Farmacologia
Aulas iniciais de Farmacologia 19/02
... E essas estruturas mais complexas podem ser obtidas de plantas... Nós temos a Heparina que é extraída de tecidos de animais (tecido suíno, tecido
bovino), temos até heparina humana , através da técnica do DNA recombinante. Temos drogas extraídas de microorganismo, até hoje, como a penicilina
que é produzida através da técnica de fermentação em grande escala, em que você produz o fungo. A oxinase(nome duvidoso) que é um fibrinolítico
extraído de células humanas, a insulina humana produzida através de biotecnologia também pela técnica do DNA recombinante. São estruturas mais
difíceis de sintetizar, porém podemos obtê-las através de plantas, animais e tecidos humanos.
Se eu quero um outro fármaco pra atuar no receptor Beta 1 cardíaco eu sei quem produz a catecolteída, então eu vou começar a sintetizar
estruturas com esse grupamento, para que se liguem e eu tenha o meu resultado. Agora GENTE, essa história é muito trabalhosa. Cada 10.000
moléculas sintetizadas no mundo, com uma possível afinidade farmacológica, uma chega ao mercado. Vamos supor que eu desenvolvi uma molécula que
se ligou perfeitamente no receptor Beta-1 cardíaco, gente nós já temos mais um anti-hipertensivo.( comentário: desenvolver anti-hipertensivo hoje em dia
é um pouco perca de tempo ¬¬) Eu sei pelos testes que essa molécula se liga muito bem no receptor Beta 1. Agora precisamos fazer experimentos préclínicos : o que essa molécula pode fazer. Primeiramente, fazem estudos em animais. Fazem experimentos farmacológicos bioquímicos, testando em
culturas de células em primeiro lugar, ver a extensão dessa ligação, depois eles vão testar em células isoladas, em órgãos isolados para ver se
ela(molécula) tem afinidade, se realmente exerce efeito benéfico que eu quero. Ae eles podem experimentar em um animal comum. Esses experimentos
podem ser feitos para testar tanto a atividade farmacológica terapêutica como também a atividade farmacológica toxicológica. Para ver se essa
molécula é muito tóxica ou se ela pode ser usada na aplicação terapêutica. Então eles vão detectar efeitos adversos agudos e crônicos no animal, vão
ver se tem alguma possibilidade de ser mutagênico, carcinogênico, teratogênico. E vão avaliar os parâmetros farmacocinéticos dessa molécula: como
que ela é absorvida; como que ela é distribuída no organismo; como que ela metabolizada; como que ela é excretada. Tudo isso é teste pré-clínco e
leva pelo menos uns 5 anos. Se por acaso o fármaco que eu desenvolvi passar pelo teste pré-clínico - não é grande sua toxicidade, tem uma boa
efetividade, uma boa ligação e é bem absorvido, eu vou para o teste clínico, e então vou para os seres humanos. O teste clínico é divido em quatro
fases: A fase I é quando eu utilizo um voluntário sadio, geralmente homens, jovens por volta dos 25 anos, vou testar uma dose deste medicamento que
eu estabeleci, se é comprimido ou líquido (que foi definido no teste pré clínico através da estabilidade da molécula). Verificar a dose necessária para
produzir o efeito que eu quero, e ainda avaliar parâmetros farmacocinéticos, nesse indivíduo sadio( quanto tempo ela leva para ser absorvida, para
ter sua compensação máxima, quanto tempo ela leva para ser excretada ou metabolizada); Teste de Fase II, agora entra um indivíduo que possui a
patologia. Por exemplo, se estou desenvolvendo um anti-hipertensivo, eu vou pegar um grupo de hipertensos, e vou testar o meu fármaco, e vou
verificar se ele é melhor que o placebo, em primeiro lugar. Vou verificar se realmente tem uma eficácia desejável, ou avaliar a dosagem novamente, as
características farmacocinéticas e a tolerância, qual que é a margem de segurança para obter o efeito e não ter toxicidade. Teste de Fase III, agora
entra um grande grupo de pacientes hipertensos, vou dividir em dois e vou comparar esse novo anti-hipertensivo com aquele que já existente no
mercado e vou verificar se ele tem alguma vantagem. Teoricamente o meu anti-hipertensivo tem que oferecer alguma vantagem daquele existente no
mercado, no mínimo sendo mais barato. Vamos supor que ele tenha a mesma eficácia, o mesmo perfil de reação adversa, mas se ele for mais barato já
é uma grande vantagem. Agora se nem isso acontecer, eu não estou contribuindo com absolutamente nada para sociedade ao lançar esse
medicamento. E o mercado está cheio desses medicamentos. A indústria farmacêutica quer sua fatia no mercado dos anti-hipertensivos que é um
medicamento muito comum, muito usado. Outro medicamento muito comum são os anti-gripais, uma medicação que não cura nada, porém quem lança
uma nova associação irracional (porque todas são!) de anti-gripais está pegando uma fatia ótima do mercado. São lançados muitos medicamentos para
tratar o sintomas da gripe. A maioria dos medicamentos que são lançados hoje,não oferecem nenhuma vantagem daqueles que já temos. É só uma
indústria farmacêutica tentando pegar aquela fatia do mercado que ela não tinha. Durante os teste clínicos, várias substâncias viram que eram inúteis.
De cada 10.000 moléculas com potencial terapêutico, uma acaba passando. Vamos supor, que minha droga passou pelos testes clíncos, a indústria
farmacêutica deu uma maquiada nos seus resultados e mostrou uma vantagem, que apareceu pela sua maquiada, nos testes de Fase III. Por exemplo,
meu anti-depressivo não leva 30 dias para iniciar o efeito como a maioria, mas 25 dias. Através dessa vantagem a indústria vai começar a fazer
propaganda. Vai mandar o propagandista no seu consultório, mostrar todos os resultados de fase III, aqueles ensaios controlados, “ olha cinco dias a
menos você pode ter o resultado”, é uma invenção, e é por ae esse tipo de propaganda que eles fazem. Oferecem amostra grátis, oferecem viagens
para congressos... Agora, quando o medicamento foi aprovado pelo ministério da saúde, começa lá no FDA, eles não aprovam nos EUA, mais aprovam
no Brasil, como o Viox (prof estava meio revoltada).
Foi para o mercado, esse medicamendo recém lançado entra em Teste clínico de Fase IV, quando eu uso de parâmetro a população em
geral. Isso porque tem pessoas com diferentes hábitos alimentares, diferentes condições sociais, culturais. E agora sim que meu medicamento está
realmente sendo testado, agora eu realmente vou saber a real eficácia e segurança desse novo medicamento. Isso segnifica que todo medicamento com
menos de 5 anos no mercado ele está em teste. Quando se prescreve um medicamento novo (menos de 5 anos), o paciente deve saber que ele é um
voluntário de pesquisa farmacológica, se você não avisar, você está sendo anti-ético. Somente depois de anos da avaliação risco/benefício admite-se
finalmente o valor terapêutico do novo medicamento.
Formas Farmacêuticas
É a maneira de como os medicamentos são preparados, apresentados e consequentemente comercializados. As formas farmacêuticas podem
ser dividas em três grandes classe: líquidas, sólidas e pastosas.
Todo medicamento tem na sua composição o fármaco ou a droga mais o excipientes ou veículo. A gente usa o termo excipiente quando o meio
em que ele está insirido é um meio sólido. A gente usa o termo veículo quando o fármaco está inserido é um meio líquido. O excipiente ou veículo é um
conjunto de substâncias ou uma substância apenas que não possui função terapêutica que tem como função dá forma e volume ao fármaco. Imagine
uma pílula de anticoncepcional se não tivesse excipiente. A quantidade que vai de hormônios é em microgramas, seria impossível de administrar sem um
excipiente. Outro exemplo o tilenol, comprimido que tem na sua fórmula o paracetamol e tem como excipiente um conjunto de substâncias que tem
finalidades da forma, volume a esse paracetamol e permitir a sua desintegração no túbulo intestinal (tem celulose, amido).
1
Vamos ver os tipos de medicamentos que existem no mercado. A forma farmacêutica é que vai defenir a via de administração. Os
medicamentos que não precisam de prescrição; os medicamentos que precisam de prescrição; os medicamentos controlados; e os medicamentos não
controlados.
Os medicamentos não controlados são aqueles que eu apresento minha receita na farmácia, pego o medicamento e recebo a receita de volta.
Os medicamentos controlados são aqueles que quando eu apresento a receita, ela ou uma cópia fica retida na fármacia para controle de entrada e
saída.
Os medicamentos que não precisam de prescrição, VENDA LIVRE, são muito mais comum nos EUA.
Todos os medicamentos que precisam de prescrição vão ter uma tarja. Essa tarja ou vai ser vermelha ou vai ser preta. Os medicamentos com
tarja vermelha quando precisam de retenção ou cópia da receita, vai está anunciado: COM RETENÇÃO DE RECEITA. Os de tarja preta são
medicamentos que o farmacêutico fica com a receita, a forma de tomar ele recebe separado. Os medicamentos de tarja preta podem induzir a
dependência, eles são psicotrópicos; alguns medicamentos de tarja vermelha são conhecidos como psicoativos atuam no SNC mas não necessariamente
levam a dependência.
Definições importantes:
Medicamentos psicoativos: são medicamentos que atuam no SNC TARJA VERMELHA com retenção de receita. Ex: Fluxetina.
Medicamentos psicotrópicos : atuam no SNC e possuem capacidade de induzir dependência. (propriedade reforçadora). Ex: diazepan, se a pessoa
não tomar ela ñ dorme, ela tem desejo de tomar.
*Atenção, o cataflan é um medicamento de tarja vermelha, mais não é um psicoativo, portanto não precisa de retenção de receita.
Qual a diferença dos medicamentos de venda livre para os medicamentos de tarja vermelha que não precisam de retenção de receita?? É a reação
adversa. Por exemplo, o cataflan pode causar hemorragia grave, gastrite, úlcera, então eu não posso tomar por conta própria. Então ele tem um perfil
diferente, ele pode fazer interação medicamentosa, não se pode vender simplesmente sem ter uma orientação médica. Os de venda livre não tem um
perfil de reação adversa importante, pode tomar sem uma prescrição médica, porém, você tem que ter uma orientação na fármacia de como tomar. A
maioria dos medicamentos de venda livre são os analgésicos e antitérmicos que não tem efeito antinflamatório. Ex: paracetamol, possui um perfil muito
baixo interação medicamentosa. No Brasil, medicamento não pode ser vendido fora da farmácia, portanto estamos na frente do primeiro mundo. Ex:
nos EUA um frasco de aspira vem com 350 comprimidos, sendo vendidos no supermercado, a professora disse que vai morrer e não terá tomado todas
as aspirinas que ela comprou lá.
O medicamento possui: o nome comercial, o nome empírico ou nome do fármaco ou o nome da drogra, a dose, a forma farmacêutica(comprimido) e a
apresentação (20 cp de 50 mg), nome do fabricante. Isso tem nos medicamentos de referência ou similares, nos genéricos não. Os genéricos tem o nome
empírico que tem que vir grandão, não tem nome comercial e tem que ter a tarja- medicamento genérico.
Se você for um médico pelo SUS, você não pode prescrever nenhum medicamento pelo nome comercial, você tem que prescrever pela DCB
denominação comum brasileira que é o nome químico de todos os fármacos.
CLASSES DE MEDICAMENTOS
Medicamento de Referêcia: é aquele que a indústria farmacêutica investiu muito dinheiro para lançá-lo no mercado. Ela começou desde do
desenvolvimento da molécula, passou pelos testes pré clínicos, foi para os teste clínicos, foi para o mercado, investiu em propaganda, patenteou o
medicamento e é um produto inovador. Primeiro que foi lançado, e teve sua eficácia e segurança comprovada. São, geralmente, medicamentos que
estão a bastante tempo no mercado, tem uma marca comercial conhecida. Ex: Novalgina, Cataflan.
Similares: Infelizmente o balconista ou farmacêutico quase sempre indica esse tipo de medicamento, pois a maioria dos balconistas tem comissão, e
também porque nesse medicamento eles tem mais lucro. São medicamentos que possuem o mesmo fármaco, a mesma drogra, o mesmo princípio ativo, a
mesma dose em forma farmacêutica, via de admistração, mesma posologia, mesma indicação terapêutica que o medicamento de referência, mas não
passou pelos testes de bioequivalência. Portanto, não possui sua bioequivalência comprovada.
*Bioequivalência: é um teste para determinar se uma nova formulação possui a mesma biodisponibilidade que uma formulação de referência.
Não sei se os veículos ou excipiente são iguais,não sei se vai ser desintegrado ou liberado ou absorvido ou então se vai atingir a mesma concentração
plasmática no mesmo tempo que o de referência. Existem siliares consagrados, como o anador. O anador possivelmente é mais conhecido que a
novalgina. Dependendo do similar a indústria nem tem interesse que vire genérico.
Os medicamentos genéricos: eles possuem tudo igualzinho de referência como o similar, porém o ministério da saúde através da Anvisa, avalia os teste
de bioequivalência entre o genérico e o seu medicamento de referência, que são apresentados pelos fabricantes para comprovação. Eu só posso
fabricar o genérico depois que venceu a patente do medicamento de referência, a não ser que é um medicamento tão essencial a saúde que eu não
posso usar a patente.
Farmacocinética x Farmacodinâmica (com muitas definições)
Farmacocinética: é o estudo do movimento do fármaco no organismo. Como esse fármaco é absorvido, distribuído no corpo, como que ele é
metabolizado e como que é excretado. De uma maneira simplista eu posso dizer o que o meu corpo faz com o fármaco.
2
Farmacodinâmica: estuda a ação do medicamento no meu organismo. O efeito que ele faz, tanto o efeito terapêutico, como o efeito tóxico. O que o
fármaco faz com meu corpo.
Farmacocinética: é o estudo da concentração das drogas nos fluídos corporais que influenciam a maneira como as concentrações variam com o tempo. É
estudo da droga durante sua permanência no corpo.
Farmacodinâmica: estudo das interações entre as drogas e as moléculas do corpo. As quais por uma série subsquência de eventos resultam em uma
resposta farmacológica.
A fase da administração até a desintegração do comprimido, até a interação do fármaco do seu veículo ou do seu excipiente, é chamada de fase
FARMACÊUTICA. (não é estudada na medicina) Definição : estuda a liberação do fármaco a partir do produto farmaceutico. É contribuído pelo
conjunto de fenômenos comprendidos entre a adminstração do medicamento e sua absorção propriamente dita. Isso vai depender da intensidade e da
velocidade que acontece para que a substância entre no organismo.
A fase farmacocinética, então, vai determinar todo o caminho do fármaco pelo corpo, todo seu processo de movimentação, até o momento que ele é
administrado, até que ele seja excretado. A farmacodinâmica vai começar estudar o fármaco quando ele se liga a alguma coisa, para produzir um
efeito. Todo fármaco precisa se ligar em alguma coisa no corpo para produzir efeito ? Não.Existe algumas exceções. Existem fármacos que não se
ligam a nada. Porém, eles alteram o pH fisiológico, como os antiácidos. Mas, a maioria dos fármacos vão te que se ligar.
Ex: Eu tomei um comprimido de Cataflan, imediatamente vai entrar na fase farmacêutica, vai se desintegrar no estômago, vai liberar o diclofenato de
potássio, esse vai para a primeira porção do intistino e vai se absorvido. Pronto, ele entrou na fase farmacocinética. Então vai ser distribuido pela
corrente sanguínea, vai para o corpo todo, e vai lá para o dedão do pé que está inflamado¬¬. Lá no dedão do pé, ele vai inibir uma enzima, a
ciclooxigenase (COX), que produz as prostoglandinas, que estão fazendo meu dedão ficar inchado, dolorido. A inibição da produção das
prostoglandinas entrou na fase farmacodinâmica, é o que ele está fazendo para o meu organismo. Então o meu dedão começa a desinchar, e esse
diclofenato de potássio começa a ser transformado quimicamente lá no fígado e conjugado perde sua atividade, então agora nós estamos de volta na
fase farmacocinética, e vai ser secretado no rim, como um conjugado e ae vai embora pela minha urina. A verdade é que todos esses eventos não
acontecem em uma reta, mais sim, tudo ao mesmo tempo.
Formas Farmacêuticas Líquidas
-soluções: são misturas homogêneas de um fármaco no solvente qualquer, preferencialmente água, mas pode ser álcool, pode ser hidroalcolico, em que
o soluto está completamente dissolvido no solvente. Essas soluções são usadas, na maioria das vezes, em injetáveis
-suspensões: são misturas heterogêneas em que um soluto muitas vezes se deposita no fundo e no momento de utilisação do medicamento é necessário
que ocorra uma difração, agitação.Ex: Amoxicilina.
-emulsões: são substâncias oleosas dispersas em meio aquoso, muitas vezes, apresentando separação de fármacos, assim, na hora de utilizar precisa
de agitação. Então são substâncias que geralmente o soluto só solúvel em meio oleoso, e eu preciso ele se disperse no meio aquoso. Ex: Scabin.
-xaropes: é forma de administração oral mais comum, em que o soluto é dissolvido em uma solução aquosa, onde o açúcar em alta concentração é
utilizado como corretivo para o sabor.
-elixires: são soluções de água e álcool, então são soluções hidrocólicas, para o uso oral, pode ser açucaradas, contendo substâncias arrómaticas.
-loções: são formas alcoolicas ou aquosas para o uso tópico, para o uso estérril.
-Linimentos: similares as loções só que com veículo oleoso.
*As mais comuns são as soluções, as suspensões e os xaropes.
Formas Farmacêuticas sólidas
-Comprimidos:formato variável (comum discóide) obtida por compressão. Fármaco (s) associados a aglutinantes e excipientes, obtidos por
compressão. Geralmente aglutinado com amido, com vários tipos de pó que tenha a capacidade de aglutinação.
- Drágeas: comprimidos com revestimento gelatinoso, que impedem a desintegração na porções superiores do trato gastro intestinal.Porque muitas vezes
o fármaco é instável em pH ácido, e pode perda a característica farmacológica do fármaco. O revestimento protetor apresenta várias camadas com
substâncias inertes ou não. Costumam ser coloridas e polidas com cera de carnaúba.
- Cápsulas:um ou mais fármacos são prensados com excipientes e colocados dentro de cápsulas gelatinosas ou amiláceas. Nós temos microesferas de
fármacos com amido ou excipientes que façam aglutinação, esses pequenos grânulos são colocados em cápsulas, se eju não quero fazer compressão, eu
posso fazer pequenos grânulos e aposicionar dentro de cápsulas.
- Pílulas:associação de princípio ativo com aglutinante viscoso. São comprimidos menores, como os anticoncepcionais.
- Supositórios:apresentações semi-sólidas para uso retal que fundem a temperatura corporal, pela presença de manteiga de cacau, glicerina ou
polietilenoglicol. São homogenizadas em substâncias serosas, para ser usados na temperatura corporal.
- Óvulos: apresentação semi-sólida para uso ginecológico (diferente do supositório na forma).
3
*O comprimido só pode ser partido, se ele tiver um sulco que o separa em duas partes, porque nesses comprimidos há uma distribuição igual de
fármaco.
*Nunca posso partir um comprimido revestido, porque se ele recebe um revestimento, ele precisa de uma proteção, e se partir no meio eu retiro essa
proteção, podendo sofrer hidrólise na saliva, no esôfago, estômago. A biodisponibilidade não está garantida.
Formas Farmacêuticas Pastosas
Preparações semi-sólidas, destinadas ao uso tópico: geléias, cremes, pomadas, ungüentos e pastas. Diferem nos veículos que são gelatinosos nas geléias,
oleosos nas pomadas e aquosos nos demais. São, geralmente, para uso tópico. Existem algumas formas para uso oral.
A forma farmacêutica vai definir a via de administração.
O que determina que via usar?
- O tipo de ação que quero: uma ação pequena, sistêmica.
-A rapidez de ação: imidiata ou posso esperar um mês.
-A natureza do medicamento
Nem sempre, mas na maioria das vezes eu quero que o fármaco atinja a corrente sanguínea. Com exceção das vias intravenosas, as outras eu preciso
que o fármaco atravesse barreiras. Em cada via vai ter um menor ou um maior número de barreiras, e isso vai determinar a velocidade de ação desse
fármaco e também a biodisponibilidade.
CLASSIFICAÇÃO
• ENTERAIS (oral, sublingual, retal)- todas as vias que vão entrar em contato com o trato gastrointestinal.
• PARENTERAIS (injetáveis)
• TÓPICAS(cremes, pomadas, colírios)
• INALATÓRIA
Via Oral
O fármaco é absorvido PRINCIPALMENTE pelo intestino, porque é o grande ógão absorvidor do nosso corpo. Por exemplo, se eu tomar um
anticoncepcional injetável eu posso passar um mês passando mal. Se eu tomar um oral, tem a vantagem de ser interrompido.
- VANTAGENS
• Auto-administração, econômica, fácil
• Confortável, Indolor
• Possibilidade de remover o medicamento
• Efeitos locais e sistêmicos
Formas farmacêuticas: cápsulas, comprimidos,xaropes, soluções etc...
ORAL - DESVANTAGENS
• absorção variável (ineficiente)
• período de latência médio a longo – Propaganda inganosa: tomou doril a dor sumiu. Em 15 min o cp não foi nem desintegrado. No mínimo 1 hra.
• ação dos sucos digestivos – importância da proteção
• Interação com alimentos
• pacientes não colaboradores (inconscientes)- idoso, crianças
• sabor
• Metabolismo de primeira passagem -PRINCIPAL DESVANTAGEM- 100% dos medicamentos administrados por via oral, pelo sistema porta, todo
medicamento absorvido pelo intestino, vai passar pelo fígado, antes de atingir a circulação sitêmica. O fármaco quando passa pelo fígado pela
primeira vez, vai sofrer ação enzimática. E isso pode acarretar em uma molécula sem atividade ou com atividade menor, parte desse fármaco vai
ser perdido.
• pH do trato gastrintestinal
LINGUAL - VANTAGENS
(mucosa oral e sub-lingual)
• Fácil acesso e aplicação
• Alta vascularização, rápido acesso a circulação sistêmica
• Não sofre MPP.
• Latência curta
• Emergência
Formas farmacêuticas: comprimidos, pastilhas, soluções, aerossois, etc...
LINGUAL - DESVANTAGENS
• Pacientes inconscientes
• Irritação da mucosa
• Sabor desagradável
• Dificuldade em pediatria
VIA
RETAL - VANTAGENS
• Alta vascularização, rápido acesso a circulação
sistêmica. Sofre pouco metabolismo de primeira
4
passagem.
• Pacientes não colaboradores (semi-conscientes,
vômitos)
• Impossibilidade da via oral
• Impossibilidade da via parenteral
Formas farmacêuticas: supositórios e enemas
RETAL - DESVANTAGENS
• Lesão da mucosa
• Incômodo
• Expulsão
• Absorção irregular e incompleta
Via parenteral
• Fármacos injetáveis
INTRA-MUSCULAR - VANTAGENS
• Efeito rápido com segurança
• Via de depósito ou efeitos sustentados
• Fácil aplicação
DESVANTAGENS
• Dolorosa
• Substâncias irritantes ou com pH diferente
• Não suporta grandes volumes
• Absorção relacionada com tipo de substância:
• sol. aquosa - absorção rápida
• sol. oleosa - absorção lenta
Formas farmacêuticas: injeções
Músculos: deltóide, glúteo.
- RECOMENDAÇÕES
• Auxílio na absorção: calor/ massagens
• Retardamento na absorção: gelo
• Locais de aplicação:deltóide, glúteo, ventroglúteo
• Posição da agulha: perpendicular ao músculo
• Aspirar antes da aplicação
• Escolha do bizel
• Pessoal treinado
• Assepsia local
VIA-MUSCULAR - RISCOS
• Trauma ou compressão acidental de nervos
• Injeção acidental em veia ou artéria
• Injeção em músculo contraído
• Lesão do músculo por soluções irritantes
• Abcessos
Via ENDOVENOSA VANTAGENS
• Efeito farmacológico imediato
• Controle da dose
• Admite grandes volumes
• Permite substâncias com pH diferente da neutralidade
VIA ENDOVENOSA - DESVANTAGENS
Material esterilizado
• Pessoal competente
• Irritação no local da aplicação
• Facilidade de intoxicação
• Acidente tromboembólico
VIA ENDOVENOSA - COMPLICAÇÕES
• Flebites, tromboflebites, acidentes embólicos
• Infecções
• Extravasamento
• Necrose
• Sobrecarga circulatória
• Reações alérgicas
VIA SUBCUTÂNEA
• Absorção constante e lenta
• Implante de Pellets (sobre a pele)
• Substâcias não irritantes (terminais nervosos)
VIA INTRADÉRMICA
• Fácil acesso
• Ações locais e sistêmicas
• Vacinas e testes alergenos
VIA VAGINAL
Preparações higiênicas femininas
Antifúngicos
Contraceptivos
Drogas para induzir trabalho de parto
Formas farmacêuticas: Supositórios vaginais ou óvulos, gel, pomadas,
soluções, emulsões
12/03/09
Fatores que podem alterar o metabolismo dos fármacos
Se a enzima que metaboliza o fármaco for polimórfica (menor atividade) pode causar toxicidade. Exemplo: CYP2B6.
Variação genética: Exemplo: propanol tem menos eficácia em pessoas negras, por outro lado os chineses têm muita sensibilidade ao propanol,
cujo principal efeito adverso é broncoconstrição.
Pode aparecer reação adversa não esperada.
Determinantes ambientais: fumaça do cigarro, carne grelhada tem efeito indutor enzimático- teofilina-> broncodilatador. Portanto, fumantes
precisam de mais teofilina para fazer o efeito.
Fatores mórbidos: disfunção hepática (dificuldade de metabolização).
Idade e sexo: extremos de vida terão dificuldades em metabolizar os fármacos. Aos 50 anos a pessoa já perdeu 25% da função hepática,
aos 75-80 já perdeu 50% da função hepática. Os recém-nascidos não têm atividade enzimática amadurecida.
Mulheres são mais sensíveis ao etanol-> o acetaldeído é tóxico para o organismo e a mulher produz menos enzima para metabolizá-lo
(acetaldeído-desidrogenase).
Álcool-> álcool desidrogenase-> acetaldeído (tóxico)->acetaldeído-desidrogenase->acetato (não tóxico).
Não é só o fígado que metaboliza os fármacos, o intestino tem enzimas do CYP450. Portanto, muitos fármacos podem ser metabolizados
antes de serem absorvidos, ou seja, ficarão menos biodisponibilizados.
Outros: cérebro, rim, placenta, pele.
5
Excreção de drogas: as drogas são principalmente, mas não exclusivamente, excretadas pelos rins. (Hang diz que excreção é a mesma coisa
que eliminação, mas isso é incorreto).
Processo pelo qual as drogas são transferidas do ambiente interno para o externo, sendo rins, pulmões, sistema biliar e intestino os principais
órgãos excretores.
O rim é o principal órgão excretor para a maioria das drogas hidrossolúveis e não voláteis.
20% o fluxo sanguíneo que chega ao rim passa pelo glomérulo.
Quem consegue atravessar as 3 membranas? A água, as moléculas com peso molecular menor que 20.000 Dáltons.
Pts não atravessam: fármacos ligados a pt não passam.
A maioria dos fármacos livres atravessa. Exceção: insulina, Heparina, porque as moléculas são muito grandes. Esse é o processo de filtração.
A maioria das drogas é lipossolúvel, porém quando metabolizadas se tornam hidrossolúveis.
A maioria dos fármacos livres consegue atravessar os túbulos renais pelo processo de filtração, secreção tubular e difusão através do túbulo
renal.
80% do fluxo sanguíneo faz contato com os túbulos contorcidos através dos capilares peritubulares, e é transportado ativamente pela
membrana para então ser excretado (ativamente, pois é contra um gradiente de concentração, com consumo de energia, pode reduzir a concentração
da substancia no plasma a quase zero – muito eficiente; depuração ou secreção de substancias ligadas a pt plasmática).
Existem grupos de pts que transportam fármacos ácidos e grupos que transportam fármacos básicos. Uma hora essas pts saturam → interação
medicamentosa. Às vezes, uma mesma pt transporta diferentes fármacos (competição) → pode-se aumentar o efeito de um fármaco (interação
farmacocinética).
Exemplo: fármacos ácidos → Indometacina, Probenecida, e todos os fármacos que se conjugam com o ácido glucurônico e glicina.
Exemplo de fármacos básicos: dopamina, estamina, morfina, dizepam (só que o diazepam pode ser conjugado com ácido glucurônico e ficar
ácido).
A secreção tubular é o mecanismo mais eficiente para eliminar fármacos. A ligação a pt plasmática não impede sua excreção quando se fala
nesse processo.
Os fármacos ligados a pt plasmática se desligam dela e se ligam as pts transmembranas → secreção.
Problemas: são as interações medicamentosas → que podem ser benéficas ou maléficas. Ex: a probenecida, aumenta a excreção renal do
ácido úrico – utilizada para gota, faz com que a penicilina não seja excretada → doses menores de penicilina.
Os fármacos podem sofrer reabsorção (difusão através do túbulo renal).
Os fármacos lipossolúveis podem voltar para a corrente sanguínea (reabsoração) e demorar mais para ser eliminado.
99% da água filtrada é reabsorvida de volta para o sangue e pode acabar levando de volta algum fármaco lipossolúvel.
Substancias altamente polares permanecem nos túbulos e são excretadas rapidamente (ex. digoxina), os que não tem essa característica
ficam sendo absorvidos (uma hora ele será eliminado).
Fármaco ionizado → mais solúvel, mais polar → mais excretado
Para acidificar a urina → ácido cítrico, ac. Acético
Para alcalinizar a urina → bicarbonato de sódio
O sangue tem sistema tampão, mas a urina não.
O fígado produz a bile e muitos fármacos acabam sendo secretados do fígado para a bile, como a bile é secretada no intestino esses
fármacos seguem esse caminho. A bile é secretada no intestino através de transporte mediado por carreadores. Conjugados glicuronídeos concentramse na bile, são transportados até o intestino, onde podem ser hidrolisados por enzimas produzidas pela flora intestinal tornando-se novamente ativos,
podendo ser absorvido pelo intestino e voltar para o sangue.
