIA......................................................................................................................... 17
2.1.5
CLASSIFICAÇÃO .......................................................................................................................... 19
2.1.6
ANOMALIAS ASSOCIADAS ......................................................................................................... 20
2.1.7
DIAGNÓSTICO PRÉ-NATAL ....................................................................................................... 21
2.1.8
DIAGNÓSTICO DIFERENCIAL ................................................................................................... 22
2.1.9
CONDUTA PRÉ-NATAL ............................................................................................................... 22
2.1.10
CONDUTA OBSTÉTRICA ........................................................................................................ 23
2.1.11
ASSISTÊNCIA NEONATAL ...................................................................................................... 24
2.1.12
PROGNÓSTICO ....................................................................................................................... 25
2.2
TRUNCUS ARTERIOSUS COMMUNIS ..................................................................................................... 28
2.2.1
DEFINIÇÃO .................................................................................................................................. 28
2.2.2
INCIDÊNCIA ................................................................................................................................. 29
2.2.3
ETIOPATOGENIA......................................................................................................................... 29
2.2.4
CLASSIFICAÇÃO .......................................................................................................................... 30
2.2.5
ANOMALIAS ASSOCIADAS ......................................................................................................... 31
2.2.6
DIAGNÓSTICO PRÉ-NATAL ....................................................................................................... 32
2.2.7
DIAGNÓSTICO DIFERENCIAL ................................................................................................... 33
2.2.8
CONDUTA PRÉ-NATAL ............................................................................................................... 33
2.2.9
CONDUTA OBSTÉTRICA............................................................................................................. 34
2.2.10
ASSISTÊNCIA NEONATAL E TRATAMENTO ........................................................................ 34
2.2.11
PROGNÓSTICO ....................................................................................................................... 35
2.3
ECTOPIA CORDIS ................................................................................................................................. 37
2.3.1
DEFINIÇÃO .................................................................................................................................. 37
2.3.2
INCIDÊNCIA ................................................................................................................................. 37
2.3.3
ETIOPATOGENIA......................................................................................................................... 37
2.3.4
CLASSIFICAÇÃO .......................................................................................................................... 38
2.3.5
ANOMALIAS ASSOCIADAS ......................................................................................................... 39
2.3.6
DIAGNÓSTICO PRÉ-NATAL ....................................................................................................... 40
2.3.7
DIAGNÓSTICO DIFERENCIAL ................................................................................................... 40
2.3.8
CONDUTA PRÉ-NATAL E OBSTÉTRICA.................................................................................... 41
2.3.9
ASSISTÊNCIA NEONATAL E TRATAMENTO ............................................................................. 41
2.3.10
PROGNÓSTICO ....................................................................................................................... 42
2.4
TUMORES CARDÍACOS ................................................................................................................. 44
2.4.1
DEFINIÇÃO .................................................................................................................................. 44
2.4.2
INCIDÊNCIA ................................................................................................................................. 44
2.4.3
ETIOPATOGENIA......................................................................................................................... 44
2.4.4
CLASSIFICAÇÃO (TIPOS) ........................................................................................................... 45
2.4.5
ANOMALIAS ASSOCIADAS ......................................................................................................... 46
2.4.6
DIAGNÓSTICO PRÉ-NATAL ....................................................................................................... 47
2.4.7
DIAGNÓSTICO DIFERENCIAL ................................................................................................... 49
9
2.4.8
2.4.9
2.4.10
CONDUTA PRÉ-NATAL E OBSTÉTRICA.................................................................................... 50
ASSISTÊNCIA NEONATAL E TRATAMENTO ............................................................................. 50
PROGNÓSTICO ....................................................................................................................... 51
3
CONCLUSÃO ............................................................................................................................................ 52
4
REFERÊNCIAS ......................................................................................................................................... 54
10
LISTA DE TABELAS
Tabela 1 - Fatores de risco para doenças cardíacas congênitas, modificada de YOO,
2008) .................................................................................................................. 15
Tabela 2 - Caracaterísticas ultrassonográficas dos tumores cardíacos (modificada
de CARVALHO, 2010) ....................................................................................... 47
11
LISTA DE GRÁFICOS
Grafico 1 - Freqüência dos tipos de ectopia cordis (CABRERA et al. 2002) ............. 38
12
LISTA DE FIGURAS
Figura 1 - Classificação de Carpentier, 1988 ............................................................ 20
Figura 2 - Rabdomioma ocupando quase totalmente o ventrículo esquerdo ............ 48
13
LISTA DE ABREVIATURAS E SIGLAS
AE = anomalia de Ebstein
CIA = comunicação interatrial
CIV = comunicação interventricular
DCC = doenças cardíacas congênitas
NYHA = New York Heart Association (Classificação)
TAC = truncus arteriosus communis
TF = Tetralogia de Fallot
EC = ectopia cordis
14
1 INTRODUÇÃO
As doenças cardíacas congênitas (DCC) são a principal causa de óbito
neonatal e infantil, não somente devido à complexidade das malformações, mas
também devido à sua incidência elevada.
Sua incidência estimada é de 7,5/1.000 nascidos vivos, se considerarmos
apenas os casos moderados e graves, sua incidência é de cerca de 6/1.000.
(Hoffman, 2002)
O diagnóstico pré-natal interfere positivamente na conduta pós-natal, menor
tempo de espera por tratamento cirúrgico, melhor prevenção de comprometimento
hemodinâmico grave, e menor período de permanência em unidade intensiva.
Essa monografia tem como objetivo fazer uma revisão (definição, incidência,
etiopatogenia,
classificação,
anomalias
associadas,
diagnóstico
pré-natal,
diagnóstico diferencial, conduta pré-natal, conduta obstétrica, assistência neonatal,
tratamento pré e pós-natal) das seguintes patologias cardíacas:
•
Anomalia de Ebstein:
•
Truncus Arteriosus Communis;
•
Ectopia Cordis;
•
Tumores Cardíacos.
Devido à associação freqüente das DCC com anomalias extracardíacas (8 a
42%) e aberrações cromossômicas (5 a 13%), estarão sempre indicados um estudo
ultrassonográfico detalhado da morfologia fetal e um estudo cromossômico.
15
A ultrassonografia cardíaca fetal pode ser realizada como rastreamento prénatal na população de baixo risco, ou como exame completo para os grupos de alto
risco, ver Tabela 1.
Tabela 1 - Fatores de risco para doenças cardíacas congênitas, modificada de Yoo, 2008)
Fetais
Anomalias
cromossômicas,
anomalias
extracardíacas, onfalocele, atresia de esôfago, atresia de
duodeno, hérnia diafragmática, associação de VACTREL,
aumento da translucência nucal, espessamento da prega
nucal, hidropisia não-imune, poli-hidrâmnio, oligoâmino,
arritmia cardíaca fetal
Paternos
Doença
caradíaca
materna,
diabetes
materna,
doença vascular do colágeno, anticorpos anti Ro/La
positivos,
fenilcitonúria,
exposição
a
àlcool,
anticonvulsivantes, lítio, ácido retinóico, rubéola, coxsakie,
citomegalovírus, parvovírus B19
Familiares
História familiar de doença cardíaca congênita ou
síndromes associadas a elas, esclerose tuberosa, síndrome
de
Nooman,
síndrome
cromossomo 22q11
de
Holt-Oram,
deleção
do
16
2 DESENVOLVIMENTO
2.1.1 ANOMALIA DE EBSTEIN
2.1.2 DEFINIÇÃO
A Anomalia de Ebstein (AE) é uma malformação cardíaca na qual ocorre uma
inserção anômala da válvula tricúspide, ou seja, a principal característica de
anomalia de Ebstein é um deslocamento apical do folheto septal da válvula
tricúspide da inserção da cúspide anterior da valva mitral, pelo menos, 8 mm/m² da
área de superfície corporal de sua inserção no ventrículo direito é mais baixa; a
válvula pode ser hipoplásica ou displásica, e incompetente ou estenótica.
