PROGRAMA DE ASSESSORIA
E TREINAMENTO
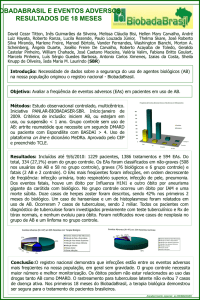
SEGURANÇA DO PACIENTE E EVENTO ADVERSO
1
OBJETIVO DA MONITORIZAÇÃO DOS
EVENTOS ADVERSOS EM PESQUISA CLÍNICA
PRIMEIRA PREOCUPAÇÃO:
Assegurar a segurança do paciente durante sua participação no
estudo clínico!
3 Propósitos Principais:
Proteger os pacientes
Proteger a indústria farmacêutica
Cumprir com as exigências regulatórias
2
OBJETIVO DA MONITORIZAÇÃO DOS
EVENTOS ADVERSOS EM PESQUISA CLÍNICA
A maioria dos estudos clínicos estuda a SEGURANÇA e EFICÁCIA da
medicação investigacional ou seja:
1. A medicação é segura?
2. A medicação funciona?
A partir do momento da assinatura do TCLE, paciente deverá ser
tratado como sujeito de pesquisa e qualquer evento adverso ocorrido
deverá ser relatado pelo investigador
3
OBJETIVO DA MONITORIZAÇÃO DOS
EVENTOS ADVERSOS EM PESQUISA CLÍNICA
A monitorização da segurança é necessária para
assegurar que a avaliação dos eventos, que podem
representar um risco inaceitável ao pacientes, seja
realizada rapidamente
Para que a detecção dos eventos adversos da
medicação investigacional seja realizada em tempo
hábil, utiliza-se um procedimento padrão em
pesquisa clínica
4
DEFINIÇÕES E TERMOS UTILIZADOS
EVENTO ADVERSO (AE):
Um evento adverso (AE) pode ser qualquer ocorrência médica
desfavorável e não desejável (incluindo um achado anormal de
laboratório), sintoma ou doença, sofrida por um paciente ou
sujeito de pesquisa, temporariamente associada ao uso de um
produto em investigação, seja ele relacionado ou não a este
produto.
Referência ICH/GCP
Exemplos:
-Evento Novo ou Piora de sintomas
-Anormalidades Laboratoriais
-Alterações nos achados do exame físico
-Reações de hipersensibilidade
-Acidentes
5
DEFINIÇÕES E TERMOS UTILIZADOS
EVENTO ADVERSO SÉRIO (SAE):
Qualquer evento adverso que:
Resulte em óbito;
Represente risco de vida;
Necessite de hospitalização, ou prolongamento de uma
hospitalização pré-existente;
Resulte em incapacitação / deficiência significativa ou
persistente;
Promova malformação / anomalia congênita.
É um evento médico importante que pode ameaçar o paciente
ou pode requerer uma intervenção para prevenir um ou mais
critérios acima mencionados.
6
DEFINIÇÕES E TERMOS UTILIZADOS
Resulte em Óbito
É necessário uma condição médica que resultou
em morte
Relatar o evento de maneira mais detalhada
possível
Óbito é o RESULTADO não o evento em si
Represente risco de vida
O evento levou ao risco de vida Imediato do
paciente (exemplo: reação hipersensibilidade com
obstrução da glote)
7
DEFINIÇÕES E TERMOS UTILIZADOS
Hospitalização
Qualquer admissão hospitalar exceto:
Hospitalização planejada
Cirurgia
eletiva
ou
procedimentos
programados ou específicos do estudo
pré
Pacientes internados a longo prazo ou com doença crônica,
uma transferência para uma UTI hospitalar será considerada
um SAE
8
RELATO DOS EVENTOS ADVERSOS
O investigador do estudo deve coletar, avaliar e
reportar quaisquer AEs que ocorram durante o
andamento de um estudo clínico.
Todos os AEs devem ser muito bem documentados, e
normalmente
as
seguintes
informações
são
geralmente solicitadas para cada evento:
Início (data/ hora)
Duração
Severidade (leve/ moderado/ grave)
Causalidade**
Sério ou não sério
Ação Tomada
9
RELATO DOS EVENTOS ADVERSOS
Causalidade
A droga do estudo
pode ter causado o evento?
NÃO
Não
relacionado
SIM
Remota
Possível
Provável
10
RELATO DOS EVENTOS ADVERSOS
Causalidade
Considerar:
Hora da administração da droga do estudo e o início do evento;
Discutir informações (o evento melhorou quando a droga do
estudo foi descontinuada, ou voltou quando a droga do estudo
foi re- administrada?);
Conhecer o padrão de resposta (eventos comum de classe da
droga suspeita, eventos comuns descritos na brochura do
investigador);
Explicações Alternativas (ex. Doença durante o estudo, outras
drogas, fatores ambientais, etc.);
11
RELATO DOS EVENTOS ADVERSOS
Na prática, todos os relatos de eventos adversos
deverão no mínimo, conter todas as informações
descritas no exemplo abaixo:
“Paciente relata que no dia 01/01 apresentou dor
de cabeça de leve intensidade. Fez uso de
aspirina
quando
necessário.
Apresentou
melhora total do sintoma após dois dias.