Alguns fármacos possuem circulação entero-hepaticas → etileno estradiol (anticoncepcional), morfina. Por isso a pessoa toma um comprimido
de microgramas e só toma outro depois de 24 h (sofre circulação entero-hepática).
Quando uma mulher tiver que tomar antibióticos, sempre aconselhar a usar outro tipo de método anticoncepcional.
6
Rifampicina (tuberculose) → o anticoncepcional não fará efeito.
Dependendo do antibiótico, pode-se ter efeito teratogênico
Reações de fase 1
OXIDAÇÃO, REDUÇÃO E HIDRÓLISE
Metabólitos → podem perder atividade farmacológica
Manter atividade farmacológica
Tornar mais tóxicos
Ficar ativos (como nos pró-fármacos)
Reações de fase 2
CONJUGAÇÃO
Fármaco mais polar, mais hidrossolúvel (mais facilmente excretado)
As enzimas que fazem as reações, principalmente as de oxidação são classificadas de CYP 450 → se diferem pelo tipo de fármaco que elas
metabolizam, pela seqüência de aa, pela capacidade de serem induzidas ou inibidas por diferentes substancias.
Essas enzimas podem ser induzidas por um grupo de fármaco e quando estou tomando um outro grupo de fármaco, o metabolismo deste
pode ser alterado → perder o efeito (se gerar metabólito inativo); aumentar o efeito (se a metabolização gerar metabólito tóxico).
Outras vias de excreção: via pulmonar (muito importante para anestésicos locais voláteis), leite materno (concentra compostos básicos), suor,
saliva e cabelo (esses três últimos têm maior importância na medicina forense; usuários de drogas → 3 meses no cabelo).
Parâmetros Farmacocinéticos
Cmax
Tmax
AUC ou ASC (área sobre a curva)
Depuração
Cinética de eliminação ordem zero
Cinética de eliminação primeira ordem
T ½ vida
IT → índice terapêutico (é determinado nos testes pré-clínicos - com animais).
IT = DL50 (dose letal para 50% dos animais)
DE50 (dose efetiva para 50% dos animais)
IT = 100 mg/Kg = 5
20 mg/Kg
Dose efetiva está bem longe da letal → isso dá uma idéia muito abrangente de que o fármaco parece ter uma janela terapêutica boa.
Esses parâmetros são importantes para os fármacos que tem um IT pequeno.
7
Janela terapêutica (também conhecida como margem de segurança) → testes clínicos - seres humanos.
O conceito de IT tem muitos pontos limitantes – para o ser humano existem várias doses tóxicas antes da letal; dose efetiva para quê? Tem
fármacos que tem vários efeitos farmacológicos → AAS é um antiinflamatório na dose de 500 mg/8 em 8 h, portanto 1,5 g/dia; dose antiplaquetária
→ 100 mg/dia.
O IT do AAS é completamente diferente para cada aplicação. IT antiplaquetário é muito maior que para a ação antiinflamatória.
Outra falha é a dose efetiva → para que eficácia se está falando?
AAS 500 mg a cada 8h → 1,5 g/dia → efeito antiinflamatório
Dose antiplaquetária → 100 mg/dia
Muito mais importante que saber o IT é saber a janela terapêutica.
Os parâmetros farmacocinéticos são muito importantes para os fármacos que apresentam janela terapêutica pequena. Grandes variações no
sangue podem levar a perda da eficácia ou a produção de toxicidade. Portanto, é muito importante definir o Cmax e o Tmax quando for definir uma
dose. Qualquer via que não seja endovenosa (Tmax = zero) tem-se o gráfico (ver desenho no caderno, o meu desenho não diz nada, ficou só uma
linha).
De maneira geral: quanto mais lipossolúvel for o fármaco menor será o Tmax e provavelmente maior será o Cmax (absorvido mais rápido e
em maior quantidade → se não for administrado por via endovenosa).
Medicamento genérico pode ter no máximo 10% de variação de Cmax e Tmax.
O Cmax tem que estar dentro da janela terapêutica.
Outro parâmetro determinado é a ASC (área sobre a curva) que avalia a exposição global de um organismo a uma dose de fármaco.
Quanto tempo o paciente ficou exposto a esse fármaco. O genérico tem que ser 90% semelhante ao de referência.
B – demora mais tempo para absorver, quantidade menor e mais tempo
para excretar. Exposição Global do fármaco é semelhante para o paciente. Desta
forma, pode usar a mesma dosagem (quem trabalha com isso são os médicos de
UTI e os farmacêuticos clínicos).
Pode ser útil quando estou tratando um paciente que tem um problema
fisiológico. Para um paciente que tem um Cmax e um Tmax totalmente diferente a
ASC pode ser semelhante.
Essa curva serve para genéricos e para pacientes com características fisiológicas e patológicas diferentes. Posso dar essa dose? Vai produzir
eficácia?
T ½ vida: tempo necessário para que a concentração plasmática de um fármaco caia pela metade.
Concentração plasmática
100 ug/ml
Tempo
0 – 5h
Concentração plasmática
50 ug/ml
8
50 ug/ml
25 ug/ml
5 – 10h
10 15h
25 ug/ml
12,5 ug/ml
Eu sei que a cada 5 h eu reduzo a concentração em 50%.
É preciso 4 ou 5 T ½ vida para que 97% seja eliminado.
Importante: Posologia
Interação medicamentosa que não pode ocorrer.
Isso só serve para fármacos com cinética de eliminação de primeira ordem
Se o T ½ vida for de 8 h, provavelmente, ele será ministrado de 8 em 8 h.
A maioria dos fármacos é tomada em múltiplas doses.
Primeira ordem – será 4 ou 5 T ½ vida
Se o tempo de ½ vida for 2 h → só daqui a 8 – 10 h é que o medicamento começara a fazer efeito, ou seja, que ele atingirá a CSS
(constante de equilíbrio dinâmico).
Se não puder esperar → dose de ataque → dose maior (máximo em cada medicamento). A dose de ataque pode ser dividida em dois. A
CSS demorará o mesmo tempo (não vai alcançar a CSS mais rápido). Vantagem: primeira dose já conseguiu o efeito, já está na janela terapêutica.
18/03/09
FARMACODINÂMICA
(a prof. sugeriu estudar pelo Rang)
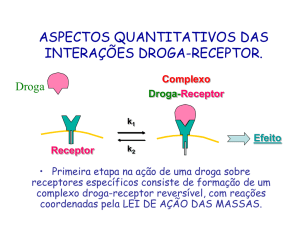
O que o fármaco faz com o organismo. Paul Ehrlich – Corpora non agunt nisi fixata (uma droga não funciona a menos que esteja ligada).
Existem fármacos que não se ligam e fazem efeitos – antiácidos
Na maioria das vezes os fármacos se ligam a pts. Exceções: antimicrobianos e antitumorais – interagem diretamente com o DNA.
O fármaco ligado a albumina não faz efeito, somente a fração livre é que irá atravessar a membrana e se ligar a pts provocando uma resposta
farmacológica.
Para que o fármaco possa interagir precisa existir especificidade, tanto da parte do fármaco quanto da pt. Ex.; angiotensina II (polipeptídeo) regula a
pressão arterial por vasoconstrição e aumento de liberação de aldosterona. A AII se liga ao receptor AT1. Se houver uma troca de AA, a pt não se liga
mais (alta especificidade).
Nenhum fármaco tem total especificidade. Ex. Antidepressivos tricíclicos – imipramina se liga a pt que faz a recaptação de neurotransmissores (NA, DA,
5HT) que estão na fenda sináptica para impedir essa recaptação. Entretanto, também reconhece o receptor muscarínico (Ach), o alfa adrenérgico (α1),
o receptor para histamina (H1). O que ocasiona os efeitos colaterais, como, boca seca, sono, alteração do apetite, retenção urinária, hipotensão.
Um aumento do grau de especificidade não garante o efeito farmacológico específico. Ex.; organofosforados – se liga na enzima acetilcolinesterase
(que degrada a Ach em acetil e colina + acetato). Os organofosforados não interagem com outras pts. São altamente específicos, contudo provocam
9
bradicardia, broncoconstrição, salivação, sudorese, lacrimejamento, aumento dos movimentos peristálticos, miose (alta especificidade com diversos
efeitos farmacológicos).
Quanto menor a potencia → maior a dose e maiores os efeitos colaterais.
AÇÃO ≠ EFEITO (importante)
Qual a ação do antidepressivo tricíclico: inibir a recaptação de neurotransmissores (antagonista do receptor).
Qual o efeito dele? Antidepressivo, boca seca, hipotensão, diminuição do apetite.
Qual a ação dos organofosforados? Inibir a AchE
Qual o efeito deles? Bradicardia, broncoconstrição, etc.
A ação é o que faz para produzir os efeitos
Principais alvos ptcos para a ligação dos fármacos: enzima, canal iônico, molécula transportadora e receptores.
Exceções: colchicina (antiinflamatório usado no tratamento da gota) se liga a tubulina, pt estrutural dos neutrófilos, impedindo sua chegada ao local da
inflamação.
VER FIGURA DO FARMACOLOGIA TEXTO E ATLAS – Célula e os quatro alvos ptcos.
Canal iônico: o fármaco pode ser ligar obstruindo o canal.
Pt de transporte: o fármaco pode ser ligar e impedir o transporte.
Enzima: o fármaco pode entrar na célula e se ligar na enzima impedindo que ela trabalhe.
Receptor no núcleo da célula: receptor para hormônio → o fármaco pode impedir a ligação do hormônio ou se ligar como se fosse o hormônio.
Receptor de membrana: receptor para neurotransmissor → o fármaco pode fazer o papel de neurotransmissor ou se ligar impedindo que o
neurotransmissor se ligue.
Enzimas: o fármaco pode inativar a enzima. Todos os AINES – diclofenaco, ibuprofeno, aspirina, paracetamol, dipirona – inibem a enzima COX
(ciclooxigenase).
Pode ser uma inibição reversível ou irreversível.
Reversível → ligações fracas (temporariamente)
Irreversível → ligações fortes (a enzima não vai mais produzir sua reação metabólica)
Ex.; todos os AINES inibem a COX de forma reversível. Somente o AAS inibe de forma irreversível.
As drogas podem atuar como substratos falsos. Ex.: fluorouracil – bloqueia a síntese de DNA, substitui a uracila na biossíntese de purinas –
antineoplásicos – e a célula não se replica.
Ex. do Rang: pró-fármaco – cortisona (nesse caso a enzima não é alvo protéico). O fármaco no receptor farmacológico é um alvo protéico.
Canais iônicos: pt de membrana que se arranja de forma oligomérica formando um poro, permitindo a passagem de muitos íons conforme a carga no
interior do poro.
Canais iônicos regulados por diferentes mecanismos: canais iônicos regulados por voltagem – onde atuam os anestésicos locais (bloqueadores) – chega
um potencial de ação e o canal iônico se abre – neurônios – canais de sódio e potássio.
10
Canais iônicos regulados por ligantes ou receptores ionotrópicos: incorporam um receptor. Ex.; receptores benzodiazepínicos (moduladores); receptores
nicotínicos (Ach) na junção neuromuscular.
Quando se falar de canais iônicos está se referindo aos canais por voltagem. Os canais por ligantes são chamados de receptores farmacológicos.
Canais iônicos – bloqueiam o canal (o fármaco).
Moduladores (canais por ligantes) – regulam a abertura e o fechamento do canal.
Molécula transportadora: se liga a pt de transporte no sitio de ação ou em outro local impedindo o transporte. Ex.: diuréticos, omeprazol.
O fármaco pode ser transportado no lugar da substancia normal.
Substrato falso – acúmulo de composto anormal. Ex. anfetamina.
Receptor farmacológico ou receptor fisiológico para o Goodman
Elementos sensoriais no sistema de comunicações químicas que coordena a função de todas as diferentes células do corpo, sendo os mensageiros
químicos representados por vários hormônios, transmissores e outros mediadores.
A grande diferença entre o receptor farmacológico é que a pt não funciona se não houver a ligação (a enzima está constantemente fazendo sua
função, a molécula de transporte e o canal iônico também funcionam constantemente).
São pts que também são altamente especializadas que podem estar na membrana ou no interior das nossas células que respondem para ligantes
endógenos, ou seja, para os neurotransmissores, hormônios.
Agonista: ligam-se ao receptor farmacológico e induz uma resposta. Ex.: Ach – agonista do receptor nicotínico na junção neuromuscular – contração;
succinilcolina: bloqueador neuromuscular despolarizante, faz a célula contrair tanto que ela pára.
Antagonista: liga-se ao receptor e bloqueia uma resposta. Ex: atropina.
A maioria dos antagonistas faz ligações reversíveis. Os que fazem ligação irreversível têm um tempo de ligação mais prolongado.
Com o M2 bloqueado, vê-se somente o efeito da adrenalina no ᵦ1 (taquicardia).
Agonista total – produz resposta máxima que o receptor é capaz de dar (mesma resposta que o mediador endógeno).
Agonista parcial – considerado de baixa eficácia – não produz efeito máximo, mesmo quando todos os receptores no tecido parecem estar ocupados.
Existem vários fármacos que são agonistas parciais, como os beta bloqueadores.
A diferença desses fármacos está na eficácia, ocupação e resposta.
Afinidade: capacidade de se ligar ao receptor.
Eficácia (ou atividade intrínseca) – capacidade de, uma vez ligado, induzir uma resposta (não confundir com eficácia clínica – está sendo falado do
receptor).
11
Potência: quantidade de droga necessária para produzir determinada resposta (quanto menor a quantidade maior a potência).
Agonista Inverso:
Gaba A: canal iônico regulado por voltagem (interior do canal é positivo). Quando o aminoácido gaba se liga, ocorre uma hiperpolarização (diminui a
excitabilidade).
Diazepam: é uma droga básica. Em caso de intoxicação – acidificar a urina
Flumazenil: usado para tratar intoxicação por diazepam – se liga no mesmo sítio – competição – antagonista.
Betacarbonila: efeito agonista inverso – ao invés de abrir o canal iônico, fecha o canal – melhora da memória – porém causa ansiedade e convulsão.
Essa droga tem eficácia negativa.
Diazepam – agonista do receptor Gaba A, porém em um outro sítio de ligação. Quando o gaba (aminoácido inibitório) se liga, ocorre uma grande
entrada de íons cloro.
No início do tratamento ocorre amnésia temporária, sono, perda de memória, diminuição da agressividade. Hoje se sabe que muitas drogas são
agonistas inversos e não antagonistas. Ex.: antihistamínicos – efeito inverso da histamina.
19/03/09
Agonistas ocasionam um nível de ativação positiva do receptor e são considerados de eficácia positiva
Agonistas inversos ocasionam um nível de ativação do receptor negativo e, portanto esses fármacos são chamados de eficácia negativa.
Os antagonistas têm eficácia zero, embora tenham afinidade. Os agonistas têm eficácia, afinidade diferente de potencia. Os agonistas inversos têm
eficácia negativa.
AFINIDADE: capacidade de se ligar ao receptor (todos têm)
O fármaco pode se ligar através de ligações reversíveis ou irreversíveis (lig. duradoura, tempo de ação maior do fármaco)
TOLERÂNCIA:
Começou usando uma dose que fazia efeito e depois de um tempo usando aquela mesma dose, houve perda da eficácia clínica. Ocorreu o fenômeno
de tolerância, é preciso aumentar a dose para ter o mesmo efeito, aquela dose que era usada não dá mais o mesmo efeito. Pq isso acontece? Acontece
por processos de dessensibilização desses receptores
Pq o paciente fica depende da nicotina? Ocorre uma alteração na conformação da pt (receptor onde a nicotina se liga no cérebro).
Down regulation: a cel começa a internalizar o receptor. Será preciso doses maiores, ou o down regulation já é algo esperado, é preciso que ele ocorra,
é o que ocorre com os antidepressivos, pois o que faz efeito é a neuroadaptação com o uso crônico.
12
INDUÇÃO FARMACOCINÉTICA: quando um fármaco induz seu próprio metabolismo; os fármacos indutores enzimáticos além de aumentarem o
metabolismo do segundo fármaco tomado ao mesmo tempo, eles podem aumentar o seu próprio metabolismo. Isso acontece com o álcool no uso crônico,
barbitúricos, alguns anticonvulsivantes como a fenitoína, a carbamazepina.
ADAPTAÇÃO FISIOLÓGICA: pode acontecer adaptação fisiológica ao efeito do fármaco, o organismo pode compensar aquela alteração na sua
função com ativação de uma outra função fisiológica. Por ex., diuréticos tiazídicos, no começo é possível perder bastante sódio com um grande volume
de urina, mas depois o corpo começa a reter esse sódio por outros mecanismos de adaptação fisiológica, daí não se não tem o mesmo efeito com a
mesma dose.
FESTA DOS CAMUNDONGOS
ÁLCOOL:
Neurotransmissão normal no cérebro do rato aparece uma sinapse inibitória, o Gaba está se ligando na célula pós sináptica no receptor Gaba A; abre
canais de íon cloreto (canal iônico regulado por ligante), hiperpolarizando e inibindo a cel.
Em outra área do cérebro e antes do uso do álcool existe uma neurotransmissão excitatória. Com liberação de glutamato que se ligam nos seus
receptores MNDA (canal iônico regulado por ligante).
O alcool é um depressor do SN, pois ele estimula a sinapse inibitória (se liga em um sitio de ação diferente do gaba, aumentando o efeito do gaba
quando este se liga ao receptor) e faz efeito antagonista sobre a sinapse excitatória (se liga ao sitio de ligação do glutamato).
O córtex frontal e o límbico são os mais inibidos pelo álcool, o que resulta em perda da coordenação motora, dificuldade de raciocínio, capacidade de
decisão e controle do impulso.
COCAÍNA:
A cocaína atua sobre uma molécula transportadora.
No cérebro do rato está mostrando uma neurotransmissão dopaminérgica, a liberação de dopamina na via da recompensa, que traz sensação de
prazer. Então, normalmente a dopamina é liberada, estimula seus receptores na via da recompensa, dá a sensação de prazer por vários motivos, como,
por ex., quando come um chocolate, no sexo, no jogo, tudo isso é liberação de dopamina. Pq as drogas de abuso fazem com que a pessoa queira usar
de novo? pq todas elas fazem com que a pessoa tenha liberação aumentada de dopamina. E quem fica dependente? Geralmente, quem não tem
sensação de prazer, não tem a via da recompensa estimulada pelos prazeres da vida.
E o que que a cocaína faz? Ela é uma das drogas que tem um poder de dependência muito grande. Ela inibe a recaptação da dopamina na via da
recompensa.
RECEPTORES FARMACOLÓGICOS - FARMCODINÂMICA
Todos as proteínas que respondem por ligantes endógenos podem ser classificados em receptores do tipo 1, 2, 3 e 4
Receptor ionotrópico: qdo um fármaco ou um ligante endógeno se liga o canal irá abrir, a cel pode ser hiperpolarizada ou despolarizada dependendo
da carga do íon que entrar, isso irá resultar nos efeitos celulares e o tempo de resposta de um receptor como este é milissegundos. Os efeitos são
inúmeros.
Receptor metabotrópico: são os mais importantes na farmacologia, devido a quantidade de receptores no organismo. Todos os receptores adrenérgicos,
dopaminérgicos, serotoninérgicos (exceção 1 da serotonina), da histamina, canabinóides, e muitos outros. Sempre que um fármaco se liga a este
receptor uma pt G será um intermediário entre o fármaco e a resposta. Ela vai levar a informação, a pt G pode ativar enzimas na membrana celular
aumentando a produção de segundo mensageiros, pode promover abertura e fechamento de canais iônicos levando a efeitos celulares. Esses receptores
levam segundos para dar uma resposta.
Receptor ligado a Tirosina Quinase: receptor com atividade enzimática, sempre que um fármaco se liga será ativado uma cascata de fosforilação ptca
nessa cel que levara a alteração da transcrição gênica e alteração da síntese ptca, portanto a resposta será mais demorada, a resposta pode levar
horas.
Receptores Intracelulares: podem estar no núcleo ou no citoplasma da cel, portanto os ligantes têm que ser altamente lipofílicas. Qdo o ocorre a ligação
do ligante ou do fármaco com o receptor, o receptor reage com o DNA da cel, - vai alterar a transcrição gênica e a síntese ptca. Todos os hormônios
esteróides, tiroideanos, a vit D, o ac retinóico, todos se ligam nesse tipo de receptor. Estudaremos em detalhe os glicocorticóides.
Como eles se diferem estruturalmente?
13
TIPO 1: 4 ou 5 subunidades que atravessam a membrana 4 ou 5 vezes, ou seja, eles se arranjam de forma oligomérica. Ex: receptor nicotínico da Ach.
TIPO 2: receptor de uma única cadeia polipeptídica, e essa pt atravessa 7 vezes a membrana celular, são conhecidos como receptores heptahelicoidais.
A terceira alça da pt faz interação com a pt G.
TIPO 3: tem uma única hélice atravessando a membrana, tem um domínio de ligação na parte externa (alias, os três têm) e seu domínio catalítico (sua
enzima) está acoplado a ele na sua parte interna. Qdo ele é ativado, ocorrerá uma cascata de fosforilação ptca. Um fármaco muito importante que se
liga nesse receptor é a insulina, que se liga a receptores como esse nas cels musculares, nos hepatócitos, nos adipócitos. Com alteração de síntese ptca
(aumento da síntese das gluts) nessa cel ocorrerá maior captação de glicose.
TIPO 4: estão totalmente dentro da cel, tanto o domínio de ligação quanto o domínio de ligação com o DNA. Qdo acontece ligação do fármaco com o
receptor acontece alteração na conformação da pt, ela expõe dedos de zinco e se liga ao DNA da cel alterando a transcrição gênica.
YOUTUBE – RECEPTOR NICOTÍNICO
Como é um receptor nicotínico na visão tridimensional
Farmacocinética, processo de absorção, distribuição
Ação de um agonista, fazendo o mesmo papel da Ach
Processo de dessensibilização do receptor – a nicotina diferente a Ach se liga no receptor e não desliga. A Ach se liga e desliga em milissegundos
devido a ação da acetilcolinesterase. Isso leva a alteração na conformação dos receptores e eles param de responder, a cel por sua vez começa a
expressar mais receptor o que faz com que seja preciso cada vez mais nicotina.
RECEPTOR DO TIPO 2
O receptor é constituído por uma única cadeia polipeptídica, essa pt atravessa 7 vezes a membrana celular, ela tem um domínio terminal extracelular
onde o fármaco se liga e tem um domínio, que é a terceira alça citoplasmática, onde tem interação com a pt G. Sempre que o receptor é ativado, essa
alça sofre mudança conformacional e ativará a pt G.
A pt G é formada por três subunidades, alfa, beta e gama. A subunidade alfa tem interação com o nucleotídeo de guanina que faz essa subunidade
ficar ativa quando a pt G é ativada, ela tem atividade enzimática, faz a hidrólise do GTP em GDP.
A pt G tem a capacidade de amplificar o sinal que ela recebe do receptor, se difundindo pela membrana, ativando outros alvos e em seguida voltar
ao repouso.
Alvo 1 e alvo 2 em uma membrana celular: pode ser uma enzima, pode ser um canal iônico.
Qdo um fármaco ou um ligante endógeno se liga ao receptor, a pt G é ativada e muda sua conformação, começa a se difundir pelo plano da
membrana, a subunidade alfa se separa da beta e da gama. Ocorre a troca do nucleotídeo GDP pelo GTP, essa subunidade se difunde e atinge uma
outra pt na membrana, que pode ser uma enzima ou um canal. As subunidades beta e gama podem atingir um segundo alvo, uma outra enzima ou um
outro canal. Depois de exercer sua ativação a pt G hidrolisa o GTP em GDP e a subunidade alfa volta a sua conformação e ao seu estado de repouso
junto com as outras subunidades. Isso tudo ocorre em milissegundos, mas a resposta do receptor leva segundos.
A cada tipo de receptor existem diferentes tipos de pts G, para que existam diferentes respostas.
GS- estimulatória- sempre estimula a adenilato ciclase - formação de AMPc (segundo mensageiro)
GI- inibitória – sempre inibe a adenilato ciclase
Gq- ativação da enzima fofolipase C (IP3 e DAG)
Pode agir sobre canais iônicos
Um novo alvo descoberto para a pt G é o Rho A/Rho quinase: controla muitas vias de sinalização, crescimento, proliferação celular, contração de
musculatura lisa e etc.
O AMPc é o 6’5’ adenosina monofosfato cíclico. A adenilato ciclase se liga ao ATP forma o AMP cíclico, o qual é degradado pela fosfodiesterase em 5’
AMP que não tem mais ação na célula. Existem substancia que são inibidoras da fosfodiesterase, a cafeína, o Viagra (ação vasodilatadora)
O AMPc está envolvido em ativação de enzimas que controlam o metabolismo energético, por exemplo, a quebra das moléculas de gordura, o AMPc
ativa as enzimas que fazem a lipólise. Está envolvido na ativação das pts contrateis do músculo liso, no transporte de íons.
14
Sempre que o AMPc for formado existira a ativação da PKA, pt quinase que ativarão outras pts.
Ex., broncodilator – beta 2 adrenérgico localizado em vários locais do corpo,porém abundante na musculatura lisa dos alvéolos pulmonares. Quem é o
receptor endógeno desse receptor? Adrenalina, que faz broncodilatação.
Quando a adrenalina se liga a esse receptor metabotrópico, ligado a uma pt G S, qdo a pt G é ativada vai ativar a adenilato ciclase, qdo a
adenilato ciclase é ativada ocorre aumento da produção de AMPc, o AMPc ativado vai ativar a PKA, a PKA vai fosforilar uma pt contrátil nessa cel,
são as miosinas de cadeia leve, inativa a pt, portanto tem um relaxamento da musculatura lisa.
Pt Gq – qdo a pt Gq é ativada ela ativa a enzima fosfolipase C que fica na membrana. Ocorre a produção de IP3 que é hidrossolúvel, se solubiliza no
citoplasma, vai no RE da cel e se liga a um canal iônico fazendo com que o Ca que estava armazenado no RE seja liberado para o citoplasma. E
sempre que um fármaco é um aumentado no citoplasma de uma cel contrátil a cel irá se contrair, de uma cel secretora ela ira se contrair. O DAG se
difunde pelo plano da membrana e vai ativar a PKC que vai ativar uma cascata de fosforilação ptca e vai dar um efeito.
Ex., alfa 1 adrenergico na musculatura do vaso que envolve vísceras e outros tecidos (não na musculatura esquelética) – na presença de adrenalina
vasoconstrição. A adrenalina se liga ao receptor acoplado a pt Gq que vai produzir DAG e IP3, liberação de Ca no citoplasma e contração da
musculatura do vaso.
Nafazolina, oximetazolina (aturgil): agonistas Alfa 1- o nariz entope devido a um processo inflamatório, vasodilatação, aumento da permeabilidade
vascular, extravasamento de líquidos dos vasos para o tecido e inchaço da mucosa. Problema: vasodilatação de rebote; não pode ser usado mais de 3
dias consecutivos; pode causar lesão e necrose do tecido.
Canais Iônicos: a pt G pode ativar canais iônicos. O receptor muscarínico 2 no coração (Ach). A pt G não ativa a produção de nenhum segundo
mensageiro, mas qdo ativa estimula a abertura de canais de potássio e o K começa a deixar a cel hiperpolarizando a cel, perdendo a força de
contração, ou seja, o coração entra em bradicardia. Os fármacos que fazem isso são a pilocarpina que é usado no tratamento do glaucoma, betanecol
para incontinência urinária, a atropina que é um antagonista deste receptor que é usado em parada cardíaca causa taquicardia, pois não deixa que a
Ach se ligue no receptor, tem se somente o efeito da adrenalina.
O terceiro tipo de receptor são os ligados a quinases: eles têm uma única hélice transmembrana e um domínio intracelular de natureza enzimática.
Receptores de fatores de crescimentos, da citocina, de insulina, de fatores natriuréticos natural.
Desenho do receptor de insulina: qdo a insulina se liga o receptor se dimeriza (é só uma hélice transmembrana antes da ligação) e ativa a tirosina
quinase que leva a uma cascata de fosforilação ptca que leva a alteração da síntese ptca.
Receptores intracelulares: tem efeito de inicio lento, todos os ligantes são lipofílicos. Ex. estrógenos, progesteronas, glicocorticóides, ac retinoico, vit D,
esteróides.
Receptor glicocorticóides: qdo ativado inibe a expressão da COX 2 que produz os mediadores da inflamação.
Quando o glicocorticóide se liga no receptor, o complexo age sobre o DNA e inibe a expressão da COX.
25/03/09
INTERAÇÃO FARMACOLÓGICA - PROVA
Como resolver o problema de interação?
Saber onde buscar a informação, o que eu vou fazer se sei que o paciente pode apresentar o problema?
As interações podem ser farmacocinéticas ou farmacodinâmicas.
Se esta tomando um fármaco que deprime o SN,a um outro com a mesma ação irá aumentar o efeito depressor.
As interações farmacocinéticas são mais difíceis de prever, são as que envolvem os processos de absorção, distribuição, metabolismo hepático e
excreção renal.(muda)
As classificações do Goodman são as mais recomendadas, pois são as mais completas.
As interações farmacêuticas, qdo um fármaco interage com outro na farmacotécnica, não serão estudadas.
Efeitos sinérgicos: um fármaco aumenta o efeito do outro na interação.
15
Efeitos antagônicos: quando um fármaco diminui o efeito do outro.
Vc pode impedir o efeito de um fármaco com o outro.
Interações farmacodinâmicas: qdo um fármaco pelo seu mecanismo de ação aumentar ou diminuir o efeito do outro, essa interferência é independente
de receptor ou no receptor. Qdo vc tiver dois fármacos brigando pelo mesmo receptor e um bloqueando o efeito do outro é uma interação
farmacodinâmica independente do receptor. Se vc usar um fármaco que aumente sua pressão e outro abaixa, sendo que eles atuem em diferentes
receptores o efeito será anulado.