Descrito pela primeira vez em 1866, por Wilhelm Ebstein em um relatório de
necropsia de um homem de 19 anos que sofria de palpitações e dispnéia desde de
criança (Radford, 1985).
Os sintomas clínicos mais comuns são: dispnéia, principalmente se o paciente
já apresenta cianose (mesmo se não está presente logo após o nascimento, vai se
constituir mais tarde), e podem apresentar sopro sistólico e diastólico na área
tricúspide principalmente com a manobra de Rivero-Carvalho.
17
2.1.3 INCIDÊNCIA
A anomalia de Ebstein é uma cardiopatia congênita complexa rara, sua
incidência é de cerca de 1 para 20.000 nascidos vivos, correspondendo a 0,5% de
todas as DCC. (Jost, 2007)
2.1.4 ETIOPATOGENIA
No coração normal, a válvula tricúspide tem três folhetos: anterior, posterior, e
septal. A AE é caracterizada por uma aderência dos folhetos septal e posterior ao
miocardio adjacente. Os folhetos se desenvolvem do endocárdio e do miocardio por
um processo de delaminação, ou seja, separação do tecido pelo descolamento da
camada interna durante o desenvolvimento embriológico, é pela falha nesse
processo que ocorre a AE. Anatomicamente, observam-se ainda:
deslocamento apical do annulus funcional;
dilatação da porção “atrializada” do ventrículo direito com variado níveis de
hipertrofia e adelgaçamento da parede;
folheto anterior geralmente se encontra redundante / deformado /
fenestrado, e sua cordoalha tendinea é curta e pouco desenvolvida,
dilatação da junção átrio-ventricular direita. (Jost, 2007).
18
Os folhetos não conseguem se alinhar adequadamente durante a sístole
ventricular, então, ocorre a regurgitação e provocando dilatação das câmaras
cardíacas direitas (cardiomegalia).
Em outras palavras, essas alterações na válvula tricúspide determinam
defeitos anatômicos que levam a repercussões hemodinâmicas: insuficiência
valvular e um fenômeno chamado de “atrialização” do ventrículo direito (que se
apresenta com paredes finas semelhantes às do átrio, e funcionalmente, perde
grande parte de sua força contrátil), que leva a uma maior resistência ao
esvaziamento do átrio direito.
Em poucos casos, as bordas do septo e dos folhetos deslocados estão
fusionadas na margem do folheto anterior, provocando uma estenose da tricúspide
com regurgitação discreta, e desta forma, apenas o átrio direito se encontra dilatado.
O espectro da malformação na AE pode variar desde um descolamento
mínimo da válvula tricúspide até casos em com imperfuração da mesma, não
permitindo passagem de fluxo do átrio direito para o ventrículo direito. (Zielinsky,
2000).
A maioria dos casos de AE são esporádicos, AE familiar é raro
Estudos de caso-controle sugerem fatores de risco com: gêmeos, história
familiar de cardiopatia congênita. A exposição materna a benzodiazepinicos e a lítio
também já foram comentadas, porém ainda foram efetivamente comprovadas por
outros estudos.
19
Há ainda raros casos descritos, da associação AE e alterações genéticas:
mutações no fator de transcriptação cardíaco NKX2.5, deleção 10p13-p14 e deleção
1p34.3-p36.11.
2.1.5 CLASSIFICAÇÃO
JOST et al, 2007, usam duas classificações para descrever a severidade da
AE.
Uma é baseada nos achados ecocardiográficos: anatomicamente leve,
moderada, severa. Esta classificação é simples, porém imprecisa. A outra é mais
detalhada e enfatiza características importantes para determinar a estratégica
cirúrgica: reparação ou substituição da válvula tricúspide. (DEARANI, 2000)
Em 1988, CARPENTIER et al. propôs a seguinte classificação para AE:
(Figura 1)
Tipo A: volume do ventrículo direito é adequado;
Tipo B: presença da dilatação da porção “atrializada” do ventrículo direito,
mas sem comprometimento do folheto anterior;
Tipo C: restrição severa do folheto anterior que pode causar obstrução do
fluxo para o ventrículo direito;
Tipo D: “atrialização” quase completa do ventrículo direito exceto por pequeno
componente infundibular.
20
Figura 1 - Classificação de Carpentier, 1988
2.1.6 ANOMALIAS ASSOCIADAS
São descritas diversas anormalidades associadas à anomalia de Ebstein, as
mais comuns são:
•
comunicação interatrial (90%);
•
atresia pulmonar anatômica ou funcional (30%); e
•
comunicação interventricular (menos comumente).
Outras malformações cardíacas associadas são: válvula aórtica bicúspide ou
atrésica, hipoplásia ou atresia da artéria pulmonar, estenose subaortica, displasia do
ventrículo esquerdo, prolapso da válvula mitral, taquicardia supraventricular e
Síndrome de Wolff-Parkinson-White.
A maioria dos pacientes com transposição dos grandes vasos tratável
cirurgicamente preenche critérios para AE em 15 a 50% dos casos.
21
Há ainda raros casos associados a trissomias do 13 e do 21, e a síndromes
como a de Turner, Cornelia de Lange e Marfan. A síndrome de Down está associada
a cardiopatia congênita em 50% dos casos, porém, as associações mais comuns
são com os defeitos septais e a tetralogia de Fallot. Em revisão da literatura dos
últimos dez anos foram encontradas 3 descrições de casos clínicos de associação
da anomalia de Ebstein com síndrome de Down e somente em um o diagnóstico foi
realizado no período pré-natal (Leite e cols, 2004).
2.1.7 DIAGNÓSTICO PRÉ-NATAL
O diagnóstico pré-natal é sugerindo em ultrassonografia obstétrica de rotina
(corte das “quatro câmaras”). Notadamente ecocardiografia fetal, em casos de
suspeito ou paciente do grupo de alto risco. (YOO, 2008)
Os achados ultrassonográficos mais importantes são:
•
Aumento da área cardíaca;
•
Átrio direito aumentado;
•
Ventrículo direito pequeno;
•
Artéria Pulmonar pequena;
•
Taquicardia supraventricular (associada)
Por se tratar de patologia congênita rara e clínica variável, não se tem na
literatura
uma
avaliação
bem
estabelecida
da
sensibilidade
do
exame
22
ultrassonográfico, uma vez que muitos casos leves sem sintomas podem passar
desapercebidos na infância, e são apenas são diagnosticados na fase adulta.
