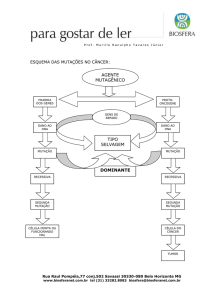
Esclerose Tuberosa
Disciplina de Genética II
Aline Bertoni S. Jorge
Introdução
Esclerose tuberosa - ou complexo esclerose
tuberosa (TSC) - doença genética rara,
multi-sistêmica que causa tumores benignos
no cérebro, rins, coração, olhos, pulmões e
pele
Herança Autossômica Dominante, com alta
penetrância e ampla variabilidade fenotípica
Histórico
1835 e 1850: dermatologistas descreveram a erupção
cutânea facial distintiva
1862: Von Recklinghausen identificou tumores no
coração e no cérebro de um RN
1880: Bourneville é creditado por ter caracterizado a
doença primeiro (doença de Bourneville )
1908: o neurologista Vogt estabeleceu uma tríade
diagnóstica de epilepsia, idiotice, e adenoma sebáceo
(um termo obsoleto para angiofibroma facial)
Histórico
Epônimos do TSC: doença de
Bourneville, síndrome de Bourneville,
doença de Bourneville-Pringle,
adenoma sebáceo de Bourneville,
adenoma sebáceo de Pringle, doença
de Bourneville-Brissaud, síndrome da
hamartomatose sistêmica múltipla,
neuromatose universal,
esponglioblastose circunscrita,
Désiré-Magloire Bourneville
neurosponglioblastose difusa ou
epilóia
Histórico
1979: Gomez descreve a doença como atualmente
entendida
1993: descoberta do envolvimento do loci TSC2
1997: descoberta do envolvimento do loci TSC1
2002: tentativas de tratamento com rapamicina para
encolher tumores associados ao TSC
Epidemiologia
Incidência: 1:6000 a 1:10000
Sem predileção por raça ou gênero
Não se pode prever a gravidade da doença
ou quais órgãos serão atingidos
Etiologia
Herança Autossômica Dominante
Mutações nos genes TSC1 e TSC2
Mutação de novo em 2/3 dos casos
70% das mutações detectadas estão no gene TSC2
e 30% no gene TSC1. Isto se deve possivelmente ao
maior tamanho do gene TSC2, aumentando a
probabilidade de sofrer uma mutação aleatória
Os Genes
TSC1 e TSC2 são ambos genes supressores
de tumor que funcionam de acordo com a
hipótese "dois golpes “
TSC1 codifica para a proteína hamartina
TSC2 codifica para a proteína tuberina
Os Genes
TSC1: Localizado no braço longo do
cromossomo 9 (9q34)
TSC2: Localizado no braço curto do
cromossomo 16 (16p13.3)
Codificam as proteínas hamartina e tuberina,
respectivamente, que atuam como
supressoras tumorais
Os Genes
Gene TSC1
Gene TSC2
Os Genes: mutações
Fisiopatologia
A mutação, nova ou herdada (1º evento mutacional),
não é suficiente para o desenvolvimento do TSC
O segundo evento mutacional ocorre em células
somáticas do alelo que já tinha uma mutação (par de
alelos inativados)
Para cada tumor, é necessário um novo evento
mutacional (somático)
Não é possível controlar quando irá ocorrer o segundo
evento mutacional (é aleatório). Desta maneira, os
pacientes desenvolvem diferentes tumores e têm
apresentações clínicas variáveis
Manifestações Clínicas
Acomete cérebro, coração, rins, pulmões,
pele, olhos
Podem experimentar nenhum (forma frustra)
ou todos os sintomas clínicos
Primeiras manifestações: tumores benignos
cardíacos, máculas hipomelanocíticas,
espasmos infantis, ataques epilépticos
Manifestações Clínicas
Manifestações no SNC
Tuberosidades corticais: malformações originando
nódulos na substância cinzenta; responsável pelas
crises convulsivas, retardo mental e distúrbios de
comportamento que podem ser observados nos
pacientes com TSC
Os distúrbios de comportamento, como irritabilidade,
agressividade, déficit de atenção, movimentos
repetitivos, exclusão social podem se relacionar ao
TSC mesmo se não se identificam as lesões
cerebrais
Manifestações no SNC
Nódulos subependimários: sofrem com
frequência calcificação nos primeiros anos de
vida
Astrocitoma gigante subependimário:
podem crescer causando obstrução no
ventrículo e hidrocefalia. Seu crescimento,
acontece mais comumente até o final da
segunda década de vida
A E, notam-se áreas mais esbranquiçadas no córtex, que correspondem aos túberes,
constituídos por células gliais ou neurônios anômalos. Alguns causam alargamento
dos giros.