Interação farmacocinética: qdo um fármaco alterar a biotransformação, distribuição, absorção e excreção do outro.
Podem ser independentes ou dependes do receptor.
Classificação dos efeitos:
O resultado da interação foi um aumento do efeito do fármaco ou foi uma diminuição?
Se for aumento pode ser:
Adição, sinergismo ou potencialização.
Se for uma diminuição de efeito:
Antagonismo (que pode ser funcional, químico, disposicional, ou receptor).
Efeito Aditivo: Diazepam causa sono, antihistamínico causa sono
Sinergismo: diazepam com álcool – depressão cardiorrespiratória –em altas doses- (atuam no mesmo receptor).O efeito de dois fármacos combinados é
mto mais forte do que simplesmente os dois fármacos separados.
Efeito potencializador: warfarina e um inibidor enzimático que inibe o metabolismo da warfarina, cimetidina – a cimetidina aumenta o efeito
hemorrágico da warfarina.Utilizo dois fármacos, quando o fármaco número dois aumento o efeito do número um.
Fármaco A e B um aumentou o efeito do outro ou os dois aumentaram.
Antagonismo: um anula o efeito do outro ou diminui.
Fisiológico:
Entre drogas: O antagonismo entre drogas surge quando o efeito de uma droga é diminuído ou abolido em presença de outra.
Farmacocinético: Diminuir ou abolir a função do outro...
Indutores enzimáticos irão acelerar o metabolismo de fármacos que são metabolizados pelo citocromo P450.
Químico: interação química dentro do organismo. Envolve uma interação química direta entre agonista (droga que faz efeito) e o antagonista (impede a
ação droga, diminui o efeito ou tóxico ou famacológico do agonista). NÃO TEM NADA A VER COM EFEITO AGONISTA E ANTAGONISTA EM
RECEPTOR.
O antagonista vai inativar o efeito agonista por reagir quimicamente com ele dentro do organismo.
Usado na prática para tratar intoxicações como chumbo ou cádimo com agentes quelantes, um deles é o dimercaprol, o dimercaprol forma um quelato,
um chumbo ou um Cádimo que diminuem a passagem desses metais pesados pelas membranas biológicas, então se eu tiver uma intoxicação por via oral
de Cádmio ou de chumbo, o que não é normal eu vou fazer uma reação de quelação no intestino com o de dimercaprol, dessa forma eu diminuo a
absorção intestinal de Cádmio e chumbo.Se ele estiver no meu sistema respiratório, eu faço uma reação de quelação e diminuo a absorção para a
corrente sanguínea.
Por que é uma interação química? Porque eles formam um relato.
Por que um antagonismo? Porque o dimercaprol impediu a ação tóxica do cadimo e do chumbo.
Antagonismo químico? Um fármaco aboliu o efeito do outro por agir quimicamente com ele dentro do organismo vivo.
Muito difícil vc perder totalmente o efeito vc vai diminuir o efeito farmacológico ou tóxico.
16
O dimercaprol não tem atividade farmacológica importante a única função dele é formar o quelato.Ele vai inativar o efeito do cádimo e do chumbo.
Mas, o pouco dele que for absorvido não me dá uma ação farmacológica significativa.
Qdo um anatagonismo é farmacocinético? Qdo um farmaco vai diminuir ou abolir a ação do outro por diminuir a absorção, por aumentar o
metabolismo ou por acelerar a excreção. Então, eu vou ter o antagonismo farmacocinético quando interferir com essas três ações. É o tipo mais comum
de antagonismo é o farmacocinético. Então, a maioria dos fármacos que são indutores enzimáticos, que a gente já estudou, como o barbitúrico, a
carbamazepina, a fenitoína, eles vão acelerar o metabolismo de fármacos que são metabolizados pelo cyp 450. E o que que vai acontecer com o
efeito desses fármacos? Tendem a diminuir. E existe alguma exceção a essa regra? Existe algum caso em que a indução enzimática leve a toxicidade e
não a perda de eficácia? Sim, qual é o exemplo clássico? O paracetamol associado ao indutor enzimático, pq o metabolito que é formado dessa
indução é tóxico, se acelerar a produção desse metabólito tóxico existirá um aumento do efeito desse metabolismo.
Como eu classificaria então a interação do paracetamol com um indutor enzimático qualquer? Interação farmacocinética, pois está acelerando o
metabolismo do paracetamol
Ocorreu um aumento do efeito do paracetamol? Qual foi o efeito? Causar uma lesão hepática. Se esse indutor enzimático não causa lesão hepática,
simplesmente aumenta a lesão hepática do paracetamol, o que é isso: adição, sinergismo, potencilização? Potencialização.
Antagonismo farmacocinético: tetraciclina associada com alimento; pq não pode tomar tetraciclina com a refeição, com leite, com antiácido (hidróxido
de alumínio ou de magnésio)? Pq esses cátions divalentes (Fe, Al, Mg, Ca) formam um quelato com a tetraciclina impedindo a absorção dela a nível
intestinal. Como impedem absorção, isso é uma interação farmacocinética. Essa também é uma interação química, é um antagonismo químico, pois forma
quelato.
O que seria um antagonismo fisiológico?
Esse antagonismo nós veremos na aula prática, na pupila do coelho, é qdo eu uso dois fármacos que tem efeitos opostos e eu anulo o efeito do outro.
Ex., administração de adrenalina, efeito vasoconstritor e ao mesmo tempo eu administrar histamina que tem efeito vasodilatador, o resultado é nulo. Nós
veremos isso na pupila do coelho, serão administradas drogas que causam miose e drogas que causam midríase, se eles não causarem esses efeitos por
atuarem no mesmo receptor, mas atuarem em receptores diferentes eu vou ter um antagonisto fisiológico.
O que é antagonismo competitivo? É um antagonismo dependente de receptor. Um fármaco A vai ser administrado junto com um fármaco B, agora sim
eu estou usando um fármaco agonista do receptor x junto com um fármaco antagonista do receptor x. Os dois fármacos vão competir para se ligar no
receptor. Se o antagonista ganhar a competição ele irá anular o efeito do agonista, essa ligação que o antagonista faz com o receptor pode ser
reversível ou irreversível.
O antagonismo reversível é o tipo mais comum e mais importante de antagonismo.
Slide 14:
Ocupação fracional (quanto que o antagonista está se ligando no receptor de determinado tecido) em diferentes concentrações de agonistas.
A primeira linha azul representa somente um agonista (sem presença de antagonista na solução). Conforme vai aumentando as doses do agonista, vai
ocupando 100% dos receptores do tecido.
Num segundo momento é dado uma dose de um de antagonista, e para ocupar 100% dos receptores é preciso aumentar a concentração dos agonistas.
Conforme a dose de antagonista aumenta, a concentração de agonista também precisa aumentar para que 100% dos receptores sejam ocupados. Isso
significa que a ligação é reversível.
Ver slide 17: antagonismo irreversível. Qdo o efeito do antagonista vai acabar? Ou qdo ele for metabolizado ou qdo o receptor for metabolizado ou
internalizado. Qual a importância prática? Se eu tiver uma intoxicação por um antagonista irreversível, eu vou ter como tratar isso com antídoto? Não,
pq a ligação é forte, não tem como desfazê-la. Se eu tiver uma intoxicação com um antagonista reversível eu tiver um agonista para competir, eu
consigo tratar a intoxicação, não só a do antagonista como a do agonista.
O benzodiazepínico é um agonista do receptor GABA A, se eu der um outro agonista ele irá competir com o benzodiazepínico e tratar a intoxicação.
O que é um antagonismo não competitivo?
Vamos supor que eu tenha uma droga A e uma droga B. essas duas drogas fazem o mesmo efeito (contração) na cel (cel de musculatura lisa em volta
de um vaso). Qdo o farmaco se liga no receptor, ocorre uma despolarização, entrada de Ca e contração celular. Só que o fármaco A se liga no
receptor A e o B no receptor B. Durante essa terapia eu acrescento o fármaco X, que compete à ligação ao receptor B. Agora eu resolvo acrescentar
um fármaco y, que impede a entrada do Ca no interior da cel. O fármaco y é um antagonista não competitivo tanto de A quanto de B (impede que o
evento aconteça). Ex. nifedipida, bloqueadora do canal de Ca, vasodilatadora, tratamento da hipertensão. A nifedipina impede a entrada de Ca, se
eu estiver tomando a nifedipida com qualquer fármaco que tenha por efeito aumentar a concentração de cálcio no citoplasma, o efeito do segundo
fármaco será abolido. É um antagonismo não competitivo pq não está ocorrendo uma competição pela ligação no receptor; estão impedindo que um
efeito decorrente do estímulo aconteça.
Qdo uma interação farmacocinética é importante?
Se essa interação depende de alterações nos níveis plasmáticos no sangue, qto mais estreita for a janela terapêutica desse fármaco mais perigosa será
essa interação. Pq? Pq se eu diminuir a concentração no sangue ele não vai fazer efeito. E se eu aumentar muito ele vai dar toxicidade. Então, fármaco
17
de janela terapêutica estreita eu tenho que me preocupar muito mais com as interações farmacocinéticas, pq vão estar alterando as concentrações dele
no sangue principalmente.
A penicilina tem uma margem de segurança muito grande. Eu posso trabalhar muito com variações de concentração plasmática que eu não vou ter
toxicidade, inclusive aumentar a concentração plasmática dela pode ser benéfica, pq eu prolongo o efeito e dou doses menores.
É o que se faz qdo associa-se penecilina com probenecida, para dar doses menores num intervalo maior entre as doses.
Janela terapêutica estreita: pode perder eficácia ou produzir toxicidade.
EXEMPLOS DE INTERAÇÕES FARMACOCINÉTICAS
Retardar ou acelerar o esvaziamento gástrico modifica a absorção do fármaco? Sim, pois o fármaco foi elaborado para permanecer um determinado
tempo dentro do organismo, se permanecer mais tempo no estomago pode ser hidrolisado, se o processo for acelerado ele pode ser excretado junto
com as fezes.
As interações químicas podem retardar a absorção, ex., colestiramina (usada no tratamento da hiperlipidemia, diminui a absorção do colesterol que
vem da dieta) e conseqüentemente, diminui a absorção da warfarina. É uma interação farmacocinética (diminuiu a absorção); classificação do efeito:
quando impede a absorção da gordura do colesterol impede a absorção da warfarina que precisa dessa gordura para ser absorvida. A warfarina vai
embora com as fezes pq ela é lipossolúvel. Que tipo de antagonismo é? Não é químico, pois não tem interação química da colestiramina com a
warfarina. É farmacocinético.
Sulfa (antibiótico) e hidrato de cloral (anestésico geral) se os dois medicamentos forem dados para uma cça, os dois vão competir por uma mesma
ligação a pt e um deles vai ficar livre, no caso o hidrato de cloral tende a ficar mais livre, ele é um depressor do SNC e tem janela terapêutica estreita.
Como ele está mais livre, pode levar a uma depressão respiratória da cça. Interação farmacocinética, pq alterou distribuição. Classifique o efeito: foi
um aumento do efeito (aumentou a depressão provocada pelo hidrato de cloral). É adição? Não, pq a sulfa não causa depressão do SNC; Sinergismo
muito menos, então só pode ser potencialização. Ela aumentou o efeito depressor do hidrato de cloral.
A indução enzimática pode causar toxicidade, ex. paracetamol.
Ex. o cloranfenicol inibe o metabolismo da fenitoína (anticonvulsivante), só que se eu aumentar muito os níveis dela no sangue eu tenho convulsão.
Interação farmacocinética. Efeito: aumento. Seria adição se o cloranfenicol também causasse convulsão. Seria sinergismo se a soma do efeito dos dois
fosse muito maior que o efeito isolado, mas aqui ele não causa. Ele só aumentou o efeito da fenitoína (potencialização).
No slide 36: as interações são farmacocinética e do tipo potencialização (a não ser que algum causa problema de estomago e seja adição).
INTERAÇÕES DE FÁRMACOS COM ÁLCOOL
É um problema de saúde pública. Doutor posso beber? Que tipo de uso o Sr (a) faz do álcool?
Álcool uso crônico: indutor enzimático (alcoolista, mas não perdeu a função hepática – é preciso aumentar a dose para ter efeito).
No uso crônico o álcool causa uma tolerância farmacocinética, ele induz o seu próprio metabolismo. Esse é um dos motivos que o paciente fica
dependente, ele precisa beber cada vez mais para ter as mesmas concentrações para o SNC de álcool. O mesmo ocorre com os fármacos que são
metabolizados pela mesma via do álcool.
Álcool uso agudo: inibidor Enzimático – Efeito aumentado de muitos fármacos.
No uso agudo o fígado não está acostumado a metabolizar grandes quantidades de álcool, desta forma as enzimas ficam saturadas e não sobra
enzima para metabolizar o fármaco que esta sendo administrado.
Álcool é depressor do SNC
O álcool pode fazer tanto interação farmacocinética quanto farmacodinâmica.
Interação farmacocinética: indução ou inibição enzimática (uso agudo ou crônico). Aqui entram os fármacos que são metabolizados pela enzima CYP
450.
Interação farmacodinâmica: interação aditiva com outros agentes depressores é a mais importante. O álcool é depressor, se meu paciente tiver que usar
um barbitúrico, um benzodizepínico, isso é grave.
Pratica: interação álcool com antihistamínico, veremos que o rato perderá toda a cordenação motora.
USO CRÔNICO DE ÁLCOOL
Interações farmacocinéticas: dar aas e paracetamol para esses pacientes é extremamente prejudicial. O paracetamol lesa mais o fígado, o aas causa
gastrite e úlcera (de todos os AINES é o que mais causa gastrite e úlcera). Para esses pacientes se prescrece dipirona, ibuprofeno,
18
E outros fármacos podem interferir com o metabolismo do álcool? (slide 45)
O álcool é metabolizado por duas enzimas importantes. A álcool desidrogenase (enzima limitante), enzimas do citocromo P 450, pela catalase, que
transformam o etanol em acetaldeído.
As mulheres são menos tolerantes ao álcool, pois a alcetadeído desidrogenase é mais lenta e acumula mais acetaldeído. O acumulo de alcetadeído leva
a taquicardia, náusea, vômitos, dor de cabeça, vermelhidão no rosto. Se eu estiver tomando um fármaco que iniba a álcool desidrogenase, irei
acumular no organismo o álcool e terei todos os efeitos do álcool aumentado.
Existem os fármacos que inibem a alcetadeído desidrogenase, daí o paciente tem a síndrome do dissulfiram. Não pode tratar o paciente com
dissulfiram sem que ele saiba, pois se ele tomar uma dose muito grande ele pode ter uma parada cardio-respiratória.
Qual é a interação do álcool com o ácido etacrínico? Farmacocinética (inibe o metabolismo do álcool), potencialização. É a mesma coisa para todos os
fármacos que inibem a acetaldeído desidrogenase, porque aumenta todo o efeito tóxico do metabólito do álcool.
26/03/2009
INTERAÇÕES MEDICAMENTOSAS
O site do drugdigest é um site para população, portanto não tem informação detalhada. A vantagem é que já chega interação com alimentos e álcool.
Entrar na página do medscape e fazer login. Entrar no Drugs e digitar o nome do medicamento.
Indicações clínicas, dosagem máxima do medicamento, outras indicações (all label), interações medicamentosas com grau de severidade, cuidados com
alguns grupos de pacientes, efeitos adversos, contra-indicações.
Pode entrar na monografia detalhada (ao lado).
NUNCA BUSCAR INFORMAÇÃO DE UM MEDICAMENTO NA BULA. O site tem que ser independente da indústria farmacêutica.
Entrar no Drug interaction Checker, selecionar o medicamento na caixa da esquerda e mandá-lo para a da direita, em seguida
Interação e mecanismo dela.
Classificar a interação: farmacocinética ou farmacodinâmica. Se o fármaco alterou processo de absorção, distribuição, metabolismo e excreção do outro
é sempre farmacocinética.
As demais são dinâmicas: o fármaco está interagindo com o mecanismo de ação.
Se houve um aumento do efeito vai ser ou adição, ou potencialização ou sinergismo.
Se houve uma diminuição de efeito vai ser antagonismo: ou antagonismo químico, fisiológico, farmacocinético, competitivo ou não competitivo.
A warfarina é um anticoagulante oral e a heparina é um anticoagulante injetável.
A warfarina é usada para tratar cronicamente um paciente que tenha uma trombose ou uma embolia, a heparina é usada para tirar o paciente do
quadro agudo de trombose ou embolia, ou seja, é usado a nível hospitalar. Depois é feito um tratamento crônico para impedir a formação de novos
trombos com a warfarina. A warfarina leva até 48h para agir e a heparina é de ação imediata.
Paciente que faz uso freqüente de warfarina tem que se proteger de acidentes.
WARFARINA ORAL X ERITROMICINA ORAL:
Interação severa. Podem interagir baseados na interação potencial que existe entre anticoagulantes seletivos e antibióticos macrolídeos seletivos (classe
química de antibióticos).
Nível 2 – interação severa.
Mecanismo de ação da interação: antibióticos macrolídeos podem inibir o metabolismo dos anticoagulantes.
Efeito Clínico: o uso corrente o antibiótico macrolídeo pode resultar num aumento do efeito de hipoprotrombinemia com possibilidade de sangramento.
19
Eritromicina inibe o metabolismo da warfarina aumentando o efeito da hemorragia.
Qual o tipo de interação? Farmacocinética – um fármaco inibindo o metabolismo do outro.
Qual o resultado? Aumento ou diminuição do efeito? Aumento.
Então é um sinergismo, potencialização ou adição? Potencialização.
Por que não é sinergismo e nem adição? Porque hemorragia é só a warfarina que causa. Eritromicina não. A eritromicina só está aumentando o efeito
da warfarina (a qual é responsável pela hemorragia).
TRANILCIPROMINA X FLUOXETINA:
A tranilcipromina é o inibidor da MAO (monoaminooxidase – enzima) – enzima que degrada a serotonina.
A fluoxetina inibe a recaptação da serotonina.
O que acontece com a quantidade de serotonina? Aumenta muito, síndrome serotoninérgica (agressividade, hipertermia que pode levar ao óbito,
convulsão, irritabilidade, etc).
Que tipo de interação? Farmacodinâmica (um fármaco é inibidor da MAO e o outro que é inibidor seletivo da recaptação de serotonina).
Qual o resultado? Aumento ou diminuição do efeito? Aumento
Então é um sinergismo, potencialização ou adição? Sinergismo
Pq não é potencializaçã? Pq os dois aumentam os níveis de serotonina. O autor classifica como sinergismo porque o efeito é muito acentuado. Se a
pessoa tomar uma caixa de tranilcipromina não terá a síndrome serotoninérgica, se tomar uma caixa de fluoxetina, também não terá a síndrome
serotoninérgica, mas se tomar a dose terapêutica de tranilcipromina e fluoxetina, a pessoa terá a síndrome serotoninérgica (é um efeito muito maior).
Interação de severidade alta: Se a pessoa estiver tomando o inibidor da MAO tem que esperar alguns dias para poder tomar fluoxetina.
TETRACICLINA E HIDRÓXIDO DE ALUMÍNIO
A Tetraciclina forma quelatos, complexos com cátions divalentes, como por exemplo, com o alumínio, com o cálcio, com o ferro.
Será formado um quelato de tetraciclina com alumínio no intestino.
Qual o resultado? Diminui a absorção da tetraciclina, portanto, diminuição do efeito da tetraciclina – Antagonismo
Que tipo de antagonismo? Químico, pois teve uma interação química, e farmacocinético, pois diminuiu a absorção.
Qual o tipo da interação? Farmacocinética (quando um fármaco diminui a absorção do outro).
FENITOÍNA E ANTICONCEPCIONAL ORAL (etilenoestradiol)
A fenitoína é exemplo clássico de anticonvulsivante (bloqueador de canal de sódio), exemplo clássico de INDUTOR ENZIMÁTICO (fenitoína, barbitúrico,
carbamazepina são exemplos de indutores enzimáticos).
A fenitoína acelera o metabolismo do etilenoestradiol, acelera sua inativação. O que vai acontecer com o efeito do anticoncepcional? Vai diminuir sua
eficácia clínica.
Que tipo de interação é essa? Farmacocinética
O resultado é o aumento ou a diminuição do efeito? Diminuição, portanto é antagonismo.
Que tipo de antagonismo? Farmacocinético, está acelerando o metabolismo.
Para analisar as interações temos sempre que fazer as seguintes perguntas:
Um fármaco está interferindo com a absorção, metabolismo, distribuição e excreção do outro?
20
Sim – farmacocinética
Não – farmacodinâmica
O efeito é aumento ou diminuição?
Aumento – adição, potencialização ou sinergismo
Diminuição – antagonismo
ÁLCOOL ETÍLICO NO USO AGUDO E DIAZEPAM
O álcool no uso agudo funciona como um inibidor enzimático. O álcool é classificado como um depressor do SNC – é um agonista do GABA.
O diazepam também é um agonista do GABA.
O álcool no uso agudo diminui o metabolismo do diazepam. Nesse sentido o efeito do diazepam será aumentado.
Que tipo de interação? Farmacocinética
Qual o resultado dessa interação? Depressão acentuada do SNC que pode levar a uma depressão cardiorrespiratória e morte.
O álcool é depressor e o diazepam é depressor. Ou Será adição, ou sinergismo ou potencialização?
Não é potencialização porque os dois são depressores.
Segundo o autor é adição, porém a prof. não concorda. Ela diz que é sinergismo, pois se a pessoa tomar um porre não fará depressão
cardiorrespiratória, se tomar uma caixa de dizepam será muito difícil fazer depressão cardiorrespiratória, mas se tomar a dose terapêutica do
diazepam com um destilado isso pode acontecer.
Por que não se usa mais barbitúricos para tratar insônia, e sim diazepam? Porque o diazepam dificilmente levará a morte, entretanto, o barbitúrico se
tomado em dose maior faz parada cardiorrespiratória.
Sinergismo: tem um efeito muito mais alto na associação do que se tomar doses isoladas.
Nesse caso, também temos a interação farmacodinâmica, pois temos dois fármacos atuando no mesmo receptor e aumentando o efeito depressor.
(Efeito final sinergismo)
Para esse tipo de paciente temos que administrar imediatamente flumazenil, que é o antagonista do benzodiazepínico ( o álcool atua num sitio de
ligação e o diazepam em outro, porém no mesmo receptor).
ÁLCOOL E DISSULFIRAM
Dissulfiram: Usado para tratar intoxicação alcoólica, inibe o metabolismo intermediário do álcool, inibe a acetaldeídodesidrogenase, que metaboliza
um subproduto do álcool – o acetaldeído. Com a associação ocorrerá o acumulo de acetaldeído.
Qual o efeito esperado? Rushing (rubor, taquicardia, náusea, vomito)
Que tipo de interação? Farmacocinética
Qual o resultado? Aumento ou diminuição do efeito? Aumento (toxicidade)
Potencialização –
Não é sinergismo, nem adição, pois o dissulfiram não causa rubor, taquicardia, náusea, vômito.
PROPANOLO E EPINEFRINA
Quando se administra adrenalina em um paciente que está usando propanolol, pode desencadear uma crise hipertensiva.
21
O propanolol é um beta bloqueador (diminui debito cardíaco; no rim diminui liberação de renina; no SNC diminui a descarga do simpático para a
periferia)
A epinefrina é um agonista alfa e beta.
Se for administrado epinefrina sozinha, a pessoa terá broncodilatação, aumento da PA.
Receptor Beta faz vasodilatação e receptor alfa faz vasoconstrição.
Agora se der epinefrina para um paciente que está fazendo uso de propanolol (beta bloqueador) – a epinefrina só atuará nos receptores alfa levando
a uma vasoconstrição e, conseqüentemente, a um aumento muito maior da PA do que se eu desse a epinefrina sozinha.
Uso de Adrenalina: Choque anafilático, parada cardíaca – se o paciente estiver fazendo uso de propanolol pode fazer crise hipertensiva.
Que tipo de interação é essa? Farmacodinâmica (interferência no mecanismo de ação).
Qual o resultado? Aumento
Potencialização, sinergismo, adição? Potencialização
Não é sinergismo, nem adição, pois propanolol não causa hipertensão, muito pelo contrário ele é um antihipertensivo.
ANFETAMINA E HIDRÓXIDO DE ALUMÍNIO
O hidróxido de alumínio é um antiácido.
Quem toma anfetamina e antiácido pode ter uma toxicidade aumentada de anfetamina, porque ela é uma substancia de natureza básica. Se eu tomar
um fármaco que vai aumentar o pH do trato gastrointestinal. O grau de ionização do fármaco vai diminuir e a lipossolubilidade vai aumentar, ou seja,
será mais absorvido. Além disso (urina básica) a excreção será diminuída.
Uma pessoa que toma antiácido todos os dias terá a urina alcalina e um pH muito mais alto no trato gastrointestinal.
Que tipo de interação é essa? Farmacocinética – acelera a absorção e diminui a excreção.
Qual o resultado? Aumento de efeito
Potencialização.
Farmacologia do sistema nervoso autônomo parassimpático 01/04
Nós temos o sistema nervoso central e o sistema nervoso periférico.
O SNC inclue: cérebro e a medula espinhal
O SNP inclue: as fibras aferentes e eferentes.
Imporância na matéria: fibras eferentes- sistema nervoso autônomo e sistema motor somático
Fibras que sai do SNC e vão fazer comunicação com a periferia. Porque muitos fármacos tem ação em musculatura lisa, músculo cardíaco, músculo
esquelético e glândulas secretoras. Essas ações podem ser compreendidas e classificadas em termos de modificações das ações dos neurostransmissores
que são liberados pelo sistema nervoso autônomo.
Sistema nervoso periférico é divido em:
-sistema nervoso autônomo (incue sistema nervoso entérrico)
-sistema eferente somático (inerva a musculatura esquelética)
-sistema eferente somático visceral (não vamos estudar agora, anéstesicos e analgésicos opióides)
Sistema nervoso autônomo é divido em:
-Sistema parassimpático
-Sistema simpático
-Sistema nervoso entérrico
Processos regulados pelo sistema nervoso autônomo: contração e relaxamento da musculatura lisa, secreções glandulares exócrinas e algumas
endócrinas, metabolismo energético, no fígado, nos músculos esqueléticos. Veremos as principais drogas que interferem com essas funções do sistema
nervoso autônomo.
Diferença das fibras do Simpático x Parassimpático:
22
No Parassimpático as fibras saem da região crânio-sacral fazendo conexão com vários órgãos e tecidos.
No Simpático as fibras saem da região torácica – lombar fazendo conexão com vários órgãos e tecidos.
As fibras do sistema nervoso parassimpático- as fibras pré-ganglionares são longas, e vão fazer conexão com uma fibra pós- ganglionar que está
próxima ao órgão alvo. A razão de fibras é de 1:1. Isso significa que a resposta do parassimpático é direta, é uma resposta localizada.
No sistema simpático, temos uma fisiologia diferente, as fibras pré-ganglionares são curtas, e as fibras pós- ganglionares são longas, e a razão entre
fibras é de 1:20, então uma fibra pré no simpático pode até fazer conexões com 20 fibras pós. Então a resposta no simpático é mais difusa.
Funções
Parassimpático – contração da pupila
Estimula salivação (erro do desenho)
Bradicardia
Bronconstrição
Contrai a musculatura lisa- aumenta o peristaltismo de todo o trato gastro intestinal
Simpático- dilatação da pupila
Estimula a salivação idem
Taquicardia
Broncodilatação
Relaxa a musculatura lisa
Para que um neurotransmissor seja considerado um neurotransmissor clássico do sistema autonomo, ele precisa seguir alguns critérios:
- precisa ter enzimas de síntese e enzimas de degradação
-precisa ser armazenado em vesículas, porque se ficar livre no citoplasma vai sofre degradação e não vai ser liberado na chagado de um potencial de
ação.
- a liberação desses neurotransmissores é cálcio-dependente, para que essa liberação ocorra é preciso que ocorra cálcio no botão sináptico, para que
ocorra liberação das vesículas e dos neurotransmissores.
-esses neurotransmissores clássicos, geralmente, tem similares exógenos, substâncias sintéticas que tem efeitos semelhantes.
Quais são os dois principais neurotransmissores do sistema nervoso autonomo?
Acetilcolina e Noradrenalina.
Existem muitos outros neurotransmissores, que também são responsáveis pela resposta do simpático e do parassimpático, são conhecidos como
neurotransmissores NANC (não adrenérgico, não colinérgico).
Porque a adrenalina não é visto dentro desse grupo, ela também não é responsável pelo parassimpático? Porque na periferia a adrenalina não é vista
como um neurotransmissor, mas sim como um hormônio, pois é liberada pela supra renal na corrente sanguínea e vai exercer suas funções longe da onde
ela é produzida.
Não tem nos slides
- O sistema motor somático: o neurônio sai da medula espinhal e vai diretamente fazer conexão com uma célula muscular esqulética. Vai fazer conexão
na junção neuro-muscular. Esse neurônio sempre libera acetilcolina, e a acetilcolina vai fazer sua contração muscular sempre por atuar nos receptores
nicotínico que estão nas células musculares esqueléticas. Então um botão sináptico é simples: é a acetilcolina atuando em receptor nicotínico.
- No sistema parassimpático eu tenho uma fibra saindo da região crânio-sacral, a fibra pré-ganglionar é curta, SEMPRE VAI LIBERAR ACETILCOLINA
NO GÂNGLIO, e vai atuar nos receptores nicotínico na fibra pós-ganglionar, e essa fibra também sempre vai liberar acetilcolina, mas dessa vez a
acetilcolina vai atuar em receptores muscarínicos em diferentes órgão inervados pelos parassimpáticos.