A ressonância nuclear magnética (axial), apesar de limitada, pode ser útil para
estudo mais detalhado das anomalias cardíacas e extracardíacas associadas. (Choil,
1994).
2.1.8 DIAGNÓSTICO DIFERENCIAL
Doenças cardíacas que provocam regurgitação da válvula tricúspide e
aumento das câmaras cardíacas direitas: displasia da válvula tricúspide (sem
descolamento apical), prolapso da válvula tricúspide, mudanças traumáticas,
cardiomiopatia arritmicogênica do ventrículo direito e endocardite.
2.1.9 CONDUTA PRÉ-NATAL
Na presença de AE, está indicada a realização de ultrassonografia detalhada
da morfologia fetal em busca de anomalias associadas. Apesar de baixa associação
com anomalias cromossômicas, também está indicado o estudo do cariótipo fetal.
O diagnóstico pré-natal e o tratamento desses pacientes, assim como a
interrupção da gestação está indicada nos casos em que há insuficiência cardíaca
23
grave e intratável (presença de sinais com hidropisia, caracterizada por ascite,
derrame pleural e pericárdico, edema de pele e de couro cabeludo).
No que se refere ao tratamento medicamentoso durante a vida fetal, são
utilizados os digitálicos e os diuréticos, por via materna, no sentido de diminuir os
efeitos da congestão sistêmica.
Quando
há
taquiarritmias
associadas
(flutter
atrial,
taquicardia
supraventricular), digitálicos e outros antiarrítmicos (amiodarona e o sotalol) devem
ser utilizados por via transplacentária (levar em conta os riscos já que este feto já
apresenta uma reserva funcional muito diminuída).
Em casos de maturidade extrema, e insuficiência cardíaca grave, há
possibilidade de intervenção intra-uterina, com a finalidade de tentar a dilatação da
valva pulmonar com cateter balão, mas o procedimento ainda é experimental e os
resultados não são animadores.
2.1.10
CONDUTA OBSTÉTRICA
A conduta obstétrica é realizada através de seguimento com exames seriados
de ultrassonografia para avaliação da vitalidade fetal e função cardíaca.
Se o
estágio da gestação permitir, fazer a utilização de corticosteróides para acelerar a
maturidade pulmonar fetal.
A via de parto por cesárea deve ser praticada, para a tentativa / programação
de terapêutica cirúrgica pós-natal.
24
2.1.11
ASSISTÊNCIA NEONATAL
Todos os pacientes com diagnóstico de AE devem se consultar rotineiramente
com um cardiologista. A profilaxia para endocardite deve ser realizada, apesar do
baixo risco desta ocorrer. Pacientes com AE leve, cardiomegalia discreta e na
ausência de arritmias podem ter uma vida normal e praticar qualquer tipo de
atividade física, e apenas acompanhamento com cardiologista é realizado.
Os pacientes com anomalia de Ebstein, que não são candidatos à cirurgia e
apresentam insuficiência cardíaca devem ser tratados com diuréticos e digoxina. A
eficácia da angiotensina em pacientes com Anomalia de Ebstein com insuficiência
cardíaca direita ainda não está bem estabelecida. O tratamento das arritmias deve
ser individualizado e combinado com o procedimento cirúrgico ou com ablação por
cateterismo.
Em crianças que sobreviveram à infância, em geral ficam por vários anos, e a
cirurgia pode ser adiada até que os sintomas apareçam: cianose se torna evidente,
ou embolia paradoxal ocorrer.
Tratamento cirúrgico deve ser cogitado se aparecerem evidências de
deterioração da função cardíaca, como o aumento progressivo do coração direito,
redução de função sistólica ventricular ou o aparecimento de arritmias.
25
No entanto, se os sintomas foram progressivos até a classe funcional III ou IV
da NYHA, o tratamento conservador tem pouco a oferecer, os riscos cirúrgicos
aumentam, e a operação é claramente indicada.
A taxa de mortalidade de pacientes com esta doença na forma grave, no
período neonatal, é alta, chegando a 85%, mesmo em grandes centros,
principalmente quando há sintomatologia precoce e necessidade de cirurgia. (Leite e
cols, 2004)
O tratamento, quando necessário, é cirúrgico: valvuloplastia com ou sem
plicatura átrio-ventricular (reconstrução biventricular é possível na maioria dos
pacientes) ou substituição da válvula Tricúspide por prótese biológica ou metálica,
Correção das anomalias associadas, ablação de feixes anômalos de condução se
presentes (S. Wolff-Parkinson-White) e implante de marca-passo átrio-ventricular se
necessário. O transplante cardíaco é reservado nos casos em que há severa
disfunção biventricular.
2.1.12
PROGNÓSTICO
Depende do grau de comprometimento e malformações associadas.
Geralmente a variedade pré-natal é mais severa que aquela descoberta em crianças
e adultos. É uma das malformações de pior prognóstico durante a vida fetal (taxa de
mortalidade de pacientes com esta doença na forma grave é de 85% no período
neonatal).
26
A evolução natural depende da gravidade da doença, já que o espectro de
anormalidades é variado. Nos casos mais leves os pacientes são assintomáticos,
enquanto que nos mais severos ocorre insuficiência cardíaca e cianose nas
primeiras semanas, com uma tendência de melhora à medida que diminui a
resistência vascular pulmonar. Com o passar do tempo acontece um retorno à
condição anterior, com dispnéia aos esforços e cianose secundária a baixo débito
e/ou shunt atrial direita-esquerda (Zielinsky, Pilla, 2000).
Embora menos de 5% dos pacientes vivam além dos 50 anos de idade, 60%
dos que sobrevivem infância e a idade adulta, conseguem viver confortavelmente em
NYHA classe I ou II. Alguns pacientes chegam aos 80 anos, e raramente, à nona
década.
Apresentação da EA Neonatal da valva tricúspide é uma rara condição
cardíaca de mau prognóstico. A maioria dos estudos de identificação de fatores de
risco a longo prazo envolvem populações mistas. Observa-se que os fetos e recémnascidos sintomáticos têm uma morbimortalidade significativamente maior do que as
crianças mais velhas (Kapusta, 2007).
Neonatos que necessitem de tratamento cirúrgico antes de 30 dias de vida
têm maior taxa de mortalidade do que os fetos submetidos a cirurgia com mais de 30
dias de vida (Yoo, 2008).
Segundo Kapusta et al., a Idade do diagnóstico menor que 12 meses foi
significativamente associada à morte, ou seja, o risco estimado de mortalidade a
curto prazo entre os pacientes diagnosticados antes da idade de um ano foi de 7,9
vezes maior do que o risco no grupo de pacientes diagnosticados após 1 anos de
27
idade.
O sexo masculino, idade gestacional e peso ao nascer não foram
significativamente
associado com o resultado deste estudo
população. A
hepatomegalia no momento do diagnóstico da EA foi significativamente associada à
morte (OR 3,4, IC 95%). Neste estudo, não houve nenhum óbito registrado após 49
meses de idade.