A D, cérebro de paciente com esclerose tuberosa cortado em plano axial (horizontal),
mostrando massa tumoral bem delimitada, acinzentada, no foramen de Monro E,
causando hidrocefalia bilateral. Trata-se de um astrocitoma subependimário de células
gigantes.
Manifestações Cardíacas
Rabdomioma: tumor do músculo cardíaco,
ocorre com maior freqüência entre o terceiro
trimestre pré-natal e o primeiro ano de vida,
podendo haver um aumento de freqüência na
puberdade. Normalmente assintomático, mas
pode causar arritmias cardíacas. Tende a
regredir e desaparecer
Tumor raro é um indicador forte de TSC na
criança
Manifestações Renais
Angiomiolipoma: tumor de células
musculares, vasos sanguíneos e adipócitos,
mais freqüente no adulto, podendo afetar
outros órgãos do abdômen
Cistos múltiplos: pequenas dilatações dos
túbulos renais, tendem a se manifestar mais
cedo, na infância. Poucos pacientes
apresentam um grande número de cistos, em
ambos rins, podendo levar à insuficiência renal
Manifestações Pulmonares
Linfangioleiomiomatose (LAM): proliferação
difusa de células musculares, vasos
sanguíneos e linfáticos, acometendo em geral
ambos pulmões de algumas mulheres em
idade reprodutiva. Pode levar à insuficiência
respiratória e óbito
Manifestações em Pele
Angiofibromas faciais: nódulos avermelhados
nas maçãs do rosto, surgindo na puberdade
Espessamento da pele na região frontal do rosto
(testa) ou na região lombar
Fibromas ungueais ou periungueais não
traumáticos
Manifestações em Pele
Máculas hipomelanóticas
Lesões “em confete” hipercrômicas
Marcas de 'peau chagrin' ou 'shagreen patch‘:
placa de fibrose subepidérmica mais
freqüente na região lombo-sacra. Área
levemente elevada, com superfície
comparada a pele de porco ou de elefante, ou
a casca de laranja
As lesões faciais características da esclerose tuberosa são pápulas e nódulos numa
distribuição em asa de borboleta, conhecidas como 'adenoma sebáceo de
Pringle'. São, na realidade, minúsculos angiofibromas. Estão presentes em mais de
90% dos pacientes acima dos 4 anos de idade e podem também envolver o queixo e a
testa.