-No simpático eu vou ter três situações diferentes: a principal e a mais comum é onde eu tenho uma fibra pré ganglionar curta liberando acetilcolina em
um gânglio simpático qualquer, a acetilcolina atuando em um receptor nicotínico e estimulando a fibra pós-ganglionar a liberar noradrenalina que é o
principal neurotransmissor do s. Simpático. A noradrenalina vai atuar em dois receptores específicos dela: alfa e beta adrenérgico. Porém, existem duas
exceções para essa regra: nas glândulas sudoríparas tem a fibra pós-ganglionar liberando acetilcolina e atuando em receptor muscarínico, na glândula
sudorípara a acetilcolina atua no receptor muscarínico estimulando a sudorese, é uma fibra simpática (só para lembrar). Outra exceção é na glândula
supra-renal que vai liberar adrenalina para corrente sanguínea, o estímulo vem do simpático, um neurônio sai da região torácica-lombar vai liberar
acetilcolina e vai atuar nos receptores nicotínicos lá na medula da supra-renal, mas a resposta disto vai ser a liberação da adrenalina que vai para
corrente sanguínea.
Onde nós temos receptores nicotínicos, muscarínicos e alfa e beta adrenergico?
Respectivamente
nos gânglios, na supra- renal;
em todos os órgãos e tecidos inervados pelo parassimpático;
todos os órgãos e tecidos inervados pelo simpático.
Receptores NANC: serotonina, ATP, dopamina, GABA, também participam das ações do simpático e do parassimpático. Eles podem funcionar como cotransmissores, como neuromodulador ou até mesmo como neurotransmiror.
23
-O neurotransmissor quando é liberado ele pode exercer suas ações na célula pós- sináptica e também pode controlar sua liberação. Essa última parte
é muito vista no sistema simpático, a noradrenalina sendo liberada e atuando em receptor inibitório para ela mesma. Isso é chamado de inibição présináptica, tem fármacos importantissimos que interferem nisso. Uma segunda coisa que pode acontecer é uma inibição pré-sináptica heterotópica, um
neurotransmissor é liberado e vai exercer sua ação na célula pós- sináptica, podendo inibir outro neurônio de liberar o seu neurotransmissor, então ele
pode ter receptor para ele em um outro neurônio que não seja o dele, por exemplo, um neurônio noradrenergico inibindo a liberação de acetilcolina em
um neurônio colinérgico.
-Outra coisa que pode acontecer é o sinergismo pós-sináptico, consiste em um neurotransmissor principal junto com um NANC qualquer. Esse
neurotransmissor NANC pode ajudar a ação do neutransmissor principal, é o que acontece com a noradrenalina que é liberada junto de um peptídeo
nos vasos sanguíneos fazendo vasoconstrição.
-Uma outra situação é um neutrasmissor principal e um NANC exercendo a mesma ação na célula, só que um por um mecanismo rápido de resposta e
outro por um mecanismo lento. Isso acontece com a acetilcolina e o GRH no gânglios.
-E uma última situação é quando eu tenho neurônio liberando um neurotransmissor principal e um NANC, só que o NANC tendo ação em um tecido
diferente do neurotransmissor principal. Ex: isso acontece nas glândulas salivares, dilatação e contração de vasos.
**Um NANC importante: óxido nítrico (NO). Ele se difunde pelas células, participa do esvaziamento gástrico, responsável pelo relaxamento da
musculatura lisa visceral do trato respiratório, digestivo e urogenital, responsável pela ereção, totalmente envolvido com vasodilatação. Vai ser muito
importante na vasodilatação que o parassimpático faz, que vai ser mediada pelo NO.
Etapas básicas de uma neurotransmissão:
Representação de uma sinapse periférica qualquer:
- Todo o neurotransmissor para ser formado precisa de um precursor. Esse precursor vai te que penetrar no neurônio por um mecanismo de transporte,
uma proteína que faz o transporte desse precursor. Dentro esse precuror vai sofrer ação enzimática, então ele vai encontrar uma enzima de síntese, que
vai transformá-lo em transmissor. Esse transmissor não pode ficar livre no citoplasma, então vai ser armazenado em vesículas sinápticas para ficar
protegido da degradação. Esse transmissor vai ser liberado através da chegada de um potencial de ação, vai ocorrer abertura de canais de cálcio, e
o cálcio que estava mais concentrado do lado de fora da célula, vai aumentar no citoplasma. O aumento de cálcio no citoplasma vai promover a
liberação das vesículas que vai liberar esses neurotransmissores armazenados. Esse transmissor vai atuar no receptor. Existem algumas formas desse
neurotrasmissor sair da sinapse e seu efeito terminar. Esse neurotrasmissor pode ser recaptado por proteína de transporte e voltar para o neurônio présinpatico, e dentro pode ser novamente armazenado em vesículas ou pode ser degradado por enzimas; esse neurotransmissor pode ser degradado na
prórpria fenda sináptica e se transformar em um metabólito inativo; esse transmissor pode atuar em receptor pré-sináptico e promover inibição de
liberação dele mesmo.
- Neurotransmissão colinérgica
O precursor da acetilcolina é a colina, metabólito de ácido graxo, a colina penetra no neurônio por um mecanismo de transporte. Dentro do neurônio
encontra uma enzima que vai sintetizá-la em acetilcolina, essa enzima vai utilizar uma coenzima chamada Acetilcoa. Essa enzima transfere o grupamente
Acetil para colina formando a acetilcolina. A enzima que faz isso se chama colina-acetil-transferase, para os íntimos CAT. A acetilcolina vai ser
armazenada por mecanismo de transporte nas vesículas. Segue os eventos de potencial de ação... A vesícula vai fundir com a membrana pós-sináptica e
então temos a liberação de neurotransmissores. Sendo acetilcolina liberada, ela vai atuar em dois receptores: nicotínicos e muscarínicos, realizando sua
ação agonista. Através da acetilcolinesterase a acetilcolina é retirada fenda sináptica. Acetilcolinesterase degrada acetilcolina em colina e acetato, e
essa colina é reaproveitada pelo neurônio.
Existem três receptores nicotínicos: os que estão no SNC; na junção neuromuscular; e nos gânglios. São os receptores ionotrópicos, ou seja, acoplados a
canais iônicos. São sempre excitatórios, a proteína se abre, a célula se despolariza, resultando em excitação, ou numa contração. Acontece na sinapse
excitatória rápida na JNM, nos gânglios.. O receptor nicotínico da acetilcolina muscular e ganglionar difere na sua estrutura molecular e a
farmacológica. Ex.: eu tenho um fármaco que tem mais afinidade pelos receptores do gânglio, do que aqueles das JNM.
Os receptores muscarínicos da acetilcolina são metabotrópicos, ou seja, acoplados a proteína G. São conhecidos cinco tipos de receptores muscarínicos:
M1- M5. Os receptores muscarínicos M1 são localizados nas glândulas secretoras, gânglio e SNC. Os receptores M2 estão no coração e em alguma
musculatura lisa. M3 e M4 musculatura lisa e glândula secretora, e o receptor M5 no SNC.
A resposta dos receptores nicotínicos é de milissegundos, enquanto a do muscarínicos é de segundos. Nos receptores muscarínicos temos resposta
excitatória como inibitória, dependendo da proteína G é que se determina o tipo de resposta.
-Ações Nicotínicas da acetilcolina: a acetilcolina vai se ligar nesse receptor vai abrir canais cálcio-iônicos, e vai produzir uma rápida despolarização.
Essa despolarização na junção neuromuscular é conhecido como potencial de placa terminal (PPT). Resulta em um potencial na fibra muscular. A
transmissão rápida nos gânglios é muito semelhante, só que é chamada de potencial pós- sináptico exitatório (PPSE); porque o gânglio pré sinápitco
quer produzir uma excitação e não uma contração. Aqui uma molécula de acetilcolina gera um potencial, e a acetilcolina é hidrolisada em questão de
milisegundos.
-Já quando a acetilcolina atua em receptor muscarínico a ação é muito mais lenta. A estrutura e a forma que ela acontece ainda não estão muito
esclarecidas. Na maioria dos casos a acetilcolina funciona mais como um modulador, vai modular uma resposta na proteína G.
Substâncias que interferem em uma transmissão colinérgica:
- Substâncias que interferem em um receptor muscarínico: O fármaco pode ser agonista desse receptor ou antagonista.
-Substâncias que afetam os gânglios do simpático quanto do parassimpático: temos substâncias que estimuladoras ganglionares e bloqueadoras
ganglionares. Ex. anti hipertensivos, quando eu quero uma queda de pressão rápida, se usa um bloqueador ganglionar.
24
-Substâncias que bloqueam a transmissão neuromuscular, são substâncias paralisantes: substâncias ñ despolarizantes e despolarizantes. Na verdade
essas substâncias são agonistas e antagonistas nicotínicos na junção neuromusucalar.
- Inibidores da síntese ou da liberação de acetilcolina: inibidores da CAT, inbidores da liberação. Não tem uma importância farmacológica nesse
momento.
- Subtâncias que intensificam a transmissão colinérgica. Pode intensificar inibindo a enzima acetilcolinesterase, ou pode intensificar estimulando a
liberação de acetilcolina. Vamos estudar a importância tanto toxicologicamente como terapeuticamente dos inibidores da acetilcolinesterase.
EU VOU COBRAR DE VCS QUATRO CLASSE DE FÁRMACOS:
-Agonista e antagonista muscarínico (2)
-bloqueadores neuromusculares: antagonista e agonista nicotínico na JNM
-e inibidor da acetilcolinesterase
Drogas parassimpaticomimeticas
São fármacos que imitam, mimetizam as ações do parassimpático. Ex.: Agonista muscarínico, inibidor da acetilcolinesterase. Esses fármacos também
podem ser chamados de agonistas colinérgicos.
Esse agonistas colinérgicos podem ser classificados nos de ação direta que são os agonista muscarínicos, pois se ligam diretamente nesse receptor. E
também podem ser classificados nos de ação indireta não se ligam no receptor, mas exarcebam a ação da acetilcolina, que são os anticolinesterasicos.
Agonistas muscarínicos, só tem um na RENAME, a pilocarpina, usada no glaucoma. Outro fármaco betamecol. Para que esses fármacos se liguem no
receptor muscarínico precisam ter duas estruturas: um gupamento éster e um amônio quartenário.
O que diferencia do agonista dos antagonistas é que nesses últimos os grupamentos éster são grupamentos de maior volume, e quando se liga na
proteína ao invez de ativar, inibe ela.
Usando um agonista muscarínico no sistema orgânico: vai diminuir a frequência e força de contração cardíaca, queda de pressão arterial
(vasodilatação mediada pelo NO), aumento do peristaltismo, contração de brônquios.
-No músculo ciliar, um agonista mucarínico, ajuda a ajustar a curvatura do cristalino. Existe também receptores muscarínicos no músculo constritor da
pupila. Quem faz isso é o receptor M3 que está presente tanto no músculo ciliar como no m. constritor da pupila. É um receptor metabotrópico que está
acoplado à proteína G, quando ativada, ativa a fosfolipase C, produzindo dois segundo messensageiros IP3, que sempre aumenta cálcio no citoplasma
da célula e diacilglicerol. Se eu aumentar cálcio no interior da célula, ela vai se contrair. A pilocarpina é usada para glaucoma de ângulo estreito. O
glaucoma é o aumento da pressão intra-ocular. A solução de humor-aquoso entre córnea e o cristalino é drenado pelos canais de Schlemm. No
glaucoma de ângulo estreito, o ângulo entre a alavanca diminui, os canais ficam obstruídos, de forma que a drenagem do humor aquoso fica
dificultada, fazendo que acumule humor aquoso e aumente a pressão intraocular. A pressão normal é 10 ml/Hg e vai para 15ml/Hg ou 20 ml/Hg.
Conforme essa pressão vai aumentando, existe a probabilidade de lesar o nervo óptico. A pilocarpina atua nos dois músculos: o ciliar e constritor da
pupila. Puxando o cristalino para as duas laterais, as alavancas tendem a descer, abrindo o espaço dos canais para drenagem do humor. Porém, a
pupila vai ficará contraída, dificultando o ajuste da pupila de acordo com a luminosidade do ambiente e dificuldade de acomodação visual, ambientes
perto e longe.
-No coração o receptor muscarínico é inibitório- M2, acoplado a proteína G inibe a adinilato ciclase que diminui a produção de AMPcíclico, que ao
mesmo tempo, abre canais de íons potássio. Tirando o potássio da célula ocorre a hiperpolarização que vai relaxar a célula, resultando em
bradicardia.
-Pode ter queda de pressão arterial pela bradicardia e pela vasaodilatação. A vasodilatação acontece por um colaborador, o NO. No vaso
sanguíneo, existem nas células endoteliais os receptores muscarínicos M3. Quando um agonista muscarínico se liga nesse receptor estimula a produção
de NO por essa célula. Esse gás vai se difundir da célula endotelial para a célula da musculatura lisa do vaso, e nessa última célula o óxido nítrico vai
fazer relaxamento. Essa célula muscular relaxando vai o vaso vai relaxar.
-Agonista muscarínico faz qualquer glândula exócrina secretar seu conteúdo (sudoríparas, salivar, lacrimal). Vai atuar nos receptores M3 e M1. Pelo
mesmo mecanismo, aumento do cálcio vai ter secreção do conteúdo da célula.
Acredita-se que existam receptores M3 nos vasos sanguíneos que sejam responsáveis pela ereção peniana. Não existe agonista muscarínico para
impotência, não deu certo (como o Viagra) devido às reações colaterais.
Agonista muscarínico para uso clínico é a pilocarpina / Betamecol que é usado para estimular a movimentação gastrointestinal e urinário para pacientes
que sofreram cirurgia, cesariana, e para pacientes que sofrem com boca seca, fazendo produção de saliva.
*A pilocarpina produz um efeito paradoxal ao invés de produzir hipotensão como esperado, causa hipertensão.
*Contra indicação para agonista muscarínico, quem tem asma, hipertiroidismo, insuficiência coronariana, para que tem problemas ácidos no estômago
porque a célula parietal possui receptores M3.
Farmacologia do Sistema Nervoso Autônomo Parassimpático 08/04
(A professora fez uma breve revisão)
Então, nós estávamos estudando transmissão colinérgica. Foi visto toda a fisiologia dessa transmissão. Vimos que na aula passada a acetilcolina atua
em dois tipos de receptores: muscarínicos e nicotínicos.
Temos receptores nicotínicos em três lugares do organismo: nos gânglios; na JNM; e no sistema nervoso central. Eles se diferem em relação a estrutura
molecular e farmacologia. Relembrando, são receptores ianotrópicos, ligados a canais iônicos, constituído por cinco subunidades, e essas subunidades
vão variar de acordo com esses três tipos de receptores.
25
Existem cinco tipos de receptores muscarínicos M1-M5. Os receptores ímpares (M1, M3, M5) são acoplados a proteína Gq, que faz a produção de IP 3 e
diacilglicerol. Os receptores pares (M2, M4)são acoplados a proteína Gs inibem a adenilato ciclase e diminui o AMPcíclico na célula. Localização: são
localizados na musculatura lisa, célula parietal, músculo constritor da pupila...
Como os fármacos podem afetar essas transmissões colinérgicas e quais são as aplicações clínicas: (tema da aula)
Substâncias que afetam os receptores muscarínicos :Podem ser agonistas desses receptores ou antagonistas: Os agonistas vão mimetizar, imitar os
efeitos do parassimpático- fármacos conhecidos como parassimpaticomimeticos. Os antagonistas vão fazer o efeito contrário do parassimpático, vão
impedir a ação da acetilcolina nos receptores muscarínicos, por causa disso são conhecidos como fármacos parassimpaticolíticos.
- Existem também substâncias que afetam os gânglios, porque a acetilcolina atua em todos os gânglios tanto do parassimpático, como do simpático,
através de sua ação em receptores nicotínicos. Essas subtâncias conhecidas como estimuladoras ganglionares e bloqueadoras ganglionares tem uma
aplicação muito restrita, para produzir uma hipotensão acentuada, por exemplo.
- Tem as substâncias que bloqueiam as transmissões neuromusculares são as substâncias não polarizantes e despolarizantes. Essas substâncias
atuam nos receptores nicotínicos na junção neuromuscular. São as drogas paralisantes, muito utilizadas em cirurgias quando um relaxamento muscular
total é necessário. Podem ser tanto agonistas como antagonistas dos receptores nicotínicos nas junções neuromusculares. As duas classes (agonista e
antagonista) causam paralisia.
- Fármacos inibidores de síntese ou da liberação de acetilcolina são aqueles que inibem CAT, inibem a proteína que faz o transporte da acetilcolina
para a vesícula, ou então aqueles que inibem o transporte da colina para dentro do neurônio. Esses fármacos não tem nenhum uso clínico. São usados
para pesquisa farmacológica. Ex.: Botox que inibe a liberação da acetilcolina.
- Substâncias que intensificam a transmissão colinérgica são substâncias que não se ligam diretamente nos receptores da acetilcolina, porém,
intensificam a ação da acetilcolina nos seus receptores. Uma classe de substância muito importante desse grupo são os anticolinesterásicos que tem
importância tanto clínica quanto toxicológica. Inibindo a acetilcolinesterase, a acetilcolina permanece mais tempo na fenda sináptica, fazendo que ela
atue mais nos seus receptores. Eles também são conhecidos como fármacos parassimpaticomimeticos.
-Estimuladores de liberação de acetilcolina são fármacos que estimulam a liberção de acetilcolina, mas não tem aplicação farmacológica.
-----------------------x----------------------------------------------x--------------------------------------------Fármacos parassimpaticomimeticos
São os fármacos agonistas muscarínicos e os anticolinesterásicos. Todos eles são classificados como agonistas colinérgicos, porque acentuam e
imitam o efeito da acetilcolina. Existem os agonistas diretos- aqueles que se ligam diretamente no receptor, os agonistas muscarínicos; e os indiretos- não
se ligam no receptor, porém, aumenta as ações da acetilcolina.
- Agonistas muscarínicos: acetilcolina, Betamecol(possui uso clínico), Pilocarpina (possui uso clínico), Carbacol.
-Anticolinesterásicos: tem os de importância toxicologica – que são os organofosforados.
**Todo fármaco para se ligar em um receptor muscarínico precisa ter duas características fundamentais: um grupamento éster e um amônio quartenário
com uma positividade. O que vai diferenciar do Agonista para o Antagonista é o fato que os radicais costumam ser mais volumosos nos Antagonistas.
Dentro dos agonistas muscarínicos existem os alcalóides naturais e sintéticos. Substâncias derivadas de plantas que também tem efeito agonista no
receptor. A muscarina que um alcalóide extraído de um cogumelo venenoso, foi o primeiro fármaco que conseguiram fazer ligação no receptor
muscarínco.
(agora a prof viu que já tinha dado essa parte da matéria ¬¬) * Transcrevi de novo, pq ela flou mais coisas que na outra aula.
Anticolinesterásicos, fármacos que intensificam a transmissão colinérgica. Existem duas formas de colinesterase: a acetilcolinesterase e a buceril
acetilcolinesterase ( que é uma pseudo-colinesterase, presente no sangue, no vaso, no fígado). Acetilcolinesterase é responsável pela rápida
hidrólise da acetilcolina, bem como a do Betamecol, Carbacol. Já a Buceril acetilcolinesterase degrada vários substratos como cocaína e outras
drogas.
-Os anticolinesterásicos são classificados de três tipos: ação curta, intermediária e os irreversíveis. Eles são divididos de acordo com a capacidade que
eles tem deinibir a acetilcolinesterase. Os fármacos que inibem de forma irreversível só tem importância toxicológica, são os organofosforados. Isso
significa que essa enzima não vai mais degradar a acetilcolina, a célula vai ter que sintetizar outra enzima para voltar a fazer a degradação. Os que
tem ligação reversível podem inibir de uma maneira rápida ou intermediária tem aplicações clínicas importantes. Eles vão aumentar a ação da
acetilcolina nos seus receptores tanto nos nicotínicos quanto nos muscarínicos. Vai depender da quantidade de bloqueio dessas enzimas.
Alguns atravessam a barreira hematocefálica e fazem a mesma ação no SNC (como os organofosforados) e causam um efeito excitatório no SNC.
Em uma intoxicação por organofosforado todos os efeitos do parassimpático estarão exacerbados: bradicardia, queda brusca de pressão arterial,
bronco constrição, aumento do peristaltismo (cólica e diarréia). Se o bloqueio for muito intenso vai ocorrer ações excessivas da acetilcolina nos
receptores nicotínicos na JNM, vai ter tremor muscular seguido de uma rigidez, uma paralisia muscular. No inicio a acetilcolina estimula demais, depois o
receptor dessensibiliza, para de responder.
26
Em uma JNM a acetilcolina se liga, promove a abertura de canais iônicos, o sódio entra, despolariza e faz a contração. Só que a acetilcolina na JNM é
degradada em questão de milissegundos pela acetilcolinesterase. Quando a acetilcolinesterase está bloqueada, acetilcolina se liga em receptor
nicotínico e não se desliga. Então ela fica ativando o receptor nicotínico em um período muito maior do que milissegundos. Esse receptor nicotínico não
vai conseguir voltar para seu estado refratário e depois ficar de repouso para polarizar de novo. O receptor nicotínico vai dessensibilizar. E mesmo
com a acetilcolina ligada ele não vai mais conseguir de novo uma despolarização. Isso se chama bloqueio por despolarização prolongada.
Existe uma doença auto-imune chamada Miastenia grave que ocorre nos receptores nicotínicos. O sistema imunológico do paciente começa a atacar os
receptores nicotínicos, destruído essa proteína na junção neuromuscular. Resulta em fraqueza muscular. Uma característica importante dessa doença é a
ptose palpebral. As manifestações da doença acontece com, primeiramente, perda da força muscular, depois tem dificulade de caminhar de subir
escadas, até , com o progredir da doença, parar em uma cadeira de rodas. Essa doença é diagnosticada com o uso de anticolinesterasico. Porque? Vai
aumentar os níveis de acetilcolina, e vai atuar com mais intensidade nos receptores nicotínicos que estão diminuídos. O paciente é tratado com
anticolinesterásico de ação intermediária. Também é necessário diminuir a atividade do sistema imunológico, administrando drogas imunossupressoras.
Revisando:
Porque usamos anticolinesterase: importância clínica: tratamento e diagnóstico da miastenia grave, tratamento do glaucoma, anestesia.
-----------------x---------------------------x--------------------------------x-------------------------x-----------------Caso clínico: Sr. Hiroyshito, 65 anos, agricultor, compareceu a emergência do pronto socorro apresentando intensa bradicardia, hipotensão,
sudorese e rigidez muscular. Diante da análise da historia do paciente, diagnosticou-se um quadro de intoxicação por inseticida organosfosforado.
Imediatamente administrou-se atropina na dose de 1 mg, via intravenosa, a cada 5 minutos, até o desaparecimento da bradicardia. Administrou-se
também pralidoxima 2g por infusão intravenosa contínua.
1)Explique farmacodinâmicamente como os organofosforados causaram os sintomas do paciente.
2) Explique farmacodinâmicamente como a atropina reverteu o quadro de intoxicação.
3) Explique porque a pralidoxima foi utillizada
A Pralidoxima (está na RENAME) é a única droga que temos no mercado que consegue reativar a ligação do organofosforado com a enzima, como
havia dito é uma ligação irreversível. Porém, ela consegue reverter a ligação do organofosforado com a enzima, porque ela tem alta afinidade pelo
fósforo do organofosforado, mas precisa ser administrado até duas horas depois da intoxicação.
Para podermos entender a ação das antropinas, precisamos conhecer os antagonistas muscarínicos (drogas parassimpaticolíticas).
-Antagonistas muscarínicos no mercado: Atropina* (atropa belladonna), hioscina ou escopolamina ( Datura stramoniumI)Buscopan , ipratópio*(muito
utilizado para inalação, broncodilatador), tropicamida*(midiatrico, dilatador pupilar), cilopentolato(midiatrico), pirenzepina (antagonista muscarínico
M1) -marcados com (* )estão na RENAME.
Atropina
Na estrutura da Atropina existem ligações de grupos volumosos, fazendo que o efeito ao se ligar no receptor seja antagonista (de bloqueio).
Os antagonistas muscarínicos tem efitos periféricos semelhantes, o que vai diferenciar é que eles tem diferente seletividade por receptores, então existe
antagonista muscarínico mais seletivo para M1 ou para M2, ou então aqueles que não são nada seletivo. A atropina é um exemplo de antagonista
muscarínico que não tem seletividade. Ela bloqueia todos os M-receptores. Todo agonista muscarínico inibe todas as secreções, faz taquicardia. Em dose
baixa a antropina faz o efeito paradoxal, em vez de produzir taquicardia, ela produz mais bradicardia, porque uma dose baixa ativa um reflexo no
SNC que ativa o parassimpático.A atropina não causa alteração da pressão arterial em indivíduos normotenos. Nas células endoteliais do vaso tem
receptores muscarínicos M3, quando esse receptor é ativado pela acetilcolina, a célula endotelial produz NO, e esse gás difunde até a célula muscular
relaxando e promovendo vasodilatação. Quando eu dou atropina eu impeço a produção de NO, então eu não vou ter vasodilatação. Na parte ocular,
o antagonista muscarínico vai fazer midríase, dilatação pupilar. Aumento da pressão ocular. Inibição da motilidade. Broncodilatação, relaxamento do
trato gastro-intestinal.
Como tratar uma intoxicação por antagonista muscarínico? Deve ser administrado um anticolinesterasico, que é uma interação farmacodinâmica de
antagonismo fisiológico.
Uma utilidade dos antagonistas muscarínicos para problemas cardíacos é no tratamento da bradicardia sinusal. Posso usar um antagonista muscarínico
para dilatar a pupila.
Os antagonistas muscarínicos são utilizados com medicação pré-anestésica para ressecar secreção. Porque muitas cirurgias eu preciso entubar o
paciente, então é necessário ter uma via área limpa, sem secreções.
Pode ajudar pessoas que sofrem de incontinência urinária, porque inibe micção.
Pode ajudar para as crianças que faz xixi na cama (enurese noturna).
27
Resposta 2 do caso clínico : a atropina bloqueou o receptores muscarínicos, a acetilcolina não se ligou e acabou revertendo a bradicardia a sudorese. A
rigidez muscular eu não reverto por antropina, porque ela ocorre pela ação da acetilcolina nos receptores nicotínico. A rigidez muscular vai continuar
acontecendo até organofosforado serem degradados ou se pralidoxina conseguir reverter essa ligação.
Resposta 3: Porque ela conseguer reverter a ligação do organofosforado com a enzima, desde que seja adm logo.
-----------------------------------------------------------x----------------------------------x-----------------------Agora vamos falar dos fármacos que são usados para bloqueadores muscular. Eles são usados em cirurgia. O anestesista é que mais vai usar esse
medicamento. São fármacos usados em cirurgia cardiaca, abdominais... São classicados em duas categorias: em bloqueadores não-despolarizantes e
despolarizantes. O que isso significa: eu tenho o fármaco que é agonista do receptor nicotínico (despolarizantes), e tem os farmacos que são
antagonistas desses receptores (não despolarizantes). Ambos vão causar paralisia, mas por mecanismo de ação diferentes no mesmo receptor.
-Não despolarizantes: foram descobertos muitos anos atrás pelos índios. Eles utilizavam alcalóides (curare) nas pontas da flecha, caçava o animal, ele
ficava paralisado e depois matavam o animal. “eu vi uma reportagem no Discovered de uma tribo indígena que ainda usa essa técnica” (Gabriel) -“O
exército brasileiro usa em combate.” O que tem nesse curare? Tem um princípio ativo chamado sucopurarina (sei lá como se escreve isso), que nada mais
é que uma antagonista de receptor nicotínico na JNM. A partir desse princípio ativo desenvolveram derivados sintéticos: Pancurônio, atracúrio, vecurônio
todos eles em uso clínico hoje. A Galamina não é só antagonista nicotínico, ela também é antagonista muscarínico M2 (coração), então a Galamina faz
muita taquicardia, por causa disso ela não é usada na clínica. Todos causam uma paralisia flácida. A ordem de paralisia muscular acontece dessa
forma: primeiro os músculos oculares, depois os músculos da face, membro e faringe, os músculos respiratórios são os últimos a serem paralisados, e os
primeiro a voltar da paralisia, por isso é necessário a entubação. Efeito indesejável: como existe receptor nicotínico nos gânglios simpático e
parassimpático vai ter queda de pressão arterial. Eles também fazem liberação de histamina. Não se ve o paciente coçando porque ele está
anestesiado, mas no camundongo vai dá
pra ver. Causa broncoconstrição por causa da liberação de histamina. E causa dor muscular pós-operatória.
-Despolarizantes: são agonistas desse receptor, vão causar paralisa por despolarização. Antes de produzir a paralisia, muita fasciculação porque
incialmente eles vão estimular o receptor nicotínico, que vai fazer o músculo contrair descordenadamente. Depois dessa primeira fase é que eu vou para
o bloqueio. Então eles vão se ligar ao receptor nicotínico, atuando como agonistas, despolarizando o receptor, porém não são hidrolisados rapidamente
pela acetilcolinesterase, vão atuar por um tempo prolongado. Explicando melhor :acontece um bloqueio de fase I, onde tem abertura do receptor,
entrada de sódio, despolarização da placa terminal, propagação e despolarização da célula muscular e contração muscular generalisada,
desorganizada, estimulando muito mais que o normal- tremor. No segundo momento, como o bloqueador neurmuscular não é degradado rapidamente,
há uma repolarização iniciada na célula, porém, a membrana é incapaz de repolarizar novamente. Não há como fazer a membrana entrar em estado
de repouso para repolarizar de novo. Isso vai fazer um bloqueio do canal de sódio e leva a dessenssibilização do receptor. Por mais que o agonista
esteja ligado o receptor não abre mais o seu canal. E paro a despolarização. Receptor nicotínico dessenssibiliza muito fácil, estimulado demais,
dessenssibiliza. Isso que acontece com a nicotina. Fármacos despolarizantes: Succinilcolina, suxametônio
Como que escolho um entre um bloqueador despolarizante e não –despolarizante: o médico vai escolher dependendo da duração do efeito. Os não
despolarizantes tem uma duração de tempo mais prolongada que os despolarizantes. Os despolarizantes é para uma cirurgia mais rápida. O
suxametônio tem uma duração de tempo de 3 min. A duração da cirurgia é o fator que leva o anestesista escolhe entre um ou outro. Além dos ñdespolarizantes durarem mais tempo, existe um antídoto para eles, que são os anticolinesterasicos.