Os fatores associados com morte já foram
avaliados no momento do
diagnóstico. Idade de apresentação (12 meses), hepatomegalia, necessidade de
ventilação mecânica e medicamentos e anormalidades cardíacas associadas foram
fortemente associados à mortalidade precoce na infância.
28
2.2 TRUNCUS ARTERIOSUS COMMUNIS
2.2.1 DEFINIÇÃO
O truncus arteriosus communis (TAC), também conhecido como tronco
arterial único ou tronco arterioso, caracteriza-se pela presença de um único tronco
arterial que emerge da base dos ventrículos através de uma única válvula arterial
para dar origem às artérias sistêmicas, coronarianas e uma ou ambas as artérias
pulmonares.
Caracteriza-se por um vaso arterial único que se origina do coração,
cavalgando o septo ventricular e supre a cirulação sistémica, pulmonar e
coronariana. Predominantemente conecta-se à :
•
Ventrículo direito 40%
•
Ventrículo esquerdo 20%
•
Igualmente dividido 40%
Basicamente
depende
da
situação
da
circulação
freqüentemente encontrados:
a) Hipodesenvolvimento estato-ponderal;
b) Infecções respiratórias de repetição;
c) Dispnéia;
d) Cianose em graus variados;
pulmonar
e
são
29
e) Insuficiência cardíaca congestiva é a causa de morte mais freqüente.
2.2.2 INCIDÊNCIA
O TAC é uma malformação congênita pouco freqüente, com uma prevalência
de 0,04-0,09 casos por 1000 nascimentos (Costa e cols, 2005), ou seja,
aproximadamente 1 : 10.000 nascimentos.
2.2.3 ETIOPATOGENIA
O truncus arteriosus communis (TAC), também conhecido como tronco
arterial único ou tronco arterioso, é o resultado de uma malformação dos esporões
embriológicos precursores da artéria pulmonar (agenesia ou atresia do seu cone e
tronco). Já a atresia da aorta não está associada ao TAC, pois a artéria ascendente
é patente no nível da válvula aórtica e origina as artérias coronarianas.
O tronco valvular freqüentemente é estenótico ou regurgitante. Uma única
valva semilunar cavalga o septo ventricular defeituoso. Há uma continuidade direta
entre uma ou duas aterias pulmonares e o tronco arterial único.
Para que o feto nasça vivo é necessária à existência de dessa comunicação
interventricular (CIV). As artérias pulmonares direita e esquerda ou o tronco não se
desenvolveram; ou o fizeram de modo anômalo em que o sangue atinge os
30
pulmões: ou saindo diretamente da aorta, ou apenas pelas artérias brônquicas como
se pode ver no tipo IV; e neste caso a hematose é insuficiente e a cianose é intensa.
2.2.4 CLASSIFICAÇÃO
A classificação mais utilizada é a de Collet e Edwards (1949):
•
Tipo I Aorta descendente e um curto tronco pulmonar nascendo do
tronco arterioso comum
•
Tipo II As artérias pulmonares (direita e esquerda) nascem próximas
uma da outra ou de um orifício comum na parede dorsal do tronco
comum.
•
Tipo III as artérias pulmonares nascem de modo independente nas
paredes laterais do tronco
•
Tipo IV Não existem artérias pulmonares, a circulação dos pulmões
é feita pelas artérias brônquicas que nascem no inicio da aorta
descendente.
Para Praahg (1970), essa classificação está incompleta, pois para ele, a
classificação acima descrita considera que TAC sempre terá uma CIV. Sua
classificação deveria ser realizada da seguinte forma:
•
Tipo A: quando há presença de CIV, e a partir daí, classificaríamos os
subtipos de TAC associada a CIV semelhante a classificação Collet e
e Edwards (1949) com algumas alterações;
31
•
Tipo B: não há presença de CIV (raríssima).
Ainda, segundo a ele, o tipo IV corresponde a atresia pulmonar associada a
CIV, e não é mais classificada com um tipo de TAC.
2.2.5 ANOMALIAS ASSOCIADAS
A principal malformação associada ao TAC, na quase totalidade dos casos, é
um defeito septal ventricular grande imediatamente abaixo da válvula arterial
comum. (Yoo, 2008). Malformações do arco aórtico associadas também são
encontradas. (Volpe, 2003)
Há relatos na literatura que o TAC esteja associado com síndrome de
DiGeorge e alguns teratogênos: bisdiamine, tedral. (Praahg, 1970).
Em estudo com 23 casos de TAC, Volpe (2003) demonstrou haver uma
associação consistente entre TAC e anomalias cromossômicas (8.7%), sendo a
trissomia do 18 a mais encontrada. Existe uma chance ainda maior de associação
com microdeleção do 22q11 (31.6%). No mesmo estudo, observou-se uma
associação freqüente com malformações extracardíacas: do sistema nervoso central
(ventriculomegalia,
holoprosencefalia),
do
sistema
urinário
(displasia
renal,
hidronefrose), do sistema digestivo (atresia de duodeno e de esôfago) entre outras.
Houve ainda, alguns casos de associação com restrição de crescimento. Outro
achado encontrado é a oligodrâmnia.
32
Alguns estudos descreveram famílias com histórico de TAC e taxa de
recorrência de 6,6%. (Lammer, 2009).
2.2.6 DIAGNÓSTICO PRÉ-NATAL
A ecocardiografia fetal é o exame de excelência para a avaliação do tipo de
truncus e outras alterações cardíacas associadas, apresentando boa acurácia
(Tometzki et al, 1999)
Para realizar o diagnóstico de TAC, deve ser visualizada a origem das artérias
pulmonares na aorta ascendente proximal, achado específico dessa patologia
cardíaca. (Tometzki et al, 1999)
Ainda podem ser visualizadas outras alterações:
no corte de três vasos, somente são identificados dois vasos (achado
encontrado na tetralogia de Fallot e na atresia pulmonar);
no corte da via de saída do ventrículo esquerdo, são identificados o defeito
do septo ventricular e a origem do tronco arterial comum (achados similares também
encotrados na na tetralogia de Fallot e na atresia pulmonar associado a CIV).
Se a avaliação cardíaca for realizada apenas com o corte de “quatro
cãmaras”, a TAC possivelmente não será diagnosticada.
33
2.2.7 DIAGNÓSTICO DIFERENCIAL
Um dos principais diagnósticos diferenciais a ser excluído é a tetralogia de
Fallot (TF), devido um achado ultrassonográfico muito similar encontrado no TAC
através do corte da via de saída do ventrículo esquerdo: quando são identificados o
defeito do septo ventricular e a origem do tronco arterial comum.
Outro
diagnóstico
diferencial
importante
é
quando
ocorrem,
concomitantemente, atresia pulmonar e um CIV.
O diagnóstico diferencial do TAC e essas patologias acima citadas poderá se
apresentar
com
maior
dificuldade
devido
a
similaridade
nos
achados
ultrassonográficos. (Volpe, 2003)
Segundo Donelly (1996), a ressonância nuclear magnética axial pode ser útil
para afastar diagnósticos diferenciais, apesar de seu estar limitado.