Manifestações Oftalmológicas
Hamartomas: nódulos na retina
Acromia: Despigmentação na retina
Manifestações Oftalmológicas
Lesões não-retinais associadas com TSC
incluem:
Coloboma
Angiofibromas das pálpebras
Papiledema (relacionada a hidrocefalia)
Prognóstico
Indivíduos com sintomas moderados geralmente
vivem bem e têm vidas produtivas, enquanto os
indivíduos com a forma mais severa podem ter
inaptidões sérias
Causas principais de morte: doença renal, tumor no
cérebro, limfangiomiomatose pulmonar, e estado
epilético ou broncopneumonia em pacientes com
impedimento mental severo
Fracasso cardíaco devido a rabdomiomas é um risco
no feto, mas um problema raramente é
subseqüentemente
Prognóstico
Complicações no rim como angiomiolipoma
(AML) e cistos são comuns, e mais freqüente
em mulheres que homens e em TSC2 que
TSC1
Linfangioleiomiomatose (LAM) é um risco para
mulheres com AML
Os nódulos subependimais ocasionalmente se
degeneram a astrocitoma subependimal de
células gigantes(ASCG). Estes podem
bloquear a circulação de líquido
cefalorraquidiano, conduzindo a hidrocefalia
Diagnóstico
Não há sinais clínicos patognomônicos para
esclerose tuberosa
Em crianças, a primeira pista é
freqüentemente a presença de ataques
apopléticos, desenvolvimento atrasado ou
remendos brancos na pele
Diagnóstico Clínico
Levantamento do histórico familiar
Examinar a pele debaixo do abajur de Wood (máculas
hipomelanóticas), os dedos do pé (fibroma ungual), a
face (angiofibroma) e a boca (covas dentais e fibroma
gengival)
TC de crânio ou, preferivelmente, RMN (tubérculo
cortical e nódulos de subependimal)
USG renal (angiomiolipoma ou cistos)
ECO em crianças (rabdomioma)
Fundoscopia (hamartomas nodular retinal)
Diagnóstico Clínico
Diagnóstico Laboratorial
Permite o aconselhamento genético e, em
alguns indivíduos, diagnóstico pré-natal
15% de falso-negativos e alta taxa de
mosaicismo somático (10% -25%)
O teste genético molecular é complicado pelo
grande tamanho dos dois genes, o que torna o
exame demorado e oneroso
Esses exames não são realizados ainda no
Brasil
Diagnósticos Diferenciais
NEM1,Síndrome de Birt-Hogg-Dube (BHDS)
Neurofibromatose tipo I
Doença Policística Renal
Autismo
Vitiligo, Exostose subungueal, Nevo do tecido
conectivo, Hiperplasia de glândulas sebáceas
Tratamento
É sintomático e dirigido a cada tipo de lesão,
em geral com efeitos benéficos ao controle do
sintoma ou mesmo da função do órgão
Alguns tumores têm indicação cirúrgica
Assistência médica durante toda vida: risco
aumentado de desenvolver os tumores
característicos da doença
Acompanhamento multidisciplinar
Aconselhamento Genético
Herança Autossômica Dominante
Cerca de um terço dos probandos com TSC
têm um pai afetado
Dois terços dos indivíduos têm o gene
alterado TSC1 ou TSC2, como resultado de
uma mutação de novo
Aconselhamento Genético
Aconselhamento Genético
Irmãos de um probando:
O risco para os irmãos do probando depende do
status genético dos pais:
Se um dos pais é afetado ou se a mutação que causa a
doença for identificada na família, o risco para os irmãos
é de 50%.
Se nenhum dos pais tem achados indicativos de TSC ou
se nenhum dos pais tem a mutação detectáveis no DNA
(somático), o risco é de 1% a 2% do risco de recidiva,
devido à possibilidade de mosaicismo germinal.
Aconselhamento Genético
Prole do probando:
Cada criança de um indivíduo com esclerose
tuberosa tem 50% de chance de herdar a mutação.
Outros membros da família do probando:
O risco para os outros membros da família
depende do status genético dos pais dos
probandos. Se um pai é afetado ou tem a mutação
causadores de doenças, os membros da sua
família estão em risco.
Espasmos Infantis
http://www.youtube.com/watch?v=MUXJbg4dI_4
Referências Bibliográficas
http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=g
ene&part=tuberous-sclerosis
http://genoma.ib.usp.br/pesquisas/doencas_escleros
e-tuberosa.php
http://www.medscape.com/viewarticle/495642
http://abet.org.br/jla2/index.php?option=com_content
&view=article&id=22&Itemid=29
http://ghr.nlm.nih.gov/gene/TSC1
http://anatpat.unicamp.br/bineuescletuber.html
http://www.scielo.br/scielo.php?script=sci_arttext&pid
=S0004-282X1998000400026&lang=pt