Entendendo o mecanismo de reversão: um fármaco anticolinesterasico junto com um fármaco antagonista muscarínico, o anticolinesterasico vai aumentar
os níveis de acetilcolina, a acetilcolina vai competir com o antagonista pela ligação do receptor, como é uma ligação reversível, a acetilcolina vai
começar a se ligar de novo, e o bloqueio é revertido. Entre a acetilcolina e o bloqueador ñ despolarizante existe um antagonismo competitivo; mais
entre o ñ despolarizante e o anticolinesterasico é um antagonismo fisiológico.
Porque prolonga o efeito quando se usa um anticolinesterasico e um bloqueador despolarizante?
Porque o despolarizante tem uma ação agonista com o receptor, e nessa ação ele está dessenssibilizando os receptores. Se aumentar a quantidade de
agonista na fenda, há um aumento da dessenssibilização do receptor, potencializando o efeito do bloqueador.
“Lembra daquele filme que eu mandei mirr vezes vocês assistirem? enCUrralad . Tem um erro de farmacologia ali. Eu vou perguntar na prova qual é o
erro desse filme.”
R: é porque o médico aplica na Courtney Love succinil colina que é um bloqueador despolarizante. Logo depois de quase matar a bandida ele a
administra um antídoto, só que não existe antídoto para bloqueador despolarizante.
28
Fármaco
06.05.09
(nos primeiros 1:50 min. da aula não dá pra ouvir o que a prof. tá dizendo pq tem muita gente falando ao mesmo tempo)
Neurotransmissão noradrenérgica (desenho)
Como a noradrenalina é sintetizada, armazenada e liberada? O precursor da noradrenalina é o neurotransmissor tirosina. A tirosina é um
aminoácido obtido pela alimentação e que penetra no neurônio por um sistema de transporte. Dentro do neurônio, a tirosina irá encontrar uma enzima
que está presente tanto nos neurônios noradrenérgicos como nos dopaminérgicos, que é a tirosina-hidroxilase (abreviatura TH). A tirosina é então
transformada em DOPA, que é um intermediário, antes da formação da noradrenalina. A DOPA vai encontrar mais uma enzima, chamada DOPAdescarboxilase, que vai transformar a DOPA em dopamina. Se fosse um neurônio dopaminérgico, a síntese terminava aqui. Mas no neurônio
noradrenérgico, esta dopamina, por um mecanismo de transporte, vai penetrar na vesícula, e dentro dela, somente nos neurônios noradrenérgicos e em
mais nenhum outro, tem mais uma enzima chamada dopamina-beta-hidroxilase. E esta dopamina é transformada em noradrenalina dentro da vesícula.
Dessa forma, a noradrenalina fica armazenada em vesículas nos neurônios noradrenérgicos.
Mas e a adrenalina? Na periferia, ela é um hormônio. Na medula da glândula supra-renal, tem um grupo de células, chamadas células
cromafins, que fazem a mesma síntese da noradrenalina. Só que, quando chega a formar a noradrenalina, existe mais uma enzima, chamada feniletanolamina-m-metil-transferase (TMNP) (não precisa decorar! =] ), que transforma a noradrenalina em adrenalina. E a adrenalina é lançada no
sangue, e pelo sangue ela faz as mesmas ações da noradrenalina. Então por isso que a adrenalina é vista como um hormônio (pq é produzida por uma
glândula, a supra-renal) e a noradrenalina é vista como um neurotransmissor (pq ela é produzida pelos neurônios noradrenérgicos).
Como a noradrenalina é liberada? Pela chegada de um potencial de ação nesse neurônio, que vai promover a abertura de canais de cálcio,
e o cálcio aqui dentro vai promover a extrusão dessas vesículas e ela vai ser liberada.
E quais são os receptores da noradrenalina que nós temos? Na periferia, os receptores alfa e beta adrenérgicos. A adrenalina e a
noradrenalina são agonistas desses receptores. Qual a diferença então entre a adrenalina e a noradrenalina? É grau de afinidade. Potencia como
agonista. A adrenalina tem mais afinidade pra um, a noradrenalina para outro...
Uma vez que elas se ligaram com seus receptores da periferia e exerceram os seus efeitos, qual é o principal mecanismo pra gente tirar
noradrenalina da fenda sináptica e acabar com o efeito dela? (porque ela tem que exercer o efeito e se desligar do receptor, terminar a sinapse)
Recaptação. Diferente da acetilcolina, que é degradação enzimática. Esse processo de recaptação também pode ser chamado de captação 1. Isso
porque ela é recaptada (ou captada) pelo neurônio que a produziu. E aqui dentro a noradrenalina pode ser novamente armazenada em vesículas, ou
então ser degradada enzimaticamente pela enzima MAO (mono-amino-oxidase). A MAO é a principal enzima que faz a degradação da
noradrenalina recaptada (pelos neurônios pré-sinápticos) que não foi armazenada em vesícula. E ela vai transformar essa noradrenalina na periferia
em um metabólito chamado VMA, o qual é eliminado pela urina. O doseamento de VMA na urina pode ser usado num paciente para saber o quanto
ele está produzindo de adrenalina e noradrenalina. Porém, a gente não tem MAO só no neurônio pré-sináptico, a adrenalina e a noradrenalina que
circularem pelo sangue vão ser degradadas pela MAO plasmática. Na fenda sináptica, a enzima que mais faz degradação é a COMT (catecol-o-metiltransferase). Então, aquela noradrenalina que não for recaptada, vai ficar livre na fenda e sofrer uma ação muito maior de COMT do que de MAO. E
a COMT também vai transformar a noradrenalina em metabólitos inativos.
A recaptação é realizada por uma proteína de transporte inespecífica na membrana da célula que recapta catecolaminas (dopamina,
noradrenalina e até a adrenalina). Há fármacos que inibem essa proteína, mas não há fármacos que acelerem a sua atividade.
A dosagem do VMA é importante para diagnosticar algumas patologias em que o paciente tem uma alta produção de adrenalina e
noradrenalina. Existe um tumor na supra-renal chamado felcromocitoma, em que as células cromafins produzem uma quantidade exagerada de
adrenalina. Como se trata? Fármacos que bloqueiam os receptores da adrenalina porque esse paciente tem uma pressão altíssima o tempo todo. Daqui
a pouco a gente vai ver esses fármacos.
Há fármacos que bloqueiam a MAO (em todos os lugares, mas principalmente nos neurônios dando, por exemplo, efeito antidepressivo). E isso
vai fazer com que haja mais noradrenalina livre e ela será mais armazenada e mais liberada quando houver um potencial de ação.
Detalhamento da formação de noradrenalina e adrenalina. A tirosina é um aminoácido que sofre a ação da tirosina-hidroxilase, que vai
acrescentar um grupamento hidroxila e transformá-la em DOPA. A DOPA-descarboxilase faz a descarboxilação da DOPA e a transforma em
dopamina. Então a dopamina sofre a ação da dopamina-beta-hidroxilase, que acrescenta uma hidroxila no carbono-beta, formando a noradrenalina
(ou noraepinefrina). A noraepinefrina sofre a ação da TNMP e vira adrenalina pelo acréscimo de um grupamento metila. Enfim, todas são muito
parecidas, por isso que há proteínas de transporte, enzimas, que são comuns para todas essas substancias. Mono-amino-oxidase, por exemplo, degrada
qualquer catecolamina.
29
Estrutura química. Essas monoaminas são chamadas catecolaminas porque tem um grupo catecol (um anel benzênico com duas hidroxilas
adjacentes) ligado a uma amina. As principais são noradrenalina, adrenalina, dopamina e isoprenalina(forma sintética). Elas tem diferenças por
afinidade nos receptores. Todas são agonistas alfa e beta, porém a noradrenalina tem uma afinidade muito maior por alfa do que a isoprenalina. E a
isoprenalina, por sua vez, tem uma afinidade muito maior por beta que a adrenalina e a noradrenalina. A adrenalina tem uma ligação intermediária
nos receptores. A isoprenalina (ou isopretenerol) é muito utilizada em parada cardíaca por ser um agonista beta muito potente. A dopamina tem uma
afinidade muito pequena por alfa e beta, e tem afinidade muito maior pelos receptores dopaminérgicos.
Receptores - ações da adrenalina e noradrenalina na periferia e em cada receptor.
Elas se ligam em receptores alfa e beta.
-
alfa: alfa-1 (A, B e D) e alfa-2 (A, B e C)
beta: beta-1, beta-2 e beta-3, com subtipos.
Todos eles são receptores metabotrópicos (acoplados a proteína G).
-
Receptores alfa -1: são acoplados a uma proteína Gq, e quando ativados, ativam a fosfolipase-C e produzem dois segundos mensageiros,
IP3 e DAG; e o resultado será sempre aumento do cálcio citoplasmático.
Receptores alfa-2: são inibitórios, e de forma geral acoplados a uma proteína Gi, que vai inibir a adenilato-ciclase, diminuir produção de
AMPc na célula e inibir a abertura de canal de cálcio.
Receptores beta: todos estimulam a adenilato-ciclase, são ligados a uma proteína Gs, que vai aumentar a produção de AMPc.
(Saber localizações e funções dos tipos – não dos subtipos. Isso serve para prever a ação dos fármacos que agem como agonistas ou antagonistas
desses receptores. Ela deixou um quadro com essas informações no portal. O quadro completo está no Rang & Dale e ela recomenda estudar por ele)
Receptor Alfa-1:
-
Localização I: musculatura lisa de vaso
Ação I: quando é ativado pela noradrenalina ou adrenalina haverá contração, ou seja, vasoconstrição. A contração ocorre porque a
ativação do receptor vai aumentar a quantidade de cálcio na célula contrátil, promovendo a contração. Aumento da RVP e da PA.
Agonistas I: descongestionantes nasais (Sorine, Afrin, Aturgil, Nafasolina, Oxinafasolina, Fenilefrina, Pseudo-efedrina...)
*obs.: Se um paciente hipertenso usar agonista alfa-1 em demasia (por via oral) pode ter uma crise hipertensiva (ler revisão “Simpaticomiméticos que
aumentam a PA” que ela deixou no portal)
-
Localização II: músculo radial da pupila (musculatura circular que envolve a pupila)
Ação II: dilatação pupilar (quando o músculo contrai, aumenta o diâmetro da pupila)
-
Localização III: trato gastrointestinal
-
Ação III: relaxamento da musculatura. Isso ocorre porque o subtipo da musculatura do TGI promove relaxamento com a ação da adrenalina
e noradrenalina (diferente dos outros subtipos).
-
Localização IV: fígado
Ação IV: glicogenólise hepática (quebra das moléculas de glicogênio nos hepatócitos), promovendo liberação de glicose para o sangue.
Receptor Alfa-2:
- Localização e ação: a noradrenalina quando é liberada age como agonista e precisa de mecanismos que diminuam a sua permanência na
sinapse. Esses mecanismos são recaptação e degradação enzimática. Mas existe um terceiro mecanismo, que ocorre graças a existência do receptor
alfa-2 na pré-sinapse. Ele é conhecido como receptor auto-inibitório. Quando há altas concentrações de noradrenalina na sinapse, por qualquer motivo,
ela vai se ligar nesse tipo de receptor. O receptor alfa-2, inibitório e acoplado a uma proteína Gi, é ativado e ocorre diminuição da produção de
AMPc no neurônio. Com isso, não há abertura de canais de cálcio e a liberação de neurotransmissor irá diminuir. Então há uma inibição da liberação de
noradrenalina regulada por ela mesma – é um feedback auto-inibitório (ela inibe a sua própria liberação).
- Agonistas: metildopa (anti-hipertensivo utilizado na gestante hipertensa). É um agonista no SNC, que, diminuindo a liberação de
noradrenalina, diminui toda a descarga do simpático na periferia. Vai fazer isso na sinapse de um neurônio simpático com a célula cardíaca, reduzindo
os batimentos cardíacos. Vai fazer isso na sinapse de um neurônio noradrenérgico com musculatura lisa de vaso, diminuindo vasoconstrição. Todas as
ações do simpático ficam diminuídas.
30
Fármaco
07/05/09
A localização e função dos receptores noradrenérgicos é extremamente importante, na aula passada havia sido visto o receptor alfa 1, que é
localizado principalmente na musculatura lisa dos vasos, quando ele é ativado sempre ele faz a contração desse vaso e temos então a vasoconstrição,
aumento da resistência vascular periférica e aumento da pressão arterial. Importante exceção é no trato gastrointestinal, não na pupila, pois na pupila
ocorre contração do músculo para ocorrer dilatação da pupila, mas de qualquer maneira o músculo contrai.A ação da Adrenalina e noradrenalina nos
receptores alfa 1 no trato gastrointestinal produz relaxamento, pois temos subtipos de receptores alfa 1, além de outro motivo que é quando a
adrenalina e noradrenalina se ligam nessas sinapses do trato gastrintestinal, há no trato gastrintestinal inervação colinérgica também, e os receptores
alfa 1 estão presentes na pós-sinapse dessas sinapses, e todos sabemos que a acetilcolina contrai essa musculatura lisa, uma vez que o receptor alfa 1
inibe essa liberação de Ach, ocorre relaxamento.O receptor alfa 2 está presente na pré-sinapse colinérgica, quando adrenalina e noradrenalina se
ligam nesse receptor, ela faz inibição da liberação de acetilcolina, com isso ocorre relaxamento, isso explica porque o simpático tem um relaxamento
do trato gastrointestinal quando ele é ativado.
Receptor alfa 1 também está presente no hepatócito, quando ele é ativado é desencadeada a glicogenólise,que é a quebra do glicogênio
em moléculas de glicose. Voltando ao alfa 2, sua localização mais importante é na pré-sinapse dos neurônios noradrenérgicos e colinérgicos, ele vai
sempre inibir a liberação de noradrenalina e acetilcolina. Receptor afla 2 é ligado a uma proteína G inibitória, vai sempre diminuir AMPc no neurônio,
diminuindo AMPc, não tem abertura de canal de cálcio, com menos cálcio entrando tem menAs liberação de neurotransmissor.Onde tiver alfa 2 em présinapse ele vai inibir a liberação de neurotransmissor, chamado de feedback auto-inibitório quando inibe Noradrenalina e heteroinibição quando inibe
liberação de acetilcolina.
Também tem receptor alfa 2 nas ilhotas pancreáticas, nas células que produzem insulina, ocorrendo por estímulo da adrenalina e
noradrenalina nesses receptores inibição da liberação de insulina, o que significa que o simpático ativado trabalha para termos glicose no sangue, pra
gente acumular energia. Esses receptores alfa 2 também estão envolvidos na contração de músculo liso vascular, pois tem esses receptores nos vasos,
igual ao alfa 1.Tem receptor alfa 2 nas plaquetas, por isso ta envolvido no processo de agregação plaquetária, e ainda tem receptor alfa 2 no sistema
nervoso central, especificamente no tronco cerebral, e ali quando ele é ativado ele inibe a liberação de noradrenalina no tranco cerebral, e sem a
liberação no tranco cerebral de noradrenalina, toda a descarga do simpático fica inibida.O alfa 2 tem essas diferentes ações de contração algumas
vezes e inibição outras devido aos diferentes subtipos.
Quando eu penso em receptor beta 1, tem que pensar em coração. Essa é a principal localização e a localização mais importante.Esse
receptor faz taquicardia, acoplado a uma proteína G estimulatória, aumenta o AMPc na célula, aumentando a contração dessa célula
cardíaca.Também tem receptor beta 1 na glândula salivar, aumenta secreção de amilase, tem receptor beta 1 no rim (muito importante), onde ele
aumenta a liberação de renina, com conseqüente maior vasoconstriçao, maior liberação de aldosterona e aumento da pressão arterial.
Receptor beta 2 tem uma localização muito importante, que é na musculatura lisa de vaso, onde ele vai dar relaxamento. Relembrando, na
musculatura lisa de vaso tem alfa 1 e beta 2, o primeiro faz contração e o segundo relaxamento.Só que quando eu dou uma injeção de adrenalina,
embora ele se ligue nos dois, o resultado final vai ser mais contração com conseqüente aumento de pressão arterial.Daí o grande problema de usar
adrenalina em paciente que usa beta bloqueador, pois será uma imensa vasoconstrição, com um aumento de pressão muito grande.Se o cara for
alérgico a adrenalina, o médico pode tentar uma isoprenalina, que é um composto sintético ou trabalha só com corticóide.O receptor beta 2 reduz
então resistência vascular periférica.
Caso clínico legal: Paciente tomando fármaco antagonista alfa 1(prazozina), e eu dou adrenalina pra ele, a adrenalina não vai ter receptor
alfa 1 pra se ligar e a pressão desse paciente vai cair, o efeito da ligação da adrenalina aos outros receptores livres não vai começar essa ausência
da ação do alfa 1, porque os efeitos hipertensivos dos outros receptores são mais a longo prazo. Além disso, tem muito mais alfa um na musculatura lisa
que alfa 2, o efeito em beta 2 que está todo livre vai ser muito maior.
Outra localização muito importante de beta 2 é na musculatura lisa dos brônquios, onde ocorre relaxamento, portanto todos os fármacos
agonistas beta 2 seletivos são broncodilatadores, são utilizados no tratamento da asma, choque anafilático, etc.Tem também beta 2 em hepatócito e
músculo, onde sempre vai aumentar a glicogenólise.
Tem receptor beta 2 na pré-sinapse do neurônio noradrenérgico, onde ele tem a ação inversa do alfa 2, pois é ligado a uma proteína G
excitatória onde aumenta AMPc, aumentando a liberação de neurotransmissores, SÓ NOS NEURÔNIOS NORADRNÉRGICOS ESSE, NÃO NOS
COLINÉRGICOS. Pergunta que o próximo premio Nobel de medicina tem que responder: porque baixas concentrações de noradrenalina estimulam beta
2.E quando está em altas concentrações de noradrenalina o alfa 2 é ativado.
Ainda tem receptor beta 2 em musculatura lisa de útero, onde também ocorre relaxamento, servem para tratar parto prematuro
(salbutanol), mas não cólicas. Também tem receptor beta 2 em músculo esquelético, o resultado da ativação nos músculos esqueléticos é o
tremor.Paciente que tremem pela sua ansiedade, posso usar um antagonista beta 2 pra diminuir tremor.O beta 3 só tem uma localização, os adipócitos,
onde faz lipólise, mas não tem ainda no mercado, pq eles fazem uma quebra de gordura muito transitória.
Os fármacos que atuam no sistema nervoso simpático vão ser fármacos muito importantes pra tratar hipertensão, pra tratar choque
cardiovascular, arritmia, asma, reações anafiláticas, e outros problemas respiratórios menores. Vamos começar com os simpaticomiméticos, que podem
31
ser classificados em: de ação direta(agonista alfa e beta) e de ação indireta(por um mecanismo qualquer, aumentam as ações da noradrenalina nos
seus receptores).
Os de ação direta são medicamentos de uso livre. Os agonistas alfa 1 seletivos são muito usados pela sua ação vasoconstritora, usados em
primeiro lugar como descongestionantes nasais.O nariz ‘’tranca’’ por uma reação inflamatória, em que são liberadas principalmente as prostaglandinas,
as quais promovem vasodilatação nesse tecido e aumentam a permeabilidade vascular, fazendo com que o conteúdo que está dentro do vaso
extravase pro tecido, a mucosa nasal fica cheia de vaso dilatado e edemaciada, isso diminui o espaço aéreo por isso que o nariz entope.Por isso é
indicado como vasoconstritor nasal, de uso local.Porém quando o efeito dele passa, a vasodilataçao que vem depois é muito pior do que a que estava
antes, isso se chama vasodilatação de rebote, por isso um vasoconstritor deve ser usado no máximo por 3 dias consecutivos, senão seu uso prolongado
pode causar uma lesão irreversível da mucosa nasal e perda de olfato, pela baixa oxigenação da mucosa.Nesse uso a absorção do medicamento é
mínima, então não causa problema sistêmicos, porém tem aqueles medicamentos que são associações antigripais que tem vasoconstritores, aí deve-se
tomar cuidado com pacientes hipertensos(revisão no portal, ‘Simpaticomiméticos que causam hipertensão’’).
Também são usados como midriáticos (fazem midríase), como a fenilefrina. Também pode usar um vasoconstritor associado a anestésico local,
é um efeito de potencialização(interação farmacocinética), vai contrair o vaso de onde o anestésico ta sendo aplicado, pra diminuir a absorção do
anestésico e ele ficar na mucosa, prolongando o efeito do anestésico local(caso para a prova).
Os agonistas alfa 2, fármacos muito importantes, com ação muito importante nos receptores alfa 2 presentes na pré sinapse. A metildopa(antihipertensivo pra gestante) e a clonidina, são conhecidos como anti-hipertensivos de ação central, pois o seu efeito se dá pela sua ação agonista alfa 2
lá no tronco cerebral, diminuindo então noradrenalina no tronco cerebral, e inibindo a descarga de todo o sistema simpático, mas também vai fazer seu
efeito na sinapse cardíaca, nos vasos sanguíneos, com diminuição da liberação de noradrenalina em todos esses lugares.A metildopa na verdade é um
pró-fármaco, ela precisa de uma metabolização pra exercer esse efeito.Eles são classificados como simpaticomiméticos de efeito simpaticolítico, uma
vez que embora seja agonista alfa 2, seu efeito é simpaticolítico, ou seja, contrário ao do simpático pois causa queda de pressão.Explica mais uma vez
que esta acoplado a uma proteína G inibitória, que diminui AMPc, não abre canal de cálcio, diminui liberação de noradrenalina.
Os agonistas beta 1, qualquer um dele vai aumentar freqüência e força de contração cardíaca, dá taquicardia. É a isoprenalina, que é um
agonista beta 1 seletivo, que tem mais afinidade por receptor beta que a própria adrenalina e noradrenalina e a dobutamina, que é um potente
agonista beta 1.Eles podem causar arritmias, com uso só em urgência, parada cardíaca e o paciente deve ser monitorado.
Agonistas beta 2 são utilizados como broncodilatadores em primeiríssimo lugar, tão na RENAME.Temos o salbutanol e fenoterol e o ipratrópio
(antagonista muscarínico), esses dois últimos tem um interação de adição farmacodinâmica, bloqueando o parassimpático e ativando o simpático.Mas
age um pouco no beta 1, pq não tem 100% de seletividade, causando taquicardia como efeito colateral característico, além do tremor.Contra-indicado
pra pacientes que tem arritmia.Como são relaxantes de musculatura lisa uterina, inibe trabalho de parto prematuro.
Os simpaticomiméticos de ação indireta também aumentam o nível de noradrenalina nas sinapses, tanto da periferia quanto do SNC. Vão ser
as anfetaminas. As anfetaminas inibem a recaptação da noradrenalina na sinapse, e entra no lugar da noradrenalina no neurônio pré-sináptico.Entra e
depleta(expulsa) o estoque de noradrenalina das vesículas, uma parte dela é liberada e outra é degradada pela MAO na sinapse.Ela aumenta os
níveis de noradrenalina na fenda por inibir a recaptaçao, e ainda estimula mais liberação de noradrenalina, por isso ela é tão estimulante do SNC.Na
periferia da taquicardia, aumento da PA, etc.Perde o efeito da anfetamina com o tempo pq acabam os estoques de noradrenalina, vai então
aumentando a dose, gerando a dependência.Síndrome de abstinência devido a neuroadaptaçao explicada na aula de antidepressivos.Drogas
psicotrópicas.
O uso clínico da anfetamina é para tratar emagrecimento irracionalmente, na verdade deve ser usada somente quem tem IMC acima de 40.
Fora anfetamina, tem cocaína, que inibe a recaptação das monoaminas.A cocaína atua na via da recompensa coma liberação de dopamina, que é o
neurotransmissor do prazer.A sensação de prazer q ela traz é muito grande, e quem não tem prazer na vida acaba usando cocaína como maneira de
obter esse prazer.A anfetamina é usada pros esportistas e camioneiros pelo seu efeito estimulante, mas depois que passa o efeito, a sensação de
cansaço é muito maior.Os antidepressivos tricíclicos também são considerados simpaticomiméticos de ação indireta pela inibição dessa recaptação de
noradrenalina também.Não se sabe pq a cocaína não tem efeito antidepressivo, e pq os tricíclicos não causam dependência.
Resumo dos principais usos clínicos dos agonistas adrenérgicos: auxiliam no caso de parada cardíaca, num choque anafilático pq são
broncodilatadores, assim como no sistema respiratório, como descongestionantes nasais, associados a anestésico local, pra inibição do trabalho de
parto, pra tratar hipertensão, pra tratar o rubor da menopausa, pra reduzir a pressão intra-ocular e na profilaxia da enxaqueca, pois vasoconstritor
diminui os sintomas dolorosos da enxaqueca.
32
Farmacologia
11/05/2009
Simpaticolíticos de ação indireta:
Eles vão inibir a ação da NA (noradrenalina) nos seus receptores, diminuindo os níveis da NA na sinapse, inibindo a síntese, o armazenamento
ou a liberação das catecolaminas.
A reserpina um fitoterápico extraído da planta Rauwolfia serpentina, até a década de 50 era o anti-hipertensivo mais usado. A reserpina
age inibindo os transportadores vesiculares das catecolaminas, bloqueando o transporte da NA e outras aminas para as vesículas sinápticas, diminuindo
a liberação de neurotransmissor. Não formando e impedindo a recaptação da NA. Com mais NA livre na fenda sináptica (pois não ocorre recaptação)
e mais degradação pela MAO. Pela degradação da MAO e por não formar NA, ocorre uma diminuição da sua concentração. Ela diminui a
concentração de DA, NA e 5-HT (serotonina).
A reserpina tem ação no SNC. O efeito psicótico esta ligado com excesso de DA (dopamina) em algumas regiões cerebrais (mesocortical), e
este fármaco diminui a concentração de DA causando o efeito antipsicótico.
Esta em desuso pelos seus efeitos colaterais (sedação e depressão pela diminuição da 5-HT no SNC), portanto este fármaco é utilizado
somente em pesquisas farmacológicas.
A α-metiltirosina inibe a tirosina hidroxilase que faz parte do primeiro processo de síntese, não formando DA, NA e adrenalina. Pois a
tirosina é precursora de todas essas catecolaminas. A α-metiltirosina é utilizada no tratamento do feocromocitoma (tumor nas células cromafins da
suprarrenal). As células cromafins produzem adrenalina. No feocromocitoma ocorre um excesso de adrenalina e a PA está alta constantemente, trata-se
esta patologia com o inibidor da tirosina hidroxilase (diminuindo a produção de adrenalina).
A carbidopa, fármaco simpaticolítico de ação indireta, inibidora da dopa-descarboxilase (enzima envolvida na síntese da DA, NA e
adrenalina), porém não é usada no tratamento da hipertensão, ela é usada no tratamento de Parkinson.
No tratamento de Parkinson utiliza-se uma associação de levodopa + carbidopa. Utiliza-se a levodopa por ser um precursor da DA, se der
somente DA ela vai ser degradada pela MAO antes de chegar ao cérebro em quantidades suficientes. Se administrar o precursor sem o inibidor da
dopa-descarboxilase vai ter a transformação de levodopa em DA na periferia, antes de chegar ao cérebro. Para não ocorrer a transformação de
levodopa em DA na periferia, associa-se levodopa com carbidopa, deixando a levodopa atravessar a BHE (Barreira Hematoencefálica), a carbidopa
não atravessa, ocorrendo no cérebro a transformação de levodopa em DA. Essa interação é farmacocinética e tem um efeito potencializador.
A guanetidina é um fármaco anti-hipertensivo de ação central. Ela é recaptada (recaptação I) para dentro do neurônio pré-sináptico e
armazenada em vesículas. Depleta os estoques de NA de dentro da vesícula, diminuindo a liberação de NA.
Guanetidina Captação I Transporte para dentro da vesícula expulsa NA de dentro da vesícula NA livre no citoplasma NA degradada
pela MAO diminuindo a liberação de NA.
Possui os mesmos efeitos da reserpina (efeito hipotensor associados a perda dos reflexos simpáticos), pode causar hipotensão postural,
diarréia, congestão nasal e incapacidade de ejaculação. O uso dele será especifico para quem não responde a tratamento aos outros tipos de
fármacos.
Simpaticolíticos de ação direta (antagonistas α e β):
São antagonistas α ou α-bloqueadores, elas atuam em dois tipos de receptores α (α1 e α2). Antagonistas α1 seletivos, α2 seletivos e fármacos
não seletivos que atuam em α1 e α2.
Nos não seletivos encontram-se dois representantes, a fenoxibenzamina e a fentolamina.
A fenoxibenzamina faz ligação covalente com o receptor (antagonista irreversível de receptor α1 e α2), o receptor nunca mais voltará a
fazer efeito. Sua ação anti-hipertensiva vai ser prolongada. Tem ação anti-hipertensiva, pois quando bloqueia α1 ocorre vasodilatação (impedindo a
constrição promovida pela NA e adrenalina). E quando bloqueia α2 (estão presentes na musc. lisa e nas pré-sinapses adrenérgicas e colinérgicas),
aumenta a produção de NA.
- Pós-sinapse vasodilatação (receptor α1 e α2 e musc. lisa).
- Pré-sinapse maior liberação de NA e Ach (colinérgico).
Mesmo que tenha um ligeiro aumento de NA não terá vasoconstrição, pois o receptor está bloqueado, tendo um efeito anti-hipertensivo. Um
efeito colateral destes antagonistas α não seletivos é a taquicardia, porque os receptores β do coração são ativados.
33
A fentolamina faz os mesmos efeitos da fenoxibenzamina, com a diferença que a fentolamina é antagonista reversível (a ligação é fraca com
o receptor e possível de desfazer). Ela apresenta um efeito anti-hipertensivo mais passageiro, o efeito colateral é igual em ambas e aumenta o débito e
freqüência cardíaca, causam hipotensão postural. Ela aumenta a liberação de NA, atuando mais nos receptores β pois α está bloqueado e o receptor β
no coração provoca taquicardia. Outro efeito colateral é a diarréia porque eles aumentam a produção de Ach e com esse aumento na musc. lisa do TGI
vai aumentar o peristaltismo provocando a diarréia. Além disso todo vasodilatador pode apresentar congestão nasal.