2.2.8 CONDUTA PRÉ-NATAL
A conduta pré-natal deve ser realizada através de ecocardiogramas seriados
a cada 2 semanas, além de procedimento para estudo do cariótipo fetal. Também é
indicada a pesquisa da microdeleção do cromossomo 22, devido a associação de
TAC com síndrome de DiGeorge. (Costa e cols,. 2005; Volpe e cols, 2003).
34
2.2.9 CONDUTA OBSTÉTRICA
A conduta obstétrica não se altera até o termo, há indicação de interrupção da
gestação se houver alguma intercorrência.
Costa e cols. (2005) descreveu um caso aonde a gravidez decorreu sem
intercorrências até 36 semanas, quando foi realizada uma cesariana eletiva por
oligoâmnio, e o feto nasceu em boas condições, sendo agendada cirurgia corretiva
com um mês de vida.
2.2.10
ASSISTÊNCIA NEONATAL E TRATAMENTO
Não há tratamento pré-natal.
A cirurgia primária para correção do TAC é amplamente aceitável e,
normalmente, apresenta bons resultados. (Brizard, 1997).
As controversas permanecem com relação a melhor época para a cirurgia
corretiva, os meios de reconstrução da via de saída do fluxo do ventrículo direito, e o
manejamento de anomalias coexistentes.
A correção se faz através da criação de um sistema de condução entre o VD
e a artéria pulmonar (translocação anterior associada a homoenxerto). Sempre é
preconizado o fechamento do septo ventricular.
35
Em um estudo com 82 casos, Brizard et al (1997) defendeu que melhor época
para a cirurgia corretiva eletiva seria entre dois a três meses de vida, apresentando
menor risco cirúrgico em relação às cirurgias realizadas com um mês de vida. Ele
também defende a substituição da válvula truncal (homoenxerto) ou invés da
reparação da mesma, como procedimento padrão ouro; apesar do risco (baixo) de
reoperação.
A correção cirúrgica é realizada no momento em que se observam sinais de
insuficiência cardíaca congestiva, e portanto, a cirurgia pode ser necessária já nos
primeiros dias de vida. Na maioria dos casos, a reparação pode ser adiada até que
a criança é de 2 a 4 meses de idade.
O homoenxerto criopreservado é geralmente preferido para o reparo inicial
devido à sua facilidade de implantação, a liberdade de linha de sutura, e baixa
incidência de estenose valvular e hemorragia (Mohamed e Ott, 1993).
A
translocação anterior da confluência pulmonar é facilmente realizada, como mostra a
experiência com a operação de Jatene para transposição. À separação da artéria
pulmonar a partir da raiz tronco é realizada com maior precisão por divisão completa
da raiz truncal.
2.2.11
PROGNÓSTICO
Os pacientes portadores TAC apresentam taxa de sobrevida relativamente
baixa: 34.8%. (Volpe, 2003). Somente a metade dos pacientes com TAC sobrevive
até um mês de vida. A morte na infância é invariavelmente devido a insuficiência
36
cardíaca congestiva. Os pouco sobreviventes a longo prazo que não se submeteram
a cirurgia reparadora, geralmente, morram devido a complicações por doença
vasculo-pulmonar
grave.
Normalmente
não
está
associado
a
alterações
hemodinâmicas, mas pode ser uma emergência neonatal (evolução para ICC em 2
semanas).
A correção cirúrgica geralmente é necessária antes de 6 meses de vida, e
com sobrevivência de 90% dos casos.
37
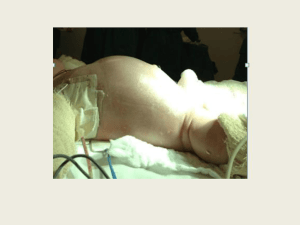
2.3 ECTOPIA CORDIS
2.3.1 DEFINIÇÃO
A ectopia cordis (EC) se caracteriza pela presença do coração localizado fora
de sua topografia habitual, geralmente, expondo-o, total ou parcialmente fora do
tórax. Esse defeito pode variar de diástase dos músculos reto-abdominal com um
coração palpável através da pele hiperpigmentada a uma larga onfalocele coberta
por uma fina membrana do intestino e que contém, fígado e ápice cardíaco.
2.3.2 INCIDÊNCIA
É uma malformação muito rara, a EC apresenta uma indicência de 5,5 – 7,9
casos por milhão de nascidos vivos. Representa cerca e 0,1% de todas as
cardiopatias congênitas.
2.3.3 ETIOPATOGENIA
A ectopia cardíaca é produzida por defeitos segmentares na desenvolvimento
mesodermal na terceira semana de vida intra-uterina, e anomalias de banda
amniótica, que produzem simultânea malformações cerebrais e toracoabdominal.
Existem algumas teorias etiológicas: (Lacanal, 2004):
38
•
Falha primária da descida e fusão da linha media;
•
Rotura precoce do cório e/ou do saco amniótico com falha de fusão;
•
Síndrome da banda amniótica.
2.3.4 CLASSIFICAÇÃO
Podemos classificar a EC em 4 tipos:
•
Tipo torácico;
•
Tipo abdominal;
•
Tipo tóraco-abdominal (mais comum e de melhor prognóstico se for da
forma parcial);
•
Tipo cervical (incompatível com a vida).
tipo cervical
3%
38%
tipo torácico
tipo toraco-abdominal
1%
tipo abdominal
58%
Grafico 1 - Freqüência dos tipos de ectopia cordis (Cabrera et al. 2002)
39
Na forma torácica completa, o esterno está ausente totalmente ou
parcialmente, não há pericárdio, e o coração se encontra totalmente fora da
cavidade torácica. Na forma incompleta, o coração está coberto pela pele, pericárdio
ou ambos.
No tipo toraco-abdominal, o defeito é inferior (ausência da metade ou dos dois
terços inferiores do esterno).
Na
forma
cervical,
há
associação
com
múltiplas
malformações
e
incompatibilidade com a vida.
A forma abdominal é muito rara (ver gráfico 1).
2.3.5 ANOMALIAS ASSOCIADAS
Cerca de 95% dos casos de EC estão associadas a outras anomalias, e em
80% dos casos há presença de uma malformação cardíaca.
As anomalias mais comumente associadas com EC são: onfalocele,
malformações cardíacas (CIA, CIV, tetralogia de Fallot) e anomalias crânio-facial e
esqueléticas (coluna e membros). Também pode estar associada com trissomia 13 e
18 ou Síndrome de Turner.
A pentalogia de Cantrell corresponde à forma de EC associada a onfalocele,
defeito da porção inferior do externo, defeito diafragmático e defeito pericárdio.
40
2.3.6 DIAGNÓSTICO PRÉ-NATAL
O diagnótico é estebelecido, através de exame ultrassonográfico (endovaginal
com 10 – 12 semanas), pela localização ectópica do coração com herniação através
de um defeito na parede anterior. O coração pode estar completamente fora do tórax
ou apenas parcialmente herniado pelo defeito esternal.
É possível identificar a patologia precocemente, com 10 semanas de
gestação (por exame via vaginal). O diagnóstico pode ser difícil, principalmente nas
formas incompletas e associadas a derrame pericárdico ou pleural, que, é
comumente presente.
Devido a evolução dos aparelhos de ultrassonografia 4D, esses tipo de
exame tem ganhado cada vez mais importância no diagnóstico dessa patologia.