Considero que este evento não está relacionado
a medicação do estudo.”
12
INTERATIVO
13
1. Um paciente após assinar o TCLE sofreu uma queda
da própria altura e apresentou fratura de MSE. O que
deve ser relatado no prontuário médico do paciente?
A) Evento Adverso de QUEDA relacionado ao estudo
B) Nada, pois o paciente sofreu um acidente que não está
relacionado ao protocolo de pesquisa
C) Evento Adverso Sério, pois fratura é um critério específico de
seriedade
D) Evento Adverso de FRATURA, com todas as informações
necessárias para o registro de um EA.
14
2. Qual é a informação faltante para o relato do evento
adverso?
“Paciente M-S retorna ao centro para realizar
a visita 1. BEG, sem alterações. Relata ter
tido um episódio de tontura leve com
melhora
espontânea
sem
uso
de
medicação.”
Adendo: paciente relata início do evento em 10/01
com melhora total no mesmo dia. Considero que este
evento
provavelmente não está relacionado a
medicação do estudo
Data de início
Data de término
Causalidade (relação com a medicação do estudo)
15
3. Qual é a alternativa errada?
A) Todo Evento Adverso Simples deve ser relatado no
prontuário médico e registrado na Ficha Clínica
B) Toda Hospitalização é um Evento Adverso Sério
C) Os sinais/sintomas pré-existentes dos pacientes serão
considerados EA caso apresentem uma piora durante o
decorrer do estudo
D) Os achados laboratoriais e de imagem poderão ser
classificados como sendo Clinicamente Significante ou Não
Clinicamente Significante pelo investigador
16
4. Qual é a informação faltante para o relato do evento
adverso?
“Paciente apresentou lombalgia em 01/01
com melhora total do sintoma após 5 dias.
Considero que este evento não está
relacionado a medicação do estudo.”
Adendo: paciente relatou lombalgia leve e não fez
uso de nenhuma medicação para tratamento do
evento.
Ação Tomada
Intensidade do Evento
17
REPORTE DOS EVENTOS ADVERSOS SÉRIOS
Todos os eventos adversos sérios (SAEs) devem ser
imediatamente comunicados ao patrocinador, exceto aqueles
especificamente identificados no protocolo ou em outro
documento (ex.: Brochura do Investigador) como não sendo
passíveis de comunicação imediata. As comunicações imediatas
devem ser seguidas de relatórios detalhados por escrito
fornecidos pelo patrocinador.
Reportar em até 24 horas após o
conhecimento do SAE !
18
RESPONSABILIDADES DO PATROCINADOR
Revisar os Dados de Segurança durante todo o período de
condução do estudo uma vez que é a única entidade a ter acesso
a todos os dados de segurança da droga em investigação;
Reportar todos os eventos sérios, relacionados a droga em
investigação e inesperados as agencias regulatórias aplicáveis;
Reportar todos os investigadores que participam de um estudo
com a droga em investigação de todos os eventos sérios,
relacionados e inesperados (SUSARs);
Relatórios Internacionais (safety reports)
19
GRAVIDEZ
Durante um estudo clínico as mulheres com potencial
de engravidar devem ser regularmente orientadas
sobre a importância de métodos contraceptivos
seguros.
Apesar de não se tratar de um SAE, por ser uma
situação especial quando uma paciente fica grávida
deve-se:
Retirar a paciente do estudo
Relatar imediatamente
Acompanhar a gravidez até seu resultado
final (resolução da gravidez)
20
CIOMS
COUNCIL FOR INTERNATIONAL ORGANIZATIONS OF MEDICAL
SCIENCES
(CONSELHO PARA ORGANIZAÇÕES INTERNACIONAIS DE
CIÊNCIAS MÉDICAS)
c/o World Health Organization
CH-1211 Geneva 27 (Switzerland)
Tel. (+41) (0) 22 791 34 67/13/06
Fax (+41) (0) 22 791 42 86
www.cioms.ch
e-mail: [email protected]
21
CIOMS
Avaliação e Monitorização dos Eventos Adversos
CIOMS I. (1990) – Reporte Internacional de Eventos Adversos
O mais valioso resultado do grupo CIOM foi a introdução do
“CIOMS I reporting form” para a normatização do reporte
internacional dos casos de eventos adversos sérios relacionados
e inesperados (SUSAR)
22
CIOMS
Reporte e Terminologia dos Eventos Adversos
Como o uso e interpretação de certos termos de Eventos
Adversos são diferentes em cada país, isso pode levar a uma
interpretação errônea dos dados ou resultar no atraso da
avaliação pelas autoridades regulatórias da droga.
A necessidade de estabelecer exigências mínimas para registrar
apropriadamente o diagnóstico dos eventos adversos e, então
descrevê-los de maneira correta, representa o tipo mais
importante de informação para levantar suspeita sobre a
segurança da droga, e quase sempre tomar-se alguma atitude.
23
24
MedWatch
É o programa do FDA para informação de segurança e Relato de
Eventos Adversos!
Alertas de segurança de produtos, recalls, alterações importantes
no rótulo a/ou qualquer alteração que possa afetar a saúde de
todos os americanos é rapidamente disseminada através do site e
do MedWatch e-list.
25
26