*Quais são os principais efeitos colaterais de um vasodilatador? Congestão nasal (todos), hipotensão postural e rubor facial.
Antagonistas α1 seletivos:
*Se for sintetizado um fármaco que é seletivo para α1? O paciente vai apresentar menos taquicardia.
Por este motivo foram desenvolvidos os antagonistas α1 seletivos (prasozina, doxazosina e terasozina) para causar vasodilatação, queda de
PA e menos taquicardia (não bradicardia, mas sim uma taquicardia que não é tão acentuada) e diarréia, porém eles continuaram causando hipotensão
postural. Os antagonistas α1 seletivos são mais utilizados do que os não-seletivos.
*Por que quando mudamos de posição não tem queda da PA? Porque temos o mecanismo reflexo de barorreceptores (receptores que estão na
parede do vaso sanguíneo e detectam quando ocorre uma mudança de posição), ocorrendo uma vasocontrição atingindo o simpático. Quando é utilizado um
vasodilatador, mesmo que não ocorra mudança de posição, ocorre perda deste reflexo e não ativa o simpático para fazer constrição.
O receptor α1 faz vasoconstrição, pois ele é acoplado a uma proteína GQ (constituída de três subunidades) que ativa a fosfolipase C ,
produzindo DAG e IP3, ambos por mecanismos diferentes vão aumentar o Ca+2 dentro da célula, e esse Ca+2 ativa o complexo actina-miosina fazendo
a contração da célula. Os seletivos para α1 tem menos reações adversas que os não-seletivos que atuam em α1 e α2.
Fenoxibenzamina e fentolamina bloqueiam α1 e α2 e prasozina, terasozina e doxasozina bloqueiam apenas receptor α1.
Antagonistas α2 seletivos:
Eles não são utilizados como anti-hipertensivos. A ioimbina aumenta a liberação de NA e aumenta o fluxo simpático. Fazendo com que a NA
aumentada atinja mais receptores α1 e β1, provocando taquicardia, aumento da PA e uma falsa sensação de energia. Ela tem receptor α2 no vaso, com
sua inibição mesmo que não iniba α1 tem vasodilatação, porém menor que antag. α1.
Usa-se α bloqueador para emergências hipertensivas por usar excessivamente os simpaticomiméticos (vasoconstritores). Ex: muito anestésico
local com vasoconstritor e deseja reverter a anestesia. Pode-se dar um vasodilatador (antagonista α1).
Na Síndrome de Raynaud a pessoa tem uma vasoconstrição periférica excessiva, pode-se administrar um antagonista α2 (vasodilatador). O
antagonista α2 também pode ser usado quando tem excesso de vasoconstritor local, obstrução urinária (relaxamento parcial da contração do músculo
liso), em caso de próstata aumentada e disfunção sexual masculina ( se associa papaverina que é um vasodilatador específico).
β-bloqueadores:
Falamos que eles são β-bloqueadores, porém isso não é totalmente verdade, pois eles podem ser antagonistas do receptor β e também
agonistas parciais. Mesmo que o fármaco seja agonista parcial ele está bloqueando o receptor β, porque ele está fazendo um efeito menor que o
agonista total, então as ações do β vão estar diminuídas. Os agonistas parciais fazem com que o receptor seja menos ativado do que se a NA ou
adrenalina se ligassem a ele.
Eles podem ter diferentes afinidades:
Antagonistas β não-seletivos bloqueia β1 e β2.
β2 seletivos nenhuma aplicação clínica (faria tanta broncoconstrição que iria asfixiar o paciente).
Os antagonistas β não-seletivos, *propranolol (RENAME), nadolol, timolol, pindolol e labetalol.
O propranolol é o β-bloqueador não-seletivo mais comum e que esta no SUS. Ele era utilizado para tratar angina. Usa-se β-bloqueador
para angina porque o coração esta recebendo menos irrigação, se diminuir o débito cardíaco diminui a necessidade de oxigenação do musc. cardíaco,
assim ele tem um efeito anti-anginoso. Porém perceberam que ocorria a queda da PA nestes pacientes que usavam propranolol para angina,
principalmente naqueles que eram hipertensivos. Sua eficácia anti-hipertensiva é melhor que a anti-anginosa e hoje é mais utilizado como antihipertensivo.
34
O propranolol é bem absorvido por V.O., a sua Cmáx é atingida entre 1 e 3 horas, tem baixa biodisponibilidade, pois sofre muito
metabolismo de primeira passagem e tem boa velocidade de absorção. Se o paciente tiver algum problema hepático a dose pode ser tóxica, pois ela
já é calculada sabendo que o medicamento irá sofrer MPP.
Entre os antagonistas β1 seletivos temos o *atenolol (RENAME), metoprolol, esmolol e acebutolol.
Não é somente o débito cardíaco que explica a diminuição da pressão, pois o organismo iria ativar o simpático e a pressão voltaria ao
normal, eles também interferem no sistema renina-angiotensina e tem uma ação central reduzindo a atividade simpática.
Tem receptor Β1 no coração e rins, no coração ele faz taquicardia e nos rins aumenta a liberação de renina. O Β2 aumenta a liberação de
NA. Então se bloquear Β1 no coração irá ocorrer a diminuição do débito cerdíaco, no rim ira diminuir a liberação de renina e bloquear Β2 na présinapse diminui a liberação de NA.
O rim libera renina que transforma angiotensinogênio em angiotensina I e a ECA transforma angiotensina I em angiotensina II. A angiotensina
II tem efeito vasoconstritor e estimula a liberação de aldosterona, por esta vasoconstrição e rejeição de sal pela ação da aldosterona vai ocorrer um
aumento da PA. O β-bloqueador vai diminuir todo esse sistema, diminuindo a produção de angiotensina II. Estes 3 mecanismos associados explicam o
mecanismo anti-hipertensivo.
Tem receptor β2 na musculatura lisa dos brônquios, ele faz a broncodilatação. Quando ocorre o bloqueio dos receptores β2 nos brônquios tem
uma broncoconstrição. Portanto outro efeito dos β-bloqueadores não-seletivos é a broncoconstrição (evitar uso em paciente asmático). Os seletivo β1
também causam broncoconstrição porém de menor intensidade.
A retirada brusca de um β-bloqueador pode causar efeitos negativos como nervosismo, taquicardia, angina e aumento da PA. Para alguns
pacientes causa diarréia, constipação, náusea, vômito e distúrbios do sono.
Farmacologia
14/05/2009
ANTIINFLAMATÓRIOS NÃO-ESTEROIDAIS (AINEs):
Os AINEs, em geral, apresentam 4 atividades terapêuticas:
Antiinflamatória
Analgésica
Antipirética
Antitrombótica
Alguns fármacos classificados como AINEs não tem atividade antiinflamatória. Dois exemplos são o paracetamol e a dipirona, que possuem
uma ação mais analgésica e antipirética.
Praticamente todos têm interferência na agregação plaquetária. Mas no uso terapêutico, só o Ácido acetilsalisílico é classificado como
antitrombótico.
A inflamação é uma reação de defesa do nosso organismo contra uma lesão, uma injúria que este organismo tenha sofrido. Pode ter uma
injuria por infecção de um vírus ou bactéria, por interação antígeno-anticorpo (ex: asma), por isquemia ou injúria física ou química.
Existem dois tipos de inflamações:
Aguda: resposta de defesa
Crônica: podem acontecer sem ter uma lesão nas células.
35
De uma maneira geral, os antiinflamatórios deveriam ser utilizados apenas para tratar de inflamações crônicas. Porém, às vezes, é necessário
tratar uma inflamação aguda porque ela pode estar trazendo prejuízo, dificultando o tratamento de um paciente.
Toda inflamação aguda tem quatro sinais cardeais:
Calor
Rubor
Dor
Edema
Quando o organismo entra em contato com o agente lesivo as células de defesa liberam mediadores químicos da inflamação como histamina,
serotonina, bradicinina, prostaglandinas, leucotrienos, fator de ativação plaquetária, interleucinas e fator de necrose tumoral.
A histamina e a prostaglandina são vasodilatadoras que causam um aumento sanguíneo no local e edema, pois ocorre um aumento da
permeabilidade do vaso e os elementos vão extravasar para o tecido acumulando liquido. A bradicinina e histamina (substâncias álgicas) estimulam os
receptores da dor (nociceptores), causando dor. As prostaglandinas são responsáveis pela dor inflamatória, elas são classificadas como hiperalgicas,
não estimulam os receptores da dor, porém aumentam a sensibilidade dos nociceptores para as substâncias álgicas. A prostaglandina sozinha não causa
dor, mas associada com histamina e bradicinina aumenta a sensibilidade destes receptores para a dor.
Os sinais secundários da resposta inflamatória são:
Adesão leucocitária ao endotélio lesado.
Quimiotaxia
Fagocitose
Na resposta inflamatória crônica não é necessário lesão para ser ativada, os sinais vão depender de onde esta ocorrendo a inflamação e
qual tipo de mediador químico está presente. Por exemplo, na asma o leucotrieno e a histamina são os mediadores que causam a broncoconstrição.
As prostaglandinas (PGI2 e PGE2) estão muito envolvidas com a inflamação, elas são responsáveis pela vasodilatação e aumento da
permeabilidade vascular. Associadas a outros mediadores são hiperalgicas.
Com um estimulo inflamatório que se liga ao mediador da inflamação e se liga a um receptor da célula, vai produzir a prostaglandina, este
mediador vai produzir a fosfolipase A2 (enzima de membrana que utiliza os fosfolipideos) para formar o precursor de mediadores que é o ácido
aracdônico. O ácido aracdônico pode sofrer ação de dois tipos de enzimas, a ação da COX (ciclo-oxigenase) que utilizam o ácido aracdônico para
formar os eicosanóides, entre eles a prostaciclina (PGI2) nas células endoteliais. A tromboxana (TXA2) formada pelas plaquetas está envolvida nos
processos de adesão plaquetária. E as prostaglandinas são formadas nos macrófagos (PGF2α).
O ácido aracdônico pode sofrer ação de outra enzima, a lipoxigenase, que produzirá os leucotrienos.
COX produz prostaciclinas, tromboxanas e prostaglandinas.
LOX produz leucotrienos.
Os AINEs inibem somente a COX elas não tem nenhum efeito sobre a LOX. Ele estará inibindo a produção de prostaglandinas e tromboxanas.
Os leucotrienos continuam a ser produzidos pela LOX.
Existem 3 tipos de COX (COX-1, COX-2 e COX-3). A COX-1 chama-se constitutiva, esta presentes nas células e regula processos fisiológicos. A
COX-2
Chama-se enzima induzida e esta presente em quantidade muito pequena, mas quando tem um processo inflamatório a COX-2 tem sua atividade
aumentada, ela é induzida nos processos inflamatórios.
36
A COX-1 que esta presente nas plaquetas é responsável pela produção das tromboxanas que são importantes para regular duas funções
fisiológicas principais, a ação agregante plaquetária e vasoconstritora.
A presença de tromboxana é muito importante para os processos de coagulação. Existe outro local em que aparece a COX-1, são as células
da mucosa gástrica. Estas células produzem PGI2 e PGE2, elas vão aumentar a produção de mucina e diminuir a produção de HCl. É preciso das PGS
para a proteção da mucosa gástrica, sem elas a mucosa fica muito suscetível a ação do HCl, pois perde a camada de muco protetor e aumenta a
secreção de HCl. Outra localização da COX-1 é nas células renais, vai promover no rim a natriurese (excreção renal de sódio).
O AINE inibe a COX-1 e a COX-2, a inibição da COX-1 pelos AINEs tradicionais leva a diminuição da produção dos subprodutos, por este
motivo que ocorrem todas as reações adversas.
Quando tem uso prolongado de AINE pode ocorrer hemorragia, pois ele esta inibindo tromboxana na plaqueta, pode ter problemas gástricos
pela inibição de PGI2 na mucosa gástrica, diminuição da função renal e aumento da PA pois está diminuindo a perfusão renal e retendo sódio no
organismo.
A COX-2 é induzida nos processos inflamatórios quando ocorre estímulo de substâncias pró-inflamatórias. A COX-2 vai produzir as
prostaglandinas que são responsáveis pelos processos inflamatórios, vão causar os 4 sinais cardeais da inflamação.
Quando os AINEs inibem a COX-2 ocorre o processo antiinflamatório e quando inibem COX-1 tem as reações adversas reduzidas.
Entre os AINEs tradicionais (não são totalmente seletivos), existe afinidade maior por COX-1 ou COX-2, porém ele inibe ambos. O AAS é mais
seletivo para COX-1, ele causa mais reações adversas que todos.
A COX-3 é responsável pela produção de substâncias antiinflamatórias e esta presente no SNC e no coração. Acredita-se que o paracetamol
tem uma seletividade maior para COX-3.
Os AINEs também tem ação antiempirética, a febre é um processo de defesa contra uma lesão. Na febre as células inflamatórias (fagócitos)
liberam mediadores e citocinas, estas citocinas vão estimular a COX, e a COX-2 induzida vai produzir as prostaglandinas que estão associadas ao
PGE2. A PGE2 desregula o ponto fixo do hipotálamo, faz com que ele tenha uma leitura errada da temperatura corporal reconhecendo que o corpo
está muito frio tentando aquecer o corpo diminuindo a perda de calor (imediata vasoconstrição), ressecando a pele e estimulando a produção de calor.
*Em um processo infeccioso para que é preciso aumentar a temperatura do corpo?
Porque em uma temperatura alta ocorre uma maior atividade das células de defesa do organismo. Na maioria das vezes a febre não precisa ser
combatida, se combate a febre quando o aumento da temperatura está causando dano. Medicamentos para a febre vão inibir a COX no SNC que vai fazer
uma diminuição das prostaglandinas e o hipotálamo volta ao normal.
Todo AINE tem efeito analgésico, pois na inflamação existe a ação das substâncias álgicas que causam dor, entre elas a bradicinina e a
histamina, elas estimulam os nociceptores. A prostaglandina por si não estimula os nociceptores, mas sensibiliza de forma que quando eles entrarem em
contato com as substâncias álgicas causam muita dor (acentuam a dor). Quando tomamos um AINE ele inibe a produção de substância hiperalgica
(substância que sensibiliza). O AINE diminui a dor, mas não inibe. Os AINEs são utilizados para tratar dor de leve a moderada, geralmente associada a
um processo inflamatório. A vantagem é que eles não causam dependência. Eles inibem a síntese de prostaglandinas que previnem o estado de
sensibilização dos receptores nervosos.
Todos AINEs causam efeitos colaterais como:
1. Ulceração gástrica intestinal.
Eles inibem a COX-1 constitutiva nas células da mucosa gástrica, diminuindo a produção de PGI2 e PGE2 que aumentam a produção de mucina
e diminuem a produção de HCl. Sem a PGI2 e a PGE2 vai diminuir a produção de muco protetor e aumentar a secreção de HCl, causando ulcerações.
37
2. Distúrbio de agregação plaquetária.
Os AINEs inibem a COX-1 na plaqueta que produz a tromboxana A2 que é uma substancia agregante plaquetária e vasoconstritora.
3. Inibição da função renal.
Inibe a COX-1 nas células renais que produzem a PGI2 e PGE2 que são vasodilatadores, aumentam a perfusão renal e promovem natriurese.
Se inibir isto por muito tempo pode ocorrer a perda da função renal e ter aumento da PA.
4. Retardo no trabalho de parto.
A gestante nos últimos três meses de gestação não pode fazer uso crônico de AINE, porque as prostaglandinas estão envolvidas com a
contração da musculatura lisa uterina. Com a diminuição das prostaglandinas pode ocorrer um retardamento no trabalho de parto e após ocorrer uma
hemorragia.
5. Reações de hipersensibilidade.
Causam muitas reações alérgicas em asmáticos. Nas células pulmonares o ácido aracdônico sofre ação de COX e LOX, ambas utilizam o ácido
aracdônico para a produção de seus subprodutos. O uso de AAS, principalmente, pode levar a um aumento da síntese de leucotrienos, pois a COX foi
inibida e a LOX, que produz leucotrienos, não tem mais competidor para usar o ácido aracdônico, e com o aumento de leucotrienos ocorre mais
broncoconstrição.
*Quanto tempo antes da cirurgia deve-se parar de tomar um antiinflamatório?
Depende de qual antiinflamatório estiver tomando, se estiver tomando AAS que é um inibidor irreversível da COX deve-se parar aproximadamente
uns 15 dias antes da cirurgia, pois as plaquetas voltam a produzir tromboxana só a partir de 14 dias. Os outros que são inibidores reversíveis deve-se parar
o uso 1 semana antes da cirurgia.
Classificação das CICLOOXIGENASES:
COX-1
COX-2
Induzida por processo inflamatório e
Interleucinas (IL1, IL2 e TNF-α)
Classificação
Essencial nos processos fisiológicos
Vasos Sanguíneos
Relaxamento vascular (PGE1, PGI), e
contração, aumento da
permeabilidade capilar (PGF, TXA)
Brônquios
Contração (PGF2, LTC, LTD, TXA) ou
relaxamento (PGE)
Rins (PGE1, PGI)
Mantém o fluxo sanguíneo renal em
pacientes com ICC, insuficiência renal
ou cirrose. Regula metabolismo de
sódio e potássio.
Aumentam na privação do sal.
Aumentam a formação de PGI2 e PGE2,
que estimulam a secreção de renina.
Plaquetas
Indução da agregação plaquetária
(TXA2) ou inibição (PGI).
Não é detectável.
Gestação/Parto
Induz a contração uterina (PGE,
PGF2α).
Possui expressão no epitélio uterino em
diferentes períodos da gestação inicial
e é importante na implantação do
embrião e na angiogênese necessária
38
para o estabelecimento da placenta.
SNC
Modulação do sistema neurovegetativo
e do processo sensorial (PGE2, PGD2,
PGH2).
Presente apenas no córtex, hipocampo,
hipotálamo e medula espinhal.
Febre
Há produção de PGE2 que ativa o
centro termorregulatório hipotalâmico.
Aumenta COX-2 no endotélio dos vasos
cranianos e micróglia.
Hiperalgesia
Potencializa a ação dos mediadores
da dor e sensibiliza os nociceptores.
Aumenta a imunorreatividade para
RNAm da COX-2.
Núcleo
Induz apoptose.
AULA 20.05 CONTINUAÇÃO DA AULAS DE AINES
A cox2 é uma enzima induzida nos processos antiinflamatórios, que se encontra em baixas concentrações nas células normalmente, e em processos
inflamatórios pode aumentar até 20 vezes mais. A cox1 é constitutiva, e está presente em quantidades importantes nas células, e produzem as
prostaglandinas e eicosanoides.
AINES Antiinflamatório não esteroidal (não hormonal).
Os AINES atuam principalmente pela inibição da cox2, que é induzida no processo inflamatório. Então seu efeito antipirético, analgésico e
antiinflamatório provavelmente se deve a essa inibição dessa cox. A inibição na cox 1 é, provavelmente, a principal causa de reações adversas
causadas pelos AINES, porque ela é constitutiva e produz aquelas substâncias para regular algumas funções fisiológicas, como: proteção da mucosa
gástrica, favorecimento da perfusão renal, favorecimento da natriurese, efeito agregante plaquetário, etc.
Todos os AINES são capazes de inibir a cox. E são divididos pela sua capacidade de fazer isso de maneira reversível ou irreversível. Somente um
deles inibe de forma irreversível, que é o AAS. Isso significa que a célula vai ter que sintetizar outra cox pra voltar a produzir os subprodutos dessa
enzima, já que a inibição é irreversível. Todos os outros AINES inibem de forma reversível, então a enzima pode voltar a produzir depois.
CLASSIFICAÇÃO DOS ANTIINFLAMATÓRIOS ANALGÉSICOS E ANTIPIRÉTICOS:
1)
Derivados do Ácido Acetilsalicílico ( Aspirina, Buferin, AAS)
CURIOSIDADE: O AAS é o único medicamento que tem o nome comercial utilizado como nome químico (aspirina), o único medicamento cujo nome
você encontra no livro de farmacologia. Esse medicamento se tornou tão consagrado que acabou ficando conhecido, embora seja uma marca
comercial.
2)
3)
4)
5)
6)
Derivados Indólicos (indometacina Indocid)
Derivados do Ácido Arilpropriônico (naproxeno, cetotofeno, ibuprofeno Profenid)
Derivados Fenâmicos (ácido mefenâmico Ponstan – utilizado para cólica menstrual)
Derivados Fenilacéticos (diclofenaco Cataflan e Voltaren)
Derivados dos Ácidos Enólicos (pirazolônicos, fenilbutazona “Tentrex”, Dipirona)
CURIOSIDADE: a Dipirona que é proibida em outros países como nos EUA e Canadá desde 1964, e é venda livre aqui.
7)
Derivados do Paraminofenol (paracetamol – acetaminofen - Tylenol)
IMPORTANTE: tem efeito apenas antipirético e analgésico, como a dipirona, e não possui eficácia antiinflamatória. (existem possíveis respostas,
não completamente aceitas)
39
8)
Sulfonanilida ( Nimisulide)
IMPORTANTE: tem também uma seletividade um pouco maior para cox2, mas não é altamente seletivo para essa cox. Esses fármacos que tem
maior seletividade para cox2, com a promessa de terem menos reações adversas gastrointestinais, não tiveram essa proteção gástrica
demonstrada na prática nos estudos clínicos.
9)
Seletivos cox-2
IMPORTANTE: Os únicos que revelaram uma proteção gástrica foram os altamente seletivos, mas apenas nos seis primeiros meses de uso. Depois
tanto faz usar um seletivo cox2 ou não seletivo que o dano gástrico é o mesmo. Agora há nova regra na prescrição desses medicamentos, visto que
causaram muitos problemas cardíacos e hepáticos. Três representantes foram retirados do mercado. E os que ainda são vendidos precisam ter
retenção de receita.
AAS: (conta o histórico do AAS, usado 400 anos antes de Cristo, com seus mecanismos descobertos quase 2000 anos depois)
Único que inibe de forma irreversível a cox1 e cox 2. E ainda é mais seletiva para cox1.
O que isso significa? Significa que se todos provocam ulceração gástrica, ela vai produzir mais. Se todos provocam problemas renais, vai produzir mais, se
produzem distúrbios de agregação plaquetária, vai produzir mais. Pelo fato de ser inibidora irreversível e ainda mais seletiva para cox1 (constitutiva), seu
perfil de reações adversas é pior.
** Usos Terapêuticos:
analgésico: para qualquer processo inflamatório (dor de leve a moderada);
antitérmico;
antiinflamatório;
Antitrombótico preventivo seu maior uso. É muito mais utilizada hoje pelo seu efeito antiagregante plaquetário do que por ser
antiinflamatório, analgésico e antitérmico.
Por que, se todos os AINES causam distúrbios de agregação plaquetária, só a aspirina é utilizada com esse fim terapêutico?
É a única usada porque é a única inibidora irreversível de cox1, então a posologia é menor e é mais seletiva a cox1.
Quem é que sintetiza a tromboxana2 (TX2)? A TX2 é sintetizada pela cox1, apenas. A cox2 não sintetiza TX2. Então a tromboxana é sintetizada pela
cox1 e a aspirina é mais seletiva para essa enzima e vai inibi-la irreversivelmente.
Perguntam à professora se não é melhor usar outro medicamento que não seja irreversível, mas que cause menos reações adversas a longo prazo (uma
vez que a aspirina produz muitos efeitos colaterais e mesmo assim é utilizada como antitrombótico):
PROFESSORA: se usasse um que não é irreversível e mais seletivo para cox2, ia ter que utilizar doses muitooo maiores do outro medicamento, que ia
produzir mais efeitos adversos do que a aspirina, que é usada em pequena dose para conseguir esse efeito antiagregante plaquetário. Se inibe cox numa
plaqueta esta não vai mais sintetizar tromboxina (na verdade vai diminuir a síntese). Então é muito vantajoso ser irreversível porque aquela plaqueta vai ter
síntese muito baixa de tromboxana durante toda sua vida. Como a plaqueta dura em média 7 a 14 dias, durante esse tempo haverá uma baixa produção de
TX2, e não há um bom efeito antiplaquetário; ou seja, será necessário produzir novas plaquetas para conseguir o efeito.
Como utilizam-se baixas doses diárias (35mg a 375mg/dia) tem-se um bom efeito antiagregante plaquetário, com a vantagem de não ter efeito
analgésico, antitérmico e antiinflamatório nessa dose. Então a possibilidade de reação adversa vai diminuir bastante. Para ter esses outros efeitos(antitérmico,
antiflamatório e analgésico) seria necessário uma dose se 1,5g/dia (que é muito acima da dose antiagregante plaquetária). Porém, como o medicamento é
administrado todos os dias(pela vida toda), como acumula no uso muito prolongado com o tempo pode aparecer problemas gastrointestinais e renais, mas
com o tempo. Se fizesse isso com o Cataflan , teria que usar a dose terapêutica dele, o problema gastrointestinal virioa antes. Na verdade, se fizesse isso
com qualquer outro AINE teríamos que administrar uma dose muito maior e as reações adversas seriam muito mais fortes.
Por isso ela é ideal entre os AINES para efeito antitrombótico. Mesmo que apareçam os problemas gastrointestinais, não se interrompe o
tratamento com a aspirina, nunca vai parar. É melhor tratar uma úlcera do que a pessoa ter um trombo ou um infarto. Se ela tiver a inibição da função renal,
comece a reter sal no organismo e aumentar a pressão, pode-se tratar com um antihipertensivo. Se ela tiver um problema gástrico, utiliza-se omeoprazol (que
é muito utilizado associado para os pacientes que utilizam as doses baixas de aspirina todos os dia, e tem problema gastrointestinal). Sempre o benefício que
ela pode trazer é maior do que os danos causados; porque qualquer risco, é possível tratar.
(Existem alguns outros antiagregantes, porém são muito mais caros, então o custo/benefício não compensa).
40
{p.s. ela fala sobre uma revisão da Lenita, sobre esses outros antiagregantes plaquetários e diz que cai na prova: Porque o AAS continua sendo o
único AINE prescrito como antiagregante plaquetário?}.
Porém o risco de causar hemorragia é maior.
Pergunta muito boa pra prova:
POR QUE A DOSE 35mg - 375mg TODO DIA DÁ UM EFEITO ANTIAGREGANTE MELHOR DO QUE TOMAR A DOSE 1,5g/dia QUE É A DOSE
ANTIINFLAMATÓRIA? Porque na dose alta (antiflamatória) inibe também a cox2 e a PGI2 (derivada de cox2), que tem efeito antiagragante plaquetário.
Na dose alta inibe a TX2, mas também a PGI2, que é antiagregante. Em dose baixa só inibe TX2 sem interferir com PGI2. Assim, inibe a produção de uma
substância antiagregante e inibe a produção da agregante também, causando mais hemorragia.
REVISANDO: Quando toma dose alta, inibe a produção de uma substância antiagregante plaquetário e ainda inibe uma substância agregante. Então diminui
o efeito agregante plaquetário. Quando tomo dose baixa eu interfiro apenas com a substância agregante, sem interferir com a antiagregante. Então a dose
baixa todo dia causa mais hemorragia do que se tomar a dose alta. Eu não quero inibir PGI2, se eu inibir PGI2 eu prejudico o efeito antiplaquetário.
Temos tambpem efeitos colaterais com uso da aspirina: hipersensibilidade cutânea, e reações alérgicas em pacientes asmáticos.
Por que a aspirina causa broncoconstrição em pacientes asmáticos? Porque quando eu uso aspirina eu aumento a produção de leucotrienos. Na célula da
mucosa pulmonar temos o ácido aracdônico sofrendo a ação da cox e lox. A cox produz prostaciclinas, tromboxanos (no caso da célula da mucosa pulmonar
não), prostaglnadinas; e a lox produz os leucotrienos. Quando uso a aspirina vou inibir somente a cox, ficando mais ac aracdonico disponível para lox. Ela
não tem mais com que competir pra produzir seus subprodutos. Então pode haver aumento da produção de leucotrienos, e o leucotrieno causa
broncoconstrição. Em paciente normal não e significativo,mas para o asmático que for hipersensível, a broncoconstrição vai piorar pode desencadear uma
crise, com o uso dos AINES, especificamente a aspirina. Ela é a que mais está relacionada com o aparecimento de broncoconstrição em asmáticos,
comparando com os outros AINES.
O paciente pode tomar aspirina, mas pra esse paciente deve-se evitar, ela pode tomar e fazer uma broncoconstrição; mas não são todos eles que
têm essa reação de hipersensibilidade. Pode piorar também para quem tem rinite, com mais coriza e mais espirros.
Criança com infecção viral não deve utilizar aspirina, por causa da Síndrome de Reye. Não se sabe porque o uso em crianças nessas
condições leva ao aparecimento da Síndrome. Mas a maior incidência da Síndrome foi em crianças até 18 meses em processo viral e que tomaram
aspirina por isso. Combinação de distúrbio hepático e encefaopatia (distúrbios de SNC), com mortalidade muito alta de 20 a 40%.é raríssima, mas
quando acontece a mortalidade é alta. Como é melhor previnir, nas bula do medicamento eles indicam a restrição para crianças de até 12 anos.
Utilizar paracetamol, dipirona.
Intoxicação aguda: maior nos EUA do que aqui no Brasil. Porque lá é vendido de maneira indiscriminada (cada frasco com 500 comprimidos)
nas prateleiras de mercado.
Na intoxicação aguda Náuseas, vômitos, hieperventilação, alterações do equilíbrio ácido-base, estimulação do SNC, hiperpirexia (aumento brusco
da temperatura corporal). Se nada for feito pode levar a depressão do centro respiratório e choque circulatório e morte. Emergência médica aguda.
Tratamento Suporte cardiovascular e respiratória. Correção das anormalidade ácido-base. Administrado bicarbonato intravenoso que ionizando o
ASS, acelerando a excreção, diminuindo a absorção (se ainda no estômago). Administração oral e tentar diminuir a absorção, no inicio. Porém quando
chega ao hospital já não dá mais e daí precisa ser de maneira intravenosa.