2.3.7 DIAGNÓSTICO DIFERENCIAL
O diagnóstico diferencial deve ser feito, principalmente, com as síndromes de
body-stalk e da banda amniótica.
41
2.3.8 CONDUTA PRÉ-NATAL E OBSTÉTRICA
Uma vez estabelecido o diagnóstico de EC, deve-se proceder com exame
ultrassonográfico morfológico e ecocardiografia fetal (20-22 semanas de gestação)
em busca de malformações associadas. O estudo do cariótipo fetal está indicado,
apesar de pouca associação dessa patologia com anormalidades cromossômicas.
O acompanhamento deve ser feito com ultrassonografias seriadas para
monitorizar o bem estar fetal e detectar presença de sinais de insuficiência cardíaca.
A via de parto é indicação obstétrica, porém em casos mais severos de EC
completa, a interrupção por parto cesariana é madatória devido o risco de ruptura
visceral em partos vaginais (em fetos viáveis).
Uma vez detectados sinais de deteriorização da função cardíaca fetal, está
indicada a interrupção da gestação.
2.3.9 ASSISTÊNCIA NEONATAL E TRATAMENTO
A única opção terapêutica é a cirúrgica corretiva pós-natal imediata. O
tratamento tem evoluído, melhorando a sobrevida desses pacientes: melhor manejo
das malformações associadas e colocação de material sintético de cobertura
(gorotex). A vantagem do enxerto autólogo sobre a prótese é devido ao fato de que
o enxerto pode crescer com o paciente e ter maior resistência à infecções.
42
Há
necessidade
de
tratar
as
malformações
associadas:
conduta
individualizada e no tempo adequado. Porém o sucesso cirúrgico ainda é pequeno,
sendo tipo de melhor prognóstico é a forma toraco-abdominal parcial.
Várias técnicas cirúrgicas têm sido descritas, podendo a doença pode ser
abordada por um ou dois estágios cirúrgicos. O primeiro estágio cirúrgico é realizado
em pacientes que necessitam de imediato tratamento e visa obter pele e tecido mole
para recobrir o coração. No entanto, nem sempre é possível, e devido à baixo débito
cardíaco o uso de prótese é necessária. O objetivo da segunda etapa é a correção
dos defeitos cardíacos congênitos associados e reconstrução do esterno.
2.3.10
PROGNÓSTICO
A presença de membrana cobrindo o coração (pericárdio, pele ou ambos)
determina o seu tratamento e prognóstico. A ausência de pericárdio e maior grau de
exposição (fenda esternal maior) causam maior contato com o líquido amniótico,
favorecendo a inflamação e o engrossamento do epicárdio, e por sua vez, pode
comprometer a função cardíaca (pior prognóstico).
Apesar das modernas opções de tratamento, o prognóstico é reservado (90%
dos recém-nascidos morrem no primeiro ano de vida), principalmente devido às
freqüentes associações com malformações cardíacas e extracardíacas (em
particular, onfalocele, hérnia diafragmática, hipoplasia pulmonar).
43
As complicações incluem: ruptura visceral durante o parto, sepse,
insuficiência cardíaca (rara), insuficiência respiratória, compressão cardíaca. O
resultado da maioria dos casos, cujo diagnóstico foi obtido no pré-natal, foi fatal após
o nascimento.
44
2.4 TUMORES CARDÍACOS
2.4.1 DEFINIÇÃO
Os tumores cardíacos primários são achados raros em pacientes de todas as
idades, sendo benignos em sua grande maioria.
2.4.2 INCIDÊNCIA
A prevalência dos tumores cardíacos primários, considerado todas as idades,
é aproximadamente de 0,01%. No feto a prevalência destes tumores é de
aproximadamente 0,14%. Cerca de 90% dos casos são benignos, e o tipo mais
comum é o rabdomioma (60%).
2.4.3 ETIOPATOGENIA
Os rabdomiomas consistem em hamartomas
proliferativos (termo
usado para designar uma proliferação celular de tecido que pertence ao
mesmo órgão de onde ele se origina).
45
2.4.4 CLASSIFICAÇÃO (TIPOS)
Os rabdomiomas consistem em hamartomas proliferativos, associados com
esclerose tuberosa em cerca de 50 a 80% dos casos. São os tumores cardíacos
primários mais freqüentes (incidência de 50 a 90%). Eles são de natureza múltipla
(característica
suficiente
para
estabelecer
seu
diagnóstico).
Suas
outras
características estão descritas a seguir (ver Tabela 2).
Os rabdomiomas possuem a tendência de aparecer no inicio do segundo
trimestre e aumentam de tamanha até o início do terceiro trimestre, e então,
regridem durante o final da gestação e nos primeiros dias de vida. Se os fetos
portadores não forem acometidos de esclerose tuberosa, apresentam bom
prognóstico na maioria das vezes.
O fibroma é o segundo tipo de tumor cardíaco mais freqüente. São massas
solitárias e heterogêneas (áreas de degeneração cística). Ocasionalmente, há
associação com carcinoma basocelular nevoide, fenda labial, fenda palatina e
síndrome de Beckwith-wiedemann (macroglossia, visceromegalia, macrossomia,
hérnia umbilical e onfalocele). A regressão espontânea do fibroma é muito rara.
O teratoma é um tumor solitário raro, heterogêneo e muito associado com
derrame pericárdico com debris que pode ser massivo e levar a tampponamento
cardíaco grave, necessitando de tratamento intra-útero (pericardiocentese ou cirurgia
fetal)
De todos os tumores cardíacos, o hemangioma é o de melhor prognóstico,
devido a sua tendência de regressão, porém pode estar acompanhado de derrame
46
pericárdico e necessitar de tratamento intra-útero. Pode estar associado a outros
hemangiomas (principalmente pele).
2.4.5 ANOMALIAS ASSOCIADAS
Os rabdomiomas apresentam associação à esclerose tuberosa em 50 a 80%
dos casos, que geralmente é diagnosticada após o nascimento. Eles raramente
estão associados a outras malformações cardíacas, mas há casos descritos de
associação com malformações cardíacas complexas (anomalia de Ebstein, tetralogia
de Fallot, dupla via de saída do ventrículo direito, transposição das grandes artérias e
síndrome de hipoplasia do coração esquerdo).
A esclerose tuberosa, doença genética autossômica dominante, que provoca
a formação de hamartomas (tumores formados por células iguais as do tecido de
origem) em diversos órgãos e sistemas, principalmente no coração, no sistema
nervoso central, e nos rins. A presença dos rabdomiomas, no feto, pode ser o sinal
mais precoce da esclerose tuberosa (cerca de 90% dos casos com esclerose
tuberosa apresentam alterações do sistema nervoso central, com manifestação
clínica de crises convulsivas e retardo mental após o nascimento).
“A presença de nódulos subependimários e/ou cistos renais é considerado sinal
patognomônico de esclerose tuberosa, ocorrendo em mais de 90% dos casos,
porém estas alterações são raramente detectadas no período pré-natal” (Carvalho,
2010). Esses achados são encontrados com mais facilidade na ressonância nuclear
magnética.