** Interação medicamentosa:
A Varfarina causa interação medicamentosa de duas maneiras: farmacodinâmica e farmacocinática.
FARMACODINÂMICA: a aspirina é um antiagregante plaquetário, e a varfarina é um anticoagulante oral; aumentando o risco de ter hemorragia.
É uma interação de adição ou sinergismo. Tem várias classificações para isso, só não é potencialização porque elas tem efeitos semelhantes.
FARMACOCINÉTICA: Tanto a aspirina quanto a varfarina são distribuídas no corpo ligadas a proteínas plasmáticas – albumina. Quando eu uso duas
juntas elas competem pela ligação com proteína, de forma que a que fica mais livre fica mais ativa e tem efeito maior. Quem fica mais livre é a
varfarina e, consequentemente mais ativa e então seu efeito anticoagulante é aumentado.
Não deve ser utilizando tratamento da gota (acúmulo de ácido úrico nas articulações) por reduzir a excreção renal de urato (interfere no
efeito de agentes uricosúricos como probenecida – no tratamento da gota, aumenta ainda mais a quantidade de ácido úrico nas articulações). A aspirina
compete com o ácido úrico pela excreção renal; a mesma proteína que excreta o ácido úrico no túbulo renal, excreta a aspirina. Quando tomo a aspirina
o ac. úrico fica no sangue, e dificulta a excreção; acumulnando mais ac. Úrico no paciente que já tem em excesso. Piorando a gota ao invés de tratar.
A aspirina e alguns outros AINES podem diminuir a eficácia de alguns hipertensivos. Tirando os bloqueadores dos canais de cálcio que são
vasodilatores e os antagonistas da angiotensina2 (losartan, candersatan), todos os outros precisam de quantidades fisiológicas de prostaglandinas no
41
organismo pra ter seu efeito antihipertensivo ótimo, porque elas são vasodilatadoras. Quando inibi-se as prostaglandinas pode se perder a eficácia do
captopril, e dos beta-bloqueadores como o propranolol.
Aspirina é um ácido fraco, sofre absorção já no estômago, mas é melhor absorvida no intestino. Rápida absorção, sendo metabolizada 75%
no fígado. Tem maior taxa de excreção em urina alcalina.
p.s. A professora pergunta se já pode perguntar tudo sobre aspirina:
Por que a aspirina causa gastrite?
Por que causa inibição da função renal?
Por que ela é a única escolhida como antiagregante plaquetário?
Por que não pode associar com varfarina? Qual o tipo de interação que ocorre?
Outras atividades terapêuticas da aspirina (além de antiagregante plaquetário):
- prevenção de câncer de cólon: em alguns cânceres de cólon, as células cancerosas têm uma produção excessiva de cox2, como a aspirina inibe cox 1 e
2 irreversivelmente, pode ser utilizada.
- tratamento de diarréia (só em diarréia que é causada por radiação, por excesso de prostaglandina atuando em musculatura lisa).
- doença de Alzheimer: reduzir o risco e retarda início (uso muito discutido); pois idosos que usavam aspirina tinham menos tendência de fazer
Alzheimer, do que pacientes que na faziam uso. 0
DERIVADO FENILACÉTICO:
DICLOFENACO (Cataflan, Voltaren)
Esse medicamento é equipotente. Isso significa que inibe tanto cox1 e cox2,com a mesma seletividade (de maneira equipotente).alguns artigos
dizem que é um pouco mais seletivo para cox2, mas na monografia do diclofenaco diz que inibe da mesma forma.
Usos terapêuticos:
Inflamações crônicas: artrite, reumatismo, gota.
Inflamações agudas: pós operatórias e odontológicas.
Possui os mesmos efeitos colaterais que a aspirina, em relação aos problemas gastrointestinais e renais, mas com menos reações de hipersensibilidade
em asmáticos (talvez porque ele inibe síntese de leucotrineos-leucócitos) (menos broncoconstritor), com rinite.
Os mais conhecidos são o Diclofenaco de sódio (voltarem); Diclofenaco de potássio (cataflam).
Existe uma crença de que o cataflan é mais eficiente para tratar as inflamações relacionadas a mucosa e tecidos moles, e o voltaren é mais
utilizado para as inflamações osteomusculares.
Dicofenaco de sódio X Diclofenaco de potássio
Não existe diferenã de indicação para a indicação deles para determinadas infamações.NÃO EXISTE ESSA DIFERENÇA DE INDICAÇÃO. Pode-se usar
tanto cataflan quanto voltaren para as duas indicações. Só existe uma diferença entre eles, é o Tmáx (tempo para atingir a dose máxima no sangue
com dose única). A Tmax do cataflan é menor que a do voltaren. Então o cataflan tem maior velocidade de absorção: porque quanto maior o Tmax
maior velocidade de absorção. Porém não muda em nada a indicação clínica, nem os efeitos colaterais. Não traz nenhuma diferença de eficácia,
apenas na posologia.
PIRAZOLÔNICOS
DIPIRONA: analgésico – antipirético. Atividade antiinflamatória não existe na clínica.
Inibe cox reversivelmente e seu efeito de dura de 24 a 48 horas. Então, apesar de ser reversível, a inibição é prolongada. Mais eficiente que a aspirina
e paracetamol para seu efeito antitérmico e analgésico (porém isso ainda é controverso).
42
p.s. tem uma revisão da Lenita sobre isso que vai cair na prova: mostra que na prática, nos ensaios clínicos a dipirona não apresentou resultados
melhores no efeito antitérmico e analgésico, embora na experiência da professora como mãe haja diferença. A Lenita diz que o paracetamol tem
reações adversas do tipo A (reação adversa comum, que é previsível nos usos terapêuticos; tem alto índice de morbidade, mas baixo de mortalidade). E
a dipirona tem reação adversa do tipo B (raras, mas são indiossincrasias – reação adversa rara que pode levar ao risco de morte e que acontece em
uma pequena parcela de indivíduos).
Reações adversas: hipotensão, robemas de coagulação sanguinea (uso prolongado), agranulocitose (grave e rara). Esse reação adversa que levou os
EUA a proibirem o uso desse medicamento lá. Retirada desde 1964.
Agranulocitose: definição: severa e seletiva neutropenia(inibição muito grande na produção de neutrófilos, e o organismo fica sem defesa, deixando o
paciente sucetível a infecções que se tornam graves nesses pacientes) .
No Brasil é venda livre, é o anador, novalgina, neosaldina. Estudos e revisões recentes discutem a segurança de dipirona. Estudos indicam que
é mais uma guerra de mercado. A indústria de paracetamol era mais forte na época. A agranulomatose é muito rara, e o paracetamol causa mais
complicações hepáticas do que a dipirona causa agranulomatose.
“Dipirona, por seus potenciais efeitos adversos, não deve será primeira escolha em pacientes febris que possam ter acesso a outras alternaticas
igualmente eficazes e mais seguras.” – Lenita, que concorda com a proibição.
Agranulocitose pode aparecer em uma pequena dose, dose teraupeutica. Não há como prever.
DERIVADOS PARAMINOFENOL
PARACETAMOL Analgésico e antipirético.
Inibe cox de forma reversível e bem mais rápida. Porém tem uma seletividade maior para cox3. A cox3, possivelmete uma variante da cox1,
encontra-se distribuída principalmente no córtex cerebral, medula e coração, sendo mais sensível ao acetaminofreno(paracetamol) do que a cox 1 e 2.
A cox3 produz substâncias antiinflamatórias (as interleucinas antiinflamatórias). Se tem um AINE mais seletivo para uma cox que produz substância
antiinflamatória, do que pra cox2 (que produz substâncias inflamatórias), provavelmente seja por isso que não tenha uma eficácia antiinflamatória tão
boa. Uma possibilidade para que o paracetamol tenha efeito apenas analgésico e antipirético.
Indicados para paciente que não podem tomar AAS (asmáticos, ulceríticos, com rinites, hemofílicos). A dose terapêutica para um adulto é de
3g/dia. Altas doses podem causar necrose hepática fatal. Acima de 4g/dia considera-se uma dose hepatotóxica de paracetamol. A prescrição é de 1
comprimido a cada 6 horas, então se tomar 4 comprimidos (750mg cada) por dia já chega aos 3g diários; dois comprimidos a mais já está
administrando a dose que pode ser hepatotóxica.
Isso pode gerar intoxicação em crianças, porque as mães confiam muito no paracetamol e podem exagerar na dose. Além disso, as
associações anti-gripais também contêm paracetamol (todas). Temos muito mais problemas com o paracetamol do que com a dipirona. A morbidade
relacionada ao paracetamol traz muito mais prejuízo. Para criança: 1 gota para kg de peso da criança, no máximo 40 gotas (até 12 anos, depois dose
de adulto).
Por que o paracetamol causa hepatotoxicidade? Se aumentar a dose as vias de conjugação não dão conta de metabolizar, vai ocorrer mais
oxidação microssomal, mais formação desse intermediário tóxico, e mais hepatotoxicidade I(mostrando figura).
Para finalizar ... os seletivos cox2!!!
INIBIDORES SELETIVOS COX2
Ver artigo da Lenita (muito importante).
Perguntam para a professora: Quem é essa Lenita?hahaha
Por que fizeram o seletivos cox2? Com a idéia de que ser tivesse maior seletividade para aquela enzima induzida nos procesos inflamatórios, haveria menos
reação adversa, maior eficácia antiinflamatória, analgésica, antitérmica. Porém a maior eficácia não aconteceu. Eles têm a mesma eficácia antiinflamatória,
analgésica e antitérmica que os outros AINES têm. Porém as reações adversas de danos gástricos, eles mostraram menores. Produzem menos lesões gástricas,
mas nos primeiros 6 meses de uso. Depois desse período tanto faz tomar um seletivo ou não seletivo.
O Vioxx foi o primeiro e ficou disponível por 4 anos. Nesse período, só nos EUA mais de 21 mil pessoas infartaram.
43
CELECOXIB E ROFECOXIB: inibidores cox2. Estudos clínicos indicaram que acentuavam problemas cardiovasculares em pacientes portadores de
cardiopatias, porém isso não foi levado como uma contra-indicação absoluta e ele foi prescrito para todo mundo. Foi retirado do mercado em 2004.
Antiagregante e vasoconstritora. Formação de trombo aumentada e infarto por isquemia. Aumento de pressão arterial, insuficiência cardíaca
congestiva, hepatotoxicidade, infarto agudo do miocárdio.
Após a proibição foi criada uma regra no Brasil, para continuar prescrevendo os seletivos cox2:
CATEME/2005: prescrição somente para pacientes com altos risco de problemas GI com uso dos não seletivos e sem história pessoal ou
familiar de problemas cardiovasculares. (muito poucos seguiram a regra)
A cox2 é induzida. Mas a gente tem em menores quantidades em algumas células do corpo. E essas quantidades produzem substância que
são importantes fisiologicamente. A gente tem prostaglandinas derivadas de cox2 (que não é induzida no processo inflamatório, porque ela está
constitutiva) que tem uma importante ação na manutenção do fluxo sanguíneo renal e na excreção de sódio. Aquele defeito inibitório crônico (visto com
o uso de AINES) dessa PGI2 derivada da cox2, na retenção de sódio e _______ pode conribuir para a retenção deste íon e consequentemente a
resistência à terapêutica antihipertensiva.
Tanto o AINE seletivo como o não seletivo inibem a cox2, então ambos podem levar a um aumento da pressão. Por que só o seletivo cox2
deu tanto infarto? Porque além dessa PGI2 no rim, a gente tem essa PGI2 nas células endoteliais do vaso, que são derivadas da cox2. e essa PGI2 é um
importante antiagregante plaquetário, e vasodilatador. Quando eu tomo um esletivo cox2 eu inibo somente essa PGI2,sem interferir com a TXO2, porque ela
é derivada de co1.
O que vai acontecer com o uso prolongado? Vai diminuir a produção de uma substancia antiagregante, sem interferir com a produção da substancia
agregante, que é também vasoconstritora. O conseqüente desequilíbrio a favor dos fatores pró-trombóticos(porque a tromboxana está sendo produzida
normalmente e só a PGI2 está inibida), pode levar a agregação plaquetária e vasoconstrição; com maior tendência para oclusão vascular e isquemia tissular.
Foi cancelado o registro: prexige 400mg retirados do mercado em 2007 na Austrália. No Brasil não foi cancelado. De lá pra cá houve
aumento significativo de hepatotoxidade por isso. Então a ANVISA cancelou em outubro de 2008 o prexige 400mg e arcóxia 120mg.
Os seletivos podem ser prescritos, mas a receita fica retida na farmácia. Passaram a ser controlados. (mostrou um vídeo).
**** VER: Possíveis razões para efeitos colaterais cardiovasculares dos seletivos cox2 (slide) não tem em nenhum livro:
•
•
•
“Prostaglandinas derivadas da COX-2 também podem ter papel critico na manutenção do fluxo sangüíneo medular renal e excreção de
sódio. A perda do efeito inibitório tônico da PGE2 (derivada da COX-2) na reabsorção de sódio em tais segmentos pode contribuir para a
retenção deste íon e conseqüente resistência a terapêuticas anti-hipertensivas, vistos com o uso de AINES (Komhoff M, 2000).
Inibidores específicos COX-2 suprimem a produção de PGI2 pelas células endoteliais, enquanto não apresentam efeito na produção de TXA2
plaquetário (Lipsky PE, 2001).
O conseqüente desequilíbrio a favor de fatores pró-trombóticos pode levar à agregação plaquetária e vasoconstrição, com maior tendência
para oclusão vascular e isquemia tissular (Emery P,2001). Apesar de ser uma preocupação teórica, ainda não existem evidências clínicas que
suportem tal risco (Schnitzer TJ ,2001).”
Transcrição da aula de farmacologia do dia 21/05 - AIES
AIES (anti-inflamatórios esteroidais)
[anti-inflamatórios hormonais/corticóides/glicocorticóides]
muito mais potentes que os anti-inflamatórios tradicionais
Não são utilizados como primeira escolha para inflamação, pois eles tem um perfil de efeitos colaterais muito maior do que os AINES .
Quando usa:
o
Uso é reservado para situações em que se comprovou real eficácia
o
Quando os AINES não tem eficácia nessas situações,
o
Exemplos: Asma (AINEs até pioram a asma), falha terapêutica com o uso dos AINES.
Adicionar à terapia um corticóide ou substituir a terapia por um corticóide.
Tal cautela se deve aos efeitos adversos generalizados desses agentes.
44
Existem certas patologias imunes inflamatórias que se não fossem os corticóides, não teríamos como tratar.
Os glicocórticoides são sintetizados no córtex da supra-renal, nas células corticais (diferente da adrenalina, sintetizada nas células cromafins
da medula da supra-renal).
O estímulo vem de um hormônio também bastante conhecido que é o ACTH, ou hormônio adrenocorticotrófico, liberado pela hipófise.
O estímulo para que a hipófise libere o ACTH vem do CRF, que é o fator liberador de corticotropina, produzido pelo hipotálamo.
Basicamente: hipotálamo libera CRF, CRF atua sobre a hipófise, hipófise libera ACTH, ACTH atua sobre a supra-renal, liberando os
glicocorticóides.
O glicocorticóide não é armazenado em vesículas, é sintetizado e liberado conforme recebe o estímulo do ACTH, ou seja, conforme a
necessidade do organismo.
Pode ter vários tipos de estímulos para o hipotálamo liberar o CRF. Um dos estímulos é o stress, nós sabemos que num momento stress nós
temos alta liberação de glicocorticóides.
Nas células do córtex da supra-renal há o receptor de ACTH, um receptor metabotrópico, ligado à proteína G e o estímulo de um agente
estimulatório vai levar à produção de ACTH.
Além dos glicorcorticóides, o córtex da supra-renal libera os mineralocorticóides, sendo que o estímulo da liberação dos mineralocorticóides é
o sistema renina-agiotensina.
A angiotensina-2 também tem receptor nas células corticais, e quando ela as estimula ela faz com que as células corticais liberem o principal
mineralocorticóide, a aldosterona.
Ação dos Glicocorticóides:
Muitas ações periféricas. Eles estão envolvidos no metabolismo de carboidratos, metabolismo de proteínas, metabolismo lipídico e em
atividades anti-inflamatórios e imunossupressoras.
Porquê que a gente precisa de um hormônio que tem ação anti-inflamatória, se a inflamação é defesa? Para que a nossa inflamação não seja
exacerbada. Se a gente não tivesse glicocorticóide, toda inflamação aguda iria evoluir para uma inflamação crônica.
Acredita-se que muitos pacientes que desenvolvem inflamações crônicas tenham ou diminuição da liberação ou falta da eficácia desses
glicocorticóides endógenos em sua ação antiinflamatória.
O glicocorticóides também tem ação imunossupressora para que o sistema imunológico também não tenha uma ação tão grande que acabe
desenvolvendo uma doença auto-imune e ele se torne descontrolado.
O glicocorticóide tem uma alça longa de retroalimentação negativa sobre o hipotálamo, isto é, quando a gente tem altas doses de
glicocorticóide no sangue, o hipotálamo é inibido de forma que ele não libere mais CRF e, assim, diminuindo a produção de glicocorticóides
endógenos. O glicocorticóide regula sua própria liberação, altos níveis sanguíneos vão diminuir sua síntese pela supra-renal.
Resumidamente, as principais ações dos glicocorticóides endógenos são: estão envolvidos no metabolismo de glicose, proteínas, etc; estão
envolvido no equilíbrio hidroeletrolítico (eles também contribuem para a quantidade de água e eletrólitos no nosso sangue); tem efeito de
retroalimentação negativa sobre a hipófise anterior e o hipotálamo; e tem os seus efeitos antiinflamatórios e imunossupressores.
o
Efeitos metabólicos: Em relação aos efeitos metabólicos e sistêmicos. Em relação aos carboidratos, podemos dizer que o
glicocorticóide tem ação contrária à da insulina. Enquanto a insulina trabalha para aumentar a captação de glicose pela célula, o
glicocorticóide diminui a captação de glicose pela célula e aumenta o nível de glicose circulante através do aumento da lise de
glicogênio (glicogenólise) e pela formação de glicose (aumenta a gliconeogênese). O uso prolongado de glicocorticóide pode levar
a uma diabetes medicamentosa [uma hiperglicemia medicamentosa]. Além disso, podemos dizer que o glicocorticóide diminui síntese
e aumenta a degração proteica. O trabalho dele é fazer com que o músculo tenha maior degradação e menor formação de
proteínas. Uso prolongado pode levar à fraqueza muscular. Quando utilizado na infância, pode prejudicar o crescimento, ficando
um adulto de baixa estatura. Altas doses por longos períodos podem levar à síndrome de Cushing, que acontece porque os
glicocorticóides têm um efeito permissivo sobre os hormônios lipolíticos, fazendo com que a gordura se redistribua de forma anormal
pelo organismo. Assim, o paciente começa a ter depósito de gordura onde normalmente não aconteceria e acaba desenvolvendo
uma característica física. O uso prolongado também faz com que atuem nos receptores dos mineralocorticóides, como aldosterona.
Nós sabemos que a aldosterona promove a retenção de sódio pelo organismo. Por isso, é comum que os pacientes achem que
engoradaram no começo do uso. Na verdade, imediatamente – se forem altas doses - pode ter um edema. Por longo período, mais
tarde pode aumentar o peso, pois pode aumentar a fome, o apetite, a pessoa passa a comer mais e pode engordar.
o
Efeito de retroalimentação negativa: o uso de glicocorticóide sintético diminui a produção de CRF e ACTH e, consequentemente,
diminui a produção de glicocorticóide endógeno, fazendo com que a supra-renal pare de trabalhar. Pode acontecer, num uso
prolongado, uma atrofia dessa supra-renal (ela não produzir mais glicocorticóide endógeno). Por isso nunca se pode interromper
uma terapia prolongada de forma brusca, uma vez que a supra-renal não está totalmente funcional e os níves de glicocorticóide no
45
sangue do paciente caem bruscamente, podendo gerar anorexia, depressão, hipotensão, fraqueza muscular generalizada e
mialgias (dores musculares). Tanto no excesso quanto na falta, os glicocorticóides fazem fraqueza muscular. No excesso, porque ele
diminui a síntese proteica e aumenta a degradação. Na falta, é por toda a diminuição energética que a redução dos níveis de
glicocorticóide causa. Nesses casos, a supra-renal fica totalmente inútil, disfuncional, para a produção de glicocorticóide sim.
Entretanto, ela pode continuar produzindo mineralocorticóide e adrenalina. Depois que você começou a diminuinr as doses, a suprarenal volta a produzir entre dois e dezoito meses
o
Em relação aos efeitos anti-inflamatórios e imunossupressores, eles inibem tanto as manifestações iniciais quanto tardias da
inflamação. Os AINEs inibem as manifestações iniciais da inflamação: a dor, o calor, o rubor e o edema, ou seja, diminuem os
sintomas. O AIES inibem, além das manifestações iniciais, as manifestações tardias. Os efeitos sobre isso são:
Cicatrização fica dificultada. É um prejuízo, a gente nunca quer que a cicatrização fique dificultada, mas fica. Ao mesmo
tempo, eles inibem as reações proliferativas da inflamação crônica. Então, eles vão inibir também aquela fibrose da
inflamação crônica que pode levar à perda funcional do órgão inflamado. Sempre o risco-benefício tem que ser muito
bem avaliada na prescrição de glicocorticóide, principalmente quando for uma prescrição de uso sistêmico. Uso tópico,
esses efeitos são bem diminuídos.
Eles afetam qualquer reação inflamatória, qualquer tipo de reação estimulada por qualquer tipo de agente lesivo.
Inflamação por patógeno invasor (viral, bacteriana), inflamação por estímulos físicos ou químicos e em repostas imunes
inadequadas. São primeira escolha para inflamação devido a respostas imunes inadequadas, principalmente no caso
da asma (lembrar que os AINES são pouco eficazes nesse tipo de inflamação). Eles vão exercer seus efeitos
antiinflamatórios através de ações sobre os vasos sanguíneos, sobre as células da inflamação e sobre praticamente todos
os mediadores da inflamação (todos eles terão sua atividade e síntese diminuída). Nas células da inflamação, a
atividade diminui pela redução da liberação de mediadores. Nos vasos sanguíneos, vão fazer a mesma coisa que os
AINES: vão diminuir a vasodilatação e a permeabilidade vascular. Consequentemente já diminui o calor, o rubor e o
edema. Em relação às células inflamatórias, eles diminuem a saída dos neutrófilos dos vasos sanguíneos e diminuem a
atividade tanto dos neutrófilos quanto dos macrófagos que estão nos tecidos. Isso é feito pelas alterações na síntese
proteica na célula, que é possível pela alteração promovida na transcrição gênica da célula. Para eles diminuirem a
saída e a atividade de neutrófilos e macrófagos, eles fazem isso alterando a transcrição dos genes que produzem fatores
de adesão celular e citocinas (que são importantes para a chegada dos neutrófilos no local de inflamação). Eles reduzem
a ativação e a expansão clonal das células T, alterando a transcrição de genes entre a Leucina-2 (?) e seu receptor, que
estão envolvidas nas funções dessas células. Eles reduzem a função dos fibroblastos, diminuem a produção de colágeno e
diminuem a cicatrização e o reparo das feridas alterando a transcrição gênica das células. Em relação aos mediadores
da inflamação, comparando com AINES que só diminuem síntese de prostaglandinas, os AIES diminuem todos os
mediadores de inflamação. A produção de todas as citocinas inflamatórias fica diminuída. Eles vão dimunuir a produção
das interleucinas 1 até 8, do fator de necrose tumoral e qualquer outra citocina envolvida na inflamação. Eles diminuem a
produção dos eicosanóides (eicosanóide é todo derivado do ácido aracdônico), então eles diminuem prostaciclina,
prostaglandina, tromboxana e leucotrienos, pois eles diminuem a expressão da Cox-2, diminuem a síntese do fator de
ativação plaquetária e diminuem até a síntese do ácido aracdônico. Reduzem o sistema complemento do sangue, reduzem
a produção de óxido nítrico, reduzem a liberação de histamina pelos basófilos e reduzem de a produção de igG, por isso
eles também são imunossupressores.
As substâncias antiinflamatórias endógenas têm a sua produção aumentada, incluindo aquelas citocinas que têm ação de
inibir a inflamação. Eles aumentam a síntese: a interleucina 10, o receptor solúvel da interleucina 1 e a anexina 1.
Resumindo: glicocorticóide diminuem tudo que contribui para a inflamação e aumenta tudo que contribui para o efeito
anti-inflamatório.
Os glicocorticóides vão agir em receptores da superfamília de receptores nucleares, os receptores intracelulares. Esses são que estão no
citoplasma. É uma proteína que está solúvel no citoplasma. Isso é possível porque o glicocorticóide é lipossolúvel, é altamente apolar. Por isso
eu tenho uma boa ação de glicocorticóide por qualquer via de administração. Esse tipo de receptor intracelular é muito parecido com os
receptores dos mineralocorticóides, esteróides sexuais, hormônios da tireóide, etc. Por isso, às vezes, em altas doses, o glicocorticóide pode
atuar num receptor de mineralocorticóide (são receptores parecidos, proteínas parecidas).
O glicocorticóide circula no sangue ligado a uma proteína ligadora de glicocorticóide, que a gente chama de GDG. Todo glicocorticóide é
distribuído no nosso organismo ligado a essa proteína, sendo que somente a fração livre é que vai exercer efeito. A fração ligada não vai
conseguir atravessar a membrana celular. A fração livre tem uma lipossolubilidade suficiente para atravessar a membrana celular e entrar no
citoplasma, por um processo de difusão passiva. No citoplasma, ele vai encontrar sua proteína receptora ali solúvel no citoplasma. Ela está
complexada com outras proteínas, chamadas de proteínas de choque térmico. Porém, quando o esteróide se liga a essa proteína, ela sai de
seu complexo com as outras proteínas e se dimeriza, se duplica e muda sua conformação. O glicocorticóide ativa essa proteína que não tinha
função importante ali no citoplasma. Essa ativação, essa mudança conformacional, leva à exposição dos dedos de zinco. Isso significa que ela
fica ativa para se complexar com um pedacinho da fita de DNA daquela célula. Então um complexo esteróide-receptor ativado penetra no
núcleo e vai se ligar na fita de DNA dessa célula. Todas as nossas células contêm receptores para glicocorticóides, em maiores ou menores
quantidades. Esse complexo esteróide-receptor ligado ao DNA da célula vai alterar a transcrição gênica, vai alterar a tradução, vai alterar
a síntese de RNA mensageiro, consequentemente, vai alterar a síntese proteica. Alterar quer dizer, ora aumenta a síntese proteica, ora
diminui a síntese proteica. Vai depender de que célula ele está se ligando e que função aquela célula regula. Se eu tiver uma doença que
diminua a síntese da GBG, eu pode haver mais toxicidade por glicocorticóide.
As respostas da administração de glicocorticóides podem levar de horas a dias para acontecer. Glicocorticóide não tem ação rápida,
diferentemente dos AINEs.
46
Recentemente descobriu-se que quando o glicocorticóide interage com essa proteína solúvel, ele já desencadeia processos de transdução de
sinal no citoplasma da célula. Esses processos de transdução de sinal levam a alguns efeitos anti-inflamatórios mais rápidos. Então às vezes a
gente tem em algumas horas efeito analgésico do glicocorticóide, diminuição do edema, do inchaço. Para esses efeitos foram descobertos que
a interação desses receptores no citoplasma já leva à produção de amp cíclico, já leva à fosforilação proteica e já leva a alguns efeitos antiinflamatórios. Essas são as ações rápidas dos glicocorticóides. Porém, as verdadeiras ações deles, as duradouras, vão acontecer com a
ligação do DNA, e aí você vai ver essas ações mais duradouras num tempo mais longo.
Para a interação do glicocorticóide com o DNA, tem-se quatro situações:
o
1º. mecanismo de transativação: Há um pedacinho da fita de DNA onde o glicocorticóide se liga. Esse pedacinho é nomeado
elemento de resposta aos glicocorticóides. O glicocorticóide ativa o receptor, tendo uma resposta positiva e levando à ativação
da maquinaria transcrição da célula, aumentando a síntese proteica.
o
2º. mecanismo de trans-repressão: O glicocorticóide se liga nesse elemento de resposta negativa, desloca o fator de transcrição e
não deixa a maquinaria de transcrição ser ativada, diminuindo a síntese proteica da célula.
o
3º. mecanismo AP1 e 4º mecanismo NF capa aberta. Existem outras duas situações, outros dois lugares da fita de DNA onde o
glicocorticoide pode se ligar e também reduzem síntese protéica. Normalmente, esses fatores de transcrição ativam a maquinaria
de transcrição. Vem o glicocorticóide, se liga nesses fatores de transcrição e não deixa a maquinaria ser ativada, diminuindo a
síntese proteica. Ou eu tenho um fator de transcrição aqui, P65 e P50, que normalmente ativam a maquinaria de transcrição,
chega o glicocorticoide, se liga, não deixa esses fatores de transcrição se ligarem nessa região (chamada NF capa aberta) e eu
não tenho ativação da maquinaria. Os exemplos 2, 3 e 4 são exemplos onde o glicocorticóide diminui a síntese proteica. O
exemplo A é quando o glicocorticóide aumenta a síntese protéica.
O receptor de glicocorticóide, ainda que com diversas ações, é só um sempre o mesmo: o GR-α. Existe outro tipo de receptor, o GR-β, mas
não tem no organismo dos mamíferos.
Exemplo prático: Os glicocorticóides diminuem a produção de todos os eicosanóides, de todos os derivados do ácido aracdônico, por diminuir
a produção dos prostanóides (que são os derivados da COX), através da inibição da COX-2. Ele inibe a síntese da COX-2 reprimindo
aqueles fatores de transcrição AP1 e NF capa aberta. Esses fatores de transcrição são importantes para ativar o gene que produz a COX.