47
2.4.6 DIAGNÓSTICO PRÉ-NATAL
O diagnóstico é facialmente feito através de exame ultrassonografico pela
observação de massa no corte de “quatro câmaras”.
O primeiro diagnóstico de tumor cardíaco diagnosticado por este meio,
ocorreu em 1982.
A ressonância nuclear magnética também pode ajudar no
diagnóstico.
O diagnóstico diferencial entre os tipos de tumores cardíacos primários pode ser
realizado através da ecocardiografia fetal, a qual nos permite caracterizar a morfologia
da massa de forma não-invasiva. (Carvalho, 2010).
A tabela 2 informa as características ultrassonográficas de cada tipo de tumor
cardíaco:
Tabela 2 - Caracaterísticas ultrassonográficas dos tumores cardíacos (modificada de Carvalho, 2010)
Localização
e
Tamanho
Ecogenecidade e
Tipo
número
de
Contornos
do derrame
textura
massas
pericárdico
Única ou múltiplas.
Hiperecogênica ou
Rabdomioma
Variável,
Regular
Ventrículo
homogênea
Mista
raro
ou
Freqüente e
Regular
Teratomas
Única. Pericárdio
heterogênea (cistos
geralmente
encapsulado
e calcificações)
Única.
Septo
Mista
grande
ou
Grande
Fibromas
interventricular
ou
heterogênea
Regular
geral
parede livre
(cistos,
em
48
calcificações
e
necrose central)
Única.
Base
do
Mista
Hemangioma
coração
ou
Variável
ou
átrio
e
Irregular
heterogênea
freqüente
direito.
Única.
Átrio
esquerdo
com
Mista
Mixomas
ou
Irregular
variável
heterogênea
pedículo
Os rabdomiomas (Figura 2) podem ser massas únicas ou múltiplas,
hipercogênicas, textura homogênea, tamanho variável e bordos bem delimitados e
regulares. É mais encontrado no miocárdio ventricular (esquerdo e/ou direito), e
comumente se expandia para o interior da cavidade; pode ainda se localizar no
septo interventricular ou na parede atrial (raro).
Figura 2 - Rabdomioma ocupando quase totalmente o ventrículo esquerdo
49
As características ecográficas do teratoma são: geralmente solitários de
grande volume (pode atingir até 15cm), encapsulado (bordas regulares), localizados
comumente no pericárdio, associado a derrame pericárdico.
Os fibromas se apresentam, assim como os teratomas, como massas
solitárias, únicas, geralmente de grande volume. Sua ecogenicidade
é mista
(calcificações, degenerações císticas e necrose na parte central do tumor) e textura
heterogênea.
Os hemangiomas e os mixomas são extremamente raros no feto. Os
hemangiomas são geralmente massas únicas, apresentando ecogenicidade mista
(contendo partes císticas e sólidas, associadas à calcificação) e localização mais
comum na base do coração e átrio direito. Os mixomas geralmente são pediculados
e se localizam no átrio esquerdo.
Além do exame ultrassonográfico, a avaliação pode ser complementada por
ressonância nuclear magnética para uma melhor definição diagnóstica.
2.4.7 DIAGNÓSTICO DIFERENCIAL
Primeiramente, há necessidade de se excluir os casos de foco hipercogênico
intracardíaco isolado (golf ball).
O diagnóstico diferencial basicamente é feito entre os tipos de tumores
cardíacos, que é estabelecido de acordo com as características ecográficas das
massas.
50
2.4.8 CONDUTA PRÉ-NATAL E OBSTÉTRICA
Diagnosticado
o
tumor
cardíaco,
está
indicada
a
realização
de
ultrassonografia detalhada para avaliação da morfologia fetal em busca de
anomalias associadas; e ainda a realização de exame de ecocardiografia fetal para
avaliar a existência de anomalias cardíacas associadas, bem com a função cadíaca.
Devido à baixa associação com anomalias cromossômicas, não está indicado o
estudo do cariótipo fetal.
Na vigência de sinais de insuficiência cardíaca, está indicado administração
de corticóides para maturação pulmonar se possível e caso não seja possível a
estabilização da função cardíaca com tratamento medicamentoso, a interrupção da
gestação por cesariana está indicada.
2.4.9 ASSISTÊNCIA NEONATAL E TRATAMENTO
A conduta neonatal é expectante devido ao crescimento lento do tumor e à
possibilidade de regressão espontânea. Na presença de rabdomiomas de grande
volume, há maior possibilidade do surgimento de complicações e a conduta deve ser
individualizada, levando-se em consideração os riscos de uma terapêutica invasiva.
O seguimento clínico pós-natal é obrigatório devido à freqüente associação à
esclerose tuberosa.
51
2.4.10
PROGNÓSTICO
Os principais fatores que determinam o prognóstico fetal são a localização e o
tamanho do tumor.
Os rabdomiomas geralmente têm boa evolução no período pré-natal, devido
ao crescimento lento dos tumores, havendo inclusive possibilidade de regressão
antes do nascimento. Porém se estiver associado à esclerose tuberosa, o
prognóstico se altera.
Em alguns casos, pode haver crescimento rápido do rabdomioma, levando a
surgimento de complicações (obstrução das vias de entrada / saída dos ventrículos,
arritmia cardíaca e, mais raramente, embolização de partes do tumor).
Os fatores de prognóstico ruim são os seguintes:
•
Obstrução do fluxo intracardíaco;
•
Função alterada da válvula com regurgitação;
•
Presença de arritmias (geralmente de difícil reversão);
•
Derrame pericárdico volumoso;
•
Hidropisia fetal.
52
3 CONCLUSÃO
Devido a evolução dos exames diagnósticos, notadamente o exame
ultrassonográfico, o diagnóstico da maioria das malformações cardíacas fetais se é
tornou possível de ser realizado sem maiores dificuldades. Pois isso, é fundamental
que o conhecimento básico do coração normal e patológico esteja ao alcance dos
ultrassonografistas obstétricos.
Na suspeita de malformação cardíaca, a gestante deve ser encaminha para
realização
de
ecocardiografia
fetal.
Com
o
diagnóstico
ultrassonográfico
estabelecido, pode-se traçar uma estratégia de seguimento pré-natal e programação
da interrupção em momento oportuno em local adequado e preparado para as
necessidades que a patalogia cardíaca possa necessitar; melhorando os resultados
de sobrevida nesse grupo de pacientes.
Entre as patologias descritas nesse trabalho, tanto a AE com o TAC são
anomalias congênitas complexas com um espectro anatômico e clínico muito
variados. Cada caso é complexo e deve ser individualizado.
O conhecimento preciso da patologia preciso é necessário sobre os diferentes
variáveis anatômicas e hemodinâmicas; e malformações associadas. Espera-se que
a sobrevida dos pacientes com essas anomalias de todas as idades continuem a
melhorar.
Os paciente portadores de EC geralmente apresentam malformação cardíaca
grave, e geralmente associadas a anomalias troncoconais. Apesar das tentativas de
53
tratamento cirúrgico, poucos doentes com estas malformações cardíacas sobreviver
e a maioria deles morrem na primeiro ano de vida.
Os tumores cardíacos são bastante raros, de fácil diagnóstico desde que
considerados, na grande maioria benignos e que cursam favoravelmente com a
extirpação cirúrgica.
Em algumas doenças cardíacas do feto é desaconselhado que o parto seja
por via vaginal. O ideal é realizar um parto cesáreo programado e, dependendo do
tipo de anomalia, que este seja realizado no próprio Hospital de Cardiologia provido
de todos os recursos médicos, cirúrgicos e de cateterismo terapêutico para o
tratamento imediato do bebê.
A grande vantagem de se ter o diagnóstico de cardiopatias congênitas ainda
na vida intra-uterina é poder programar o parto e o atendimento neonatal imediato
prevenindo que a situação clínica do bebê se torne muito grave ou até irreversível.
54
4 REFERÊNCIAS
AGUADOA, Gutiérrez-Larraya; IZIQUIERDOB, Alberto Galindo; LLODIOB, José
Ignacio Olaizola et al. Tumores cardíacos fetales. Rev Esp Cardiol. 1997;50:18791.
ARNAIZ, Pilar; TOLEDO, Isabel; BORZUTZKY, Arturo. Comportamiento clínico de
los tumores cardíacos desde el feto hasta el adulto: serie multicéntrica de 38
pacientes. Rev Méd Chile 2006; 134: 1135-1145
BEHL Raj, BLESOVSKY. Ebstein's anomaly: sixteen years' experience with
valve replacement without plication of the right ventricle. Thorax 1984;39:8-13.
BRIZARD Christian P, COCHRANE Andrew, AUSTIN Conal, et al. Management
strategy and long-term outcome for truncus arteriosus. Eur J Cardiothorac Surg
1997;11:687-695.
BRUN M, MAUGEY-LAULOM B, RAUCH-CHABROL F, GRIGNON A, DIARD F.
Prenatal Ultrasound Diagnosis of Anterior Abdominal Wall Defects. J Radio l
998; 79:1461-1468.
CABRERA A, ROGRIDO D, LUIS MT, et al. Anomalías cardíacas en la ectopia
cordis. Rev Esp Cardiol 2002;55:1209-12.
CARPENTIER A, et al. A new reconstructive operation for Ebstein’s anomaly of
the tricuspid valve. J Thorac Cardiovasc Surg. 1988;96: 92–101.
CARVALHO Sandra Regina Marques, MARCOLIN Alessandra Cristina, CAVALLI
Ricardo Carvalho et al. Rabdomiomas cardíacos fetais: análise de cinco casos.
Rev Bras Ginecol Obstet. 2010; 32(4):156-62;
CHOIL Young Hi, el al. MR Imaging of Ebstein’s Anomaly of the Tricuspid Valve.
AJR 1994;163:539-543.
COSTA Patrícia, CARRIÇO Ana, MONTERROSO José, MATIAS Alexandre,
AREIAS José. Truncus Arteriosus, Diagnóstico Pré-Natal. Rev Port Cardiol
2005;24 (1) :135-136
DEARANI,
Joseph;
DANIELSON, Gordon. Congenital Heart Surgery
Nomenclature and Database Project: Ebstein’s anomaly and tricuspid valve
disease. Ann Thorac Surg. 2000;69(suppl):S106 –S117;
DONNELY Lane F, HIGGINS Charles B.
Abnormalities. AJR:166, April 1996
MR Imaging of Conotruncal
PRAAHG Van. Truncus arteriosus: what is it really and how should it be
classified? Eur J Cardio-thorac Surg (1987) 1:65-70;
55
HOFFMAN, Julien; KAPLAN, Samuel. The incidence of congenital heart disease.
J Am Coll Cardiol 39:1890,2002.
HUMPL Tilman, HUGGAN Paul, HORNBERGER Lisa el al. Presentation and
outcomes of ectopia cordis. Can J Cardiol Vol 15, No 12, 1999.
JOST, Christine H. Attenhofer; CONNOLLY Heidi M; et al. Ebstein’s Anomaly.
Circulation 2007;115;277-285.
KAPUSTA Livia et al. Ebstein’s anomaly: factors associated with death in
childhood and adolescence: a multi-centre, long-term study. European Heart
Journal (2007) 28, 2661–2666 doi:10.1093/eurheartj/ehm398.
KIRBY Margaret L. Pulmonary Atresia or Persistent Truncus Arteriosus: Is It
Important to Make the Distinction and How Do We Do It? Circ Res. 2008 August
15; 103(4): 337–339.
LACANAL Escuro Ruiz, HEREDIA Maese, PEIRO Cuenca et al. Ectopia cordis
torácica
no asociada a cardiopatia. An Pediatr (Barc) 2004;60(2):184-93;
LAMMER Edward, CHAK Jacqueline S, IOVANNISCI David M. Chromosomal
abnormalities among children born with conotruncal cardiac defects. Birth
Defects Res A Clin Mol Teratol. 2009 January ; 85(1): 30–35.
LEITE, Maria de Fátima, GIANISELLA, Roberto; ZIELINSKY Paulo; Anomalia de
Ebstein detectada in utero e síndrome de Down. Diagnóstico pré-natal de uma
combinação rara. Arq. Bras. Cardiol. vol.82 no.4 São Paulo Apr. 2004
LEITHISIER, Richard; FYFE, Derek; WEATHERBY III, Ellsworth et al. Prenatal
Sonographic Diagnosis of Atrial Hemangioma. AJR:147, December 1986
MATAMALA Ortigato; GARCIA, Garcia. Anomalía de Ebstein y exposición al litio
en el embarazo. An Pediatr (Barc). 2006;65(6):626-42;
MOHAMED, Gamal; OTT, David. Anterior Translocation of the Pulmonary
Confluence in the Surgical Treatment of Truncus Arteriosus. Texas Heart
Institute Journal 1993;20:285-7
RADFORD, Dorothy; R F GRAFF, G H NEILSONRadford DJ, Graff RF, Neilson GH:
Diagnosis and natural history of Ebstein's anomaly. Br Heart J 1985; 54: 517-22.
TOMETZKI et al.Accuracy of Prenatal Echocardiographic Diagnosis and
Prognosis of Fetuses With Conotruncal Anomalies. JACC Vol. 33, No. 6, 1999
Tometzki et al. 1697
May 1999:1696–701
56
VOLPE P, PALLADINI D, BUONADONNA A L, et al. Common arterial trunk in the
fetus: characteristics, associations, and outcome in a multicentre series of 23
cases. Heart 2003;89:1437–1441
YOO, Shi-joon; JAEGGI Edgar. Avaliação ultrassonográfica do coração fetal. In:
CALLEN: Ultrassonografia em ginecologia e obstetrícia, pg 512. 5ª Edição, 2008.
ZIELINSKY Paulo; PILLA Carlo. Anomalia de Ebstein Com Valva Tricúspide
Imperfurada. Diagnóstico Pré-Natal. Arq Bras Cardiol. volume 75, (nº 1), 2000
ZIELINSKY Paulo. Malformações Cardíacas Fetais. Diagnóstico e Conduta. Arq.
Bras. Cardiol. vol.69 n.3 São Paulo Sept. 1997.