Se inibe esses fatores, não ativa o gene que expressa a COX na célula e sem a COX-2 na célula eu não tenho todos os derivados dela, todas
as prostaglandinas que estão envolvidas no processo inflamatório. Ao mesmo tempo, ele induz a formação da anexina 1 (glicocortina), que é
uma proteína que inibe a fosfolipase A2, ou seja, uma substância anti-inflamatória. A fosfolipase A2 precisa ser ativada para transformar
fosfolipídeo em ácido aracdônico por estímulo inflamatório. A anexina 1 inibe essa enzima, não deixa essa enzima transformar fosfolipídeo
em ácido aracdônico e sem ácido aracdônico eu não tenho leucotrieno, prostaglandina, prostaciclina, tromboxana... São dois exemplos
mostrando que o aumento da síntese proteica leva a ação antiinflamatória e a diminuição da síntese de outra proteína leva a ação
antiinflamatória.
Efeitos de glicocorticóides sobre os ossos: degrada o osso, aumentando a atividade dos osteoclastos e diminuem a síntese das células que
formam a matriz óssea, os osteoblastos. Para piorar, eles aumentam a excreção renal de cálcio e diminuem a absorção intestinal de cálcio.
Isso significa que o uso prolongado leva à osteoporose, que é tida como osteoporose muito grave.
Tanto pessoas esperando por transplante renal quanto aqueles já sofreram o transplante usam glicocorticóide de maneira excessiva. Aquele
que não foi transplantada, para não perder totalmente a função do rim e aquela que foi transplantada, para não ter rejeição.
Como os glicocorticóides são imunossupressores, eles deixam o paciente suscetível a infecções, tendo uma diminuição da sua resposta de
defesa num processo infeccioso (tanto que o paciente transplantado usa máscara o tempo, pois ele está suscetível a contrair infecções).
Síndrome de Cushing. É uma síndrome que é facilmente reconhecida, devido a suas características físicas: redistribuição anormal da
gordura (a pessoa tem depósito de gordura na face, deixando a face em lua cheia; depósito de gordura na coluna cervical, levando à
corcova de búfalo; aumento da gordura abdominal) e ao mesmo tempo, como tem degradação de proteína, leva ao afinamento de
membros superiores e inferiores, braços e pernas finos. Os pacientes de Cushing também estão muito mais suscetíveis a pegar gripes,
resfriados, infecções fúngicas. Na mucosa oral, eles tem muita cândica, pois eles estão com o sistema imune comprometido. Além de a maioria
deles ter atrofia da supra-renal. Eles podem desenvolver equimoses pelo corpo (porque, se diminui a produção de ácido arcdônico, a
tromboxona é derivada do ácido aracdônico através da COX-1, tem menos ácido aracdônico, então eu tenho agregação plaquetária
prejudicada, aparecendo as manchas roxas). Pode acontecer hipertensão, pois começa a atuar em receptor da aldosterona e a reter sal no
organismo e consequentemente aumentar a pressão. Pode ter um aumento do apetite, podendo levar à obesidade. Pode ainda aparecer
cataratas. Se parar de usar, o estado não é totalmente reversível. Algumas coisas sim, outras não. A pessoa pode desenvolver síndrome de
Cushing por uma superprodução glicocorticóide endógeno (ela está produzindo em excesso). Aí o que que eu tenho que utilizar? Não existe
um antagosnista de receptor de glicocorticóide, então tem que descobrir a causa do excesso da produção de glicocorticóide. Se for por um
excesso na produção de ACTH, eu tenho antagonistas de receptores de ACTH. Se for um tumor na supra-renal, aí vai ter que usar agentes
anti-neoplásicos para reverter o tumor, cirurgia.
INDICAÇÕES PARA O USO DE GLICOCORTICÓIDES:
Menos indicado [aqui a professora se contradisse]: para a terapia de reposição. Existem pessoas que tem insuficiência da supra-renal,
não produzem glicocorticóides endógenos e tem que repor. Para esses pacientes, o aparecimento de reações adversas diminui bastante,
já que eles têm falta de glicocorticoide.
47
Para todas as outras patologias, os efeitos adversos aparecem mais. Na terapia anti-inflamatória e imunossupressora, eles são indicados
na asma (primeira escolha) (?)imunodeficiencia (?), uso tópico em várias condições inflamatórias da pele, olhos, ouvidos e nariz. Por
exemplo, eczema, aquela alergia que o médico não sabe o que é, dá glicocorticóide que 70% dos casos vão sumir – se não sumir, ele
vai procurar o que é. Conjuntivite, rinite alérgica – aliás, o glicocorticóide é muito utilizada na forma nasal para pacientes com rinite –
estados de hipersensibilidade (aquelas reações alérgicas graves a picadas de insetos e a medicamentos, o glicocorticóide está em
primeira escolha)
Doenças com componente auto-imune envolvido (aquelas doenças como artrite reumatóide, que pode ser uma doença auto-imune),
Prevenção rejeições de órgãos transplantados
Doenças neoplásicas.
Em caso de choque anafilático: não usa só o glicocorticóide. Nesse caso, são duas aplicações: adrenalina imediatamente, para fazer a
broncodilação e o glicocorticóide, para que todos aqueles mediadores da resposta que estão causando o edema, como a histamina, que
estão causando broncoconstrição, comecem a ter sua síntese diminuída.
DETALHES DOS GLICOCORTICÓIDES:
Tem quatro coisas que todo glicocorticóide precisa ter:
o
1º. Precisam ter 21 átomos de carbono.
o
2º. Entre o carbono C4 e C5 tem que ter ligação dupla.
o
3º. No carbono C3, eu tenho que ter um grupo cetona.
o
4º. E no C21, esse grupamento hidroxila
São apresentados como ésteres, acetonidos ou sais. Têm-se os ésteres de glicocorticóides, acetanos, benzoatos, butiratos, diacetatos (acetato
de betamesona, benzoato de dexametasona, etc, etc). Eles podem ser acetonidos ou sais: fosfato sódico de dexametasona, succinato sódico
de tal tal tal..). A diferença é os ésteres e os acetonidos são os pró-corticóides, que é um pró-corticoide o pró-fármarco, de forma que ele só
vai se tornar ativo depois que sofrer o metabolismo no organismos.
o
Os ésteres e os acetonidos tem que sofrer hidrólise no organismo para liberar o corticoide ativo.
o
Os sais já são o corticoide ativo.
A diferença na prática é um maior tempo de ação dos pró-corticóides do que dos corticóides já ativos.
RENAME 2007: Meticortem (que é a prednisona) e o Predsin (prednisolona). A prednisolona é um fármaco ativo, a prednisona é um prófármaco. Metil-prednisolona é a forma injetável e está na Rename. A prednisona é a forma em compromido. A dexametasona é o que mais
tem no SUS, em comprimido, pomada e xarope. A hidrocortisona, muito utilizada na terapia de reposição para quem tem insuficiência da
supra-renal. Atriancinolona é utilizado na forma de pomada, muitas vezes para infecções bucais. A betametasona (Celestamine), muito
utilizada para quem tem asma na forma de xarope e injetável. Beclometasona, que é o Clenil. Budesonida, que é a bombinha para quem tem
asma. Esses são todos os medicamentos disponíveis no mercado, sendo que os dois primeiros os que constam na RENAME (medicação
essencial).
As diferenças entre eles são a via de administração, potência anti-inflamatória e tempo de ação.
o
Via de administração: qualquer via eu posso usar para glicocorticóide (via oral, via tópica, via parenteral). Os glicocorticóides
endógenos se ligam a globulina de ligação de glicocorticóide, como eu falei. Os sintéticos se ligam à albumina. De qualquer
forma, eles vão ser distribuídos ligados a proteína. Se houver desequilíbrio nessa ligação, eu posso ou ter perda de eficácia ou
aumento de efeito. Eles penetram na célula por difusão (porque são altamente lipossolúveis) e são altamente metabolizados no
fígado.
o
Interação medicamentosa: Cuidar com o uso concomitante ao daqueles fármacos que forem indutores ou inibidores enzimáticos.
Os antiácidos, por mecanismo desconhecido, diminuem o efeito farmacológico dos corticóides administrados por via
oral.
Antifúngicos azóis(?), como o traconazol, fluconazol, que são inibidores enzimáticos, podem aumentar a toxicidade
do corticosteróide, porque vão inibir seu metabolismo, como qualquer outro inibidor enzimático. Ex: Alcoolismo
agudo, cimetidina, eritromicina.
Indutor enzimático: Acelera o metabolismo e pode perder eficácia. No caso dos barbitúricos, que são inibidores
enzimáticos, eles podem tanto diminuir o efeito dos corticosteróides, como ter seu efeito diminuído pelos corticóides.
Anticoncepcional hormonal: duas interações.
48
o
o
Naturalmente, os anticoncepcionais já causam edema e os corticóides também. O risco de aumento do edema e
da pressão arterial é maior – isso é uma relação farmacodinâmica.
Mas, ao mesmo tempo, reduz-se o metabolismo do corticóide pelo uso do estrógeno e posso ter as
concentrações de corticóide no sangue aumentadas, então também vai aumentar o risco de todos os efeitos
tóxicos.
Nesses casos, troca-se o fármaco ou se acrescenta um diurético (um terceiro fármaco) aos fármacos já prescritos resolve o
problema do edema, mas não resolve os outros problemas e ainda acrescenta os efeitos colaterais do diurético (poderia
causar um desequilíbrio eletrolítico nessa paciente, que só está precisando do diurético porque está usando dois
hormônios. É mais racional tirar um hormônio do que acrescentar o diurético).
Tempo de ação:
ação curta são os que tem ação que duram entre 8 a 12 horas
ação intermediária duram de 12 a 36 horas
ação longa, de 36 a 72 horas.
Potência anti-inflamatória e na capacidade de causar edema, de reter líquido. Os que têm maior potência antiinflamatória e não
atuam em receptor mineralocorticóide (quer dizer, não retem líquido no organismo). São eles: betametasona, dexametasona e a
triancinolona. Já a hidrocortisona tem a capacidade máxima de causar edema e uma baixa potência antiinflamatória, é mais
utilizada em terapia de reposição para aquele paciente que não produz glicocorticóides endógenos. Betamesona e dexametasona
são muito utilizadas como anti-inflamatórios, na forma de pomada, de xarope, de comprimido. “Esses aqui” em vários usos: na asma,
como anti-inflamatórios, terapia de reposição. O médico vai avaliar a indicação baseado nesses fatores.
Farmacologia
27/05/2009
ANTI-HISTAMÍNICOS E ANTI-ULCEROSOS
Só se chama de anti-histaminico os antagonistas H1, os outros são chamados de antiulcerosos.
HISTAMINA
Detectada a 1ª vez em esporões de centeio. Fazia contração uterina e outros músculos lisos e ao mesmo tempo tinha ação vasodilatadora.
Perceberam que pessoas expostas a antígenos apresentavam sintomas semelhantes à intoxicação por esporão de centeio.
Isolaram a histamina e descobriram 4 receptores: H1, H2, H3 e H4.São todos metabotrópicos. Existem fármacos antagonistas para todos os
receptores de histamina.
A histamina tem 3 funções:
1-
Está envolvida nas respostas alérgicas. Mediador principal dos sintomas alérgicos, principalmente aqueles relacionados à rinite alérgica e à
urticária.
2-
Está envolvida na secreção de HCl pela célula parietal. É o principal estimulante.
3-
Na periferia e no SNC funciona como neurotransmissor regulando a liberação de outros neurotransmissores.
O mastócito armazena a histamina nos tecidos e o basófilo no sangue. Os mastócitos que liberam a histamina responsável pelos sintomas de
hipersensibilidade. Essas reações atingem primariamente 3 tecidos, devido à grande quantidade de mastócitos neles presente:
1 - pele (prurido, hiperemia, pápulas e eritemas)
2 – brônquios (broncoconstrição)
3 – intestino (cólicas, aumento do peristaltismo)
49
A histamina é sintetizada a partir da histidina por uma descaboxilase presente em todos os tecidos que contém histamina. Ela é degradada
por dois tipos de enzimas: tiamina oxidase e n-metil-transferase. Seus metabólitos são inativos. Existem algumas células que sintetizam e liberam
constantemente a histamina (sem armazenar). Na epiderme, na mucosa gástrica e nos neurônios histaminérgicos.
Os anti-histamínicos influenciam na resposta alérgica e na liberação de HCl.
Papel da histamina na resposta alérgica
Ela é o principal mediador e estimula a liberação de outros mediadores que vão contribuir para os sinais e sintomas. O complexo
antígeno/anticorpo se liga na superfície dos mastócitos e basófilos. O receptor ao qual ele se liga é metabotrópico e a sua proteína G ativa a
fosfolipase C com aumento de IP3 e DAG, o que, consequentemente, aumenta a entrada de cálcio (intra ou extracelular), causando a liberação da
histamina. A proteína G também ativa fosfolipase A2, responsável pela produção do ácido araquidônico, o que leva à liberação de outros mediadores
inflamatórios (fator de agregação plaquetária, leucotrienos, eicosanóides, PGs, cininas, entre outros). Os anti-histamínicos, portanto, diminuem a
produção de histamina e de outros mediadores.
Existem dois tipos de reações de hipersensibilidade imediata: local e sistêmica. A anafilaxia acontece se houver uma liberação de histamina
rápida e em todos os tecidos, pode levar ao edema de glote e broncoconstrição (tto: adrenalina e corticóide).
Alguns antibióticos, bloqueadores neuromusculares, contrastes radiológicos, morfina e outras substâncias podem provocar a liberação de
histamina sem formação de complexo antígeno/anticorpo, podendo levar a uma anafilaxia.
Então, grande liberação de histamina tem como sintomas: prurido (principalmente na palma das mãos, face e couro cabeludo), hipotensão,
aumento da F.C, edema, urticária, cólicas, náuseas, hipersecreção gástrica e broncoconstrição. O efeito se torna menos intenso com o tempo porque vão
se acabando os estoques de histamina dos tecidos.
O teste alérgico utiliza a histamina como controle positivo, para comparar a região onde a histamina foi aplicada com as outras regiões onde
foram administrados complexos antígeno/anticorpo variados. Única aplicação clínica da histamina.
Os anti-histamínicos são metabolizados no fígado (interação).
Receptores
H1
Ligado à proteína G, ativa fosfolipase C – IP3 e DAG – aumenta cálcio intracelular.
- Presente no SNC (hipotálamo), quando ativados pela histamina aumenta o estado de vigília, por isso os anti-histamínicos causam sono.
- Presente na musculatura lisa dos brônquios e do TGI. A histamina causa contração.
- Presente nas células endoteliais dos vasos. A histamina estimula a liberação de óxido nítrico, por isso a histamina causa vasodilatação.
- Presente nas terminações nervosas, por isso causa prurido quando histamina é liberada na epiderme.
H2
Acoplado à proteína Gs – aumenta AMPc
- Presente nas células parietais do TGI. Essas células são responsáveis pela produção de HCl, têm receptores que ativam a bomba de prótons que por
sua vez jogam H para a luz gástrica. Esses íons reagem com o Cl e formam HCl. O AMPc formado pela ativação dos receptores H2 ativa essa bomba
de prótons. Essa bomba pode ser ativada também pela acetilcolina e gastrina, mas os antagonistas H2 são os mais utilizados como anti-ulcerosos
(CIMETIDINA), mesmo sem interferir com a acetilcolina e gastrina geram uma diminuição de 90% na secreção de HCl. Isso prova que a histamina é o
50
principal estimulante para a secreção gástrica estomacal. Os inibidores de bomba de prótons (OMEPRAZOL), são ainda mais eficientes (95%) porque
inibem de forma irreversível e inibem os estimulo vindo da acetilcolina e gastrina também.
- Presentes na musculatura lisa dos vasos, faz vasodilatação.
- Presentes no coração gerando taquicardia.
H3
Acoplado a uma proteína G inibitória que diminui AMPc.
- Presente no SNC. É um auto-receptor inibitório. Quando ativado pela histamina inibe a liberação da própria histamina e de outros neurotransmissores.
Antagonistas H3 em testes podem aumentar a liberação desses neurotransmissores, podendo um dia ser utilizados para melhorar a memória e estado
de vigília.
H4
Acoplado a uma proteína Gi inibe adenilato ciclase e imobiliza o Cálcio armazenado.
- Presente em eosinófilos
- Presente em células do TGI
- Presente em células do SNC
A ativação periférica pela histamina resulta em quimiotaxia, recruta células inflamatórias. Antagonistas desse receptor podem ser úteis como
antiinflamatórios, estão em ensaios pré-clínicos.
ANTI-HISTAMÍNCOS
1ª GERAÇÃO:
PROMETAZINA
DEXCLOFENIRAMIDA (Histamin)
HIDROXIZINA
Antagonistas reversíveis H1. Descobriu-se que são na verdade agonistas inversos desses receptores. A proteína G tem atividade sem ativação
do receptor. Dessa forma, o antagonista se ligando ao receptor, diminui essa atividade constitutiva do receptor. Os efeitos farmacológicos são os
mesmos.
Principal reação adversa dos de 1ª geração: sedação em doses terapêuticas. Não devem ser ingeridos com álcool. (Prometazina + álcool).
Possuem também ação antagonista nos receptores muscarínicos, causando todos os efeitos anticolinérgicos: boca seca, visão turva, taquicardia, retenção
urinária. São utilizados para as reações de hipersensibilidade imediata. São mais eficazes na prevenção (rinite sazonal) que no tto. No resfriado comum
o anti-histamínico não é indicado por ser um fármaco ressecante, uma vez que o muco é importante para eliminar o vírus. Possuem também propriedade
anti-hemética diminuindo os efeitos da cinetose pela ação nos receptores nos núcleos vestibulares.
2ª GERAÇÃO:
LORATADINA
ALEGRA
51
Causam menos sedação porque atravessam menos a BHE.
1ª GERAÇÃO
2ª GERAÇÃO
- atravessam BHE
- atravessam menos a BHE
- baixo peso molecular
- alto peso molecular
- lipossolúveis
- menos lipossolúveis
- maior afinidade H1 no hipotálamo
- menor afinidade por H1 no hipotálamo
* só os de 1ª geração são eficazes para efeito anti-hemético e
sedativo.
- maior afinidade H1 na periferia (ação antialérgica melhor)
- maior cardiotoxicidade
* doses acima das terapêuticas podem gerar depressão do SNC
FARMACOLOGIA
28/05/09
ANTI-HIPERTENSIVOS
Sistema Cardiovascular:
-Função: fornecer um aporte de oxigênio e de nutrientes para os tecidos, e ao mesmo tempo remover os produtos de desgaste, que as células não
precisam mais. Para que ele possa cumprir essa função de maneira adequada é necessária uma adequada perfusão tecidual. Então, a gente precisa
que o aporte sanguíneo chegue de maneira adequada aos tecidos, para que esses processos ocorram de maneira adequada também.
-Componentes: esses componentes vão trabalhar juntos pra que esses tecidos e essas células tenham uma perfusão adequada. O coração é uma bomba
propulsora. Os vasos são dutos fechados que são classificados em artérias e veias -- as artérias levando o sangue do coração para os tecidos, as veias
fazendo o caminho inverso. O próprio sangue, elemento fundamental do sistema cardiovascular. E o rim, responsável por manter um volume adequado
de sangue (ou de liquido circulante) nesse sistema, e também responsável pela composição desse liquido circulante (quantidade de sais e eletrólitos).
Fármacos que atuam sobre o rim para controlar a pressão arterial- Diuréticos
Fórmula da regulação da pressão arterial: PA = DC x RVP
Débito cardíaco: DC = VS x FC
52
VS = volume sistólico: volume de liquido que o coração ejeta durante a sístole
FC = freqüência cardíaca
Então a força de contração, e a quantidade de vezes que o coração bate (se contrai) durante o tempo vai resultar no debito cardíaco. Claro que
quanto maior for esse debito cardíaco maior será a pressão arterial. Mas não é só o debito cardíaco que controla a PA, mas também a resistência
vascular periférica. A RVP é determinada pelo tônus vascular.
Regulação do tônus vascular: nós temos vários sistemas que interferem com a contração e o relaxamento dos vasos sanguíneos.
- Sistemas neurais: o Sistema Nervoso Simpático interfere com contração.
- Sistemas humorais: hormônios que são liberados na corrente sanguínea e vão regular também o tônus vascular, como a adrenalina e a angiotensina II,
que faz parte do sistema renina-angiotensina. E há os hormônios locais, que regulam o fluxo sanguíneo em determinadas regiões dos tecidos: hormônios
locais vasodilatadores, como prostaglandina e histamina; e vasoconstritores, como a tromboxana e a própria angiotensina II quando ela atua em
receptores AT1.
Então, a PA é regulada pelo DC e RVP. Esses dois fatores são regulados pelo Sistema Nervoso Simpático. O SNS tem conexão com artérias, vênulas,
coração e rins. SNS ativado faz, de maneira geral, vasoconstrição (via receptores alfa-1) . Via receptor beta-2, o SNS faz vasodilatação. No entanto,
quando os dois receptores estão ativados, o resultado será vasoconstrição e conseqüentemente aumento da PA. No coração, o SNS aumenta o debito
cardíaco porque aumenta tanto a força quanto a freqüência de contração cardíaca via ação do receptor beta-1. Nos rins, o SNS estimula a liberação
de renina, via receptores beta também. Liberando renina, todo o sistema renina-angiotensina será ativado, então como resposta haverá aumento de PA.
Então, sistema nervoso simpático trabalha, de uma maneira geral, para aumentar a pressão arterial. Há uma classe de fármacos que irá interferir com
a ação do simpático nessas regiões, os simpaticolíticos.
Mecanismo Arco-reflexo Barorreceptor: são receptores de pressão nos vasos sanguíneos. Quando a gente tem uma vasodilatação esses receptores
detectam que o vaso está vasodilatado. Para que a gente não tenha uma queda de pressão exagerada, os barorreceptores irão ativar, de forma
reflexa, o Sistema Nervoso Simpático. Então, o SNS faz a vasoconstrição e a PA é regulada. Não há uma classe de fármaco que aja sobre esse sistema
com aplicações terapêuticas. Porém, esse sistema é importante pra se explicar reações adversas de alguns anti-hipertensivos. Muitas reações adversas
causam como efeito colateral a hipotensão postural, que é aquela hipotensão que se tem quando se muda de posição, e a vista escurece. Isso acontece
porque, quando eu uso vasodilatador, esse mecanismo dos barorreceptores dos vasos fica comprometido.
Mecanismo humoral Sistema Renina-Angiotensina: porque os fármacos que interferem com o sistema renina-angiotensina provocam queda de
pressão? Será que é só porque diminui síntese de angiotensina II? Ou existem outras coisas decorrentes dessa diminuição que também explicam o efeito
anti-hipertensivo?
Hormônios locais: regulam a perfusão tecidual em determinadas regiões do organismo. Tromboxana, como vasoconstritora; o óxido nítrico, potente
vasodilatador; fármacos vasodilatadores que sao doadores de NO para os vasos; e as prostaglandinas, vasodilatadoras também. Quando eu inibo a
síntese dessas prostaglandinas, mesmo que localmente, dependente da região, eu posso perder o efeito anti-hipertensivo de alguns medicamentos,
porque a síntese normal de prostaglandinas em determinadas regiões é muito importante.
(Figura: vcs todos tem acesso a ela pelo site do ministério da saúde. Eu deixei pra vcs o sexto consenso brasileiro sobre o tratamento da hipertensão
arterial anexado na aula de hoje. Se vcs entrarem no site do Ministério pra pegar esse censo vcs vão ter acesso a figura. Ela dá uma visão geral do
controle da hipertensão arterial e eu consigo colocar a ação de qualquer fármaco que a gente for estudar nessa figura.)
A figura mostra, em primeiro lugar, o mecanismo reflexo dos barorreceptores. Então a gente tem os receptores de pressão na parede do vaso, quando
esse vaso tem uma dilatação esses receptores detectam que houve uma vasodilatação e eles estimulam uma região do sistema nervoso central, chamado
núcleo do trato solitário, a ativar o sistema nervoso simpático. O SNS irá fazer três coisas. No vaso, vai fazer vasoconstrição, via receptores alfa-1. No
rim, vai estimular a liberação de renina. E no coração, vai aumentar o debito cardíaco. Quando eu estimulo a liberação de renina eu ativo todo o
sistema renina-angiotensina, aumentando a produção de angiotensina II. A angiotensina II tem dois mecanismos pra aumentar a pressão arterial: vai
fazer a vasoconstrição, via receptor AT1, e vai estimular liberação de aldosterona pela supra-renal. Nós vimos que esse mineralocorticóide tem dois
estímulos para a sua liberação: um do ACTH e outro da angiotensina II. Liberando a aldosterona, ela vai ter ação no rim, promovendo reabsorção de
sódio e excreção de potássio. Isso vai fazer com que eu acumule liquido e vai aumentar a pressão arterial também. Então tudo está relacionado. Aqui
mostra aqueles hormônios locais vasodilatadores e vasoconstritores. Angiotensina II e tromboxana é vasoconstritor; oxido nítrico, bradicinina, PGI II e a
angiotensina II quando se liga no receptor AT2 é vasodilatador. Os fármacos que vão atuar sobre o rim, nós não temos aqui os mecanismos e vamos
detalhar pra vocês. Todos os fármacos anti-hipertensivos vão interferir de alguma forma nesses processos. Os inibidores da ECA vão interferir aqui(?), os
antagonistas de receptores da angiotensina II vão bloquear receptor AT1, os beta bloqueadores vão atuar no coração, no rim e no SNC, os antihipertensivos de ação central vão atuar no SNC e os diuréticos vão atuar no rim.
Bom, qual que é a principal patologia predominante do sistema vascular? é a hipertensão. Ela é definida como uma elevação persistente da PA. E o
que é considerado uma pressão alta? é uma pressão que se mantém constantemente em valores acima de 139/89 mmHg. Segundo o Goodman, o valor
ideal, saudável para todos nos deveria se manter em torno de 120/80. Qual é a conseqüência de ter a PA acima desses valores o tempo todo? Eu não
vou conseguir garantir a perfusão tecidual adequada. E se alguns tecidos chave no meu organismo não receberem uma perfusão adequada eu posso
53
trazer prejuízo para esses tecidos. Os três tecidos importantíssimos de se manter a perfusão são cérebro, coração e rim. Se eu tiver uma perfusão
inadequada para eles eu posso ter conseqüências graves. Quais? Os acidentes vasculares cerebrais (AVC); no coração, as cardiopatias, fulminando num
infarto do miocárdio; e no rim eu posso ter desde uma insuficiência renal aguda até uma insuficiência renal crônica, com perda da função renal. A
hipertensão arterial é grave porque ela não apresenta sintomas. Se você tiver constantemente uma pressão de 15/9, você não sente nada. Pode sentir
uma dor de cabeça, mas esse é um sinal de tantos outros problemas. Só há sintoma da PA alta quando algum desses órgãos já foi tingido e dai já é um
pouco tarde.
Existem dois tipos de hipertensão.
-
Hipertensão Secundária, quando ela tem causas especificas (primárias) : felcromocitoma (tumor na drenagem), problema congênito impedindo
a função renal adequada. Corresponde a 10% dos casos.
Hipertensão Primária ou Essencial: sem causas especificas -> pode ser fator genético, stress, hábitos e costumes da vida moderna
(sedentarismo, dita rica em sal e gorduras, hábitos de fumar e beber). Corresponde a 90% dos casos.
(A segunda doença mais prevalente na vida moderna é a depressão que também está associada com tudo isso.)
Hipertensão é assintomática ate que um órgão alvo seja atingido.
Como tratá-la? Duas maneiras. Abordagem terapêutica e não terapêutica. Independente de os medicamentos serem utilizados ou não, sempre deve-se
utilizar a abordagem não terapêutica: evitar tabagismo, álcool, fazer prática de exercício físico, reduzir peso corporal, dieta e reduzir stress. Tudo isso
deve ser acompanhado junto com o tratamento farmacológico. Se eu trabalhei com o paciente a abordagem não farmacológica e não adiantou então
eu vou auxiliar com a abordagem farmacológica. É claro que a maioria dos pacientes já procuram o médico quando a abordagem não farmacológica
não adianta sozinha, e o médico vai ter que entrar com as duas abordagens (fármaco+mudança de estilo de vida). Mas nunca um fármaco controla a
hipertensão arterial sozinho se não for associado a mudanças de hábitos de vida errados. Por isso o médico e a equipe de saúde devem estar em
contato com o doente pra verificar essa mudança.
Bom, vamos falar de medicamentos então. Os fármacos que vão controlar a pressão arterial podem ter ação sobre 1, 2, 3 ou 4 dos locais que
controlam a PA. E eles são classificados de acordo com o seu mecanismo de ação.
-
Diuréticos: ênfase nos tiazidicos, diuréticos de alça e poupadores de potássio
Agentes Simpaticolíticos: antagonistas beta-adrenérgicos, ou beta-bloqueadores; antagonistas alfa-adrenérgicos; e agentes de ação
central (ênfase na metildopa que é muito utilizada na hipertensa grávida).
Bloqueadores de canal de cálcio: os vasodilatadores verapamil, holandipino e etc.
Inibidores da ECA: captopril e inalapril.
Antagonistas do receptor da angiotensina II: toda a família losartan, tandesartan, ibesartan e etc.
Vasodilatadores inespecíficos: os arteriais e os arteriais e venosos.
Diuréticos
(Vamos lembrar coisas básicas que eu já falei na aula de excreção de drogas)
Os diuréticos são fármacos muito antigos que tem sido usados há muito tempo como tratamento de várias afecções (edema, mulheres que
querem emagrecer). Os diuréticos são os medicamentos de primeira escolha (é a primeira tentativa) para tratar hipertensão leve e moderada, quando
o paciente possui poucos fatores de risco. O primeiro diurético que foi lançado no mercado sinteticamente, como fármaco diurético, foi lançado em
1957. Antes disso se usavam chás e plantas diuréticas. A clorotiazida foi o protótipo de todos os diuréticos. É bom a gente definir e diferenciar aqui
dois termos. Na verdade esses fármacos são tanto diuréticos quanto natriuréticos. Natriurese é a capacidade de excretar sal (sódio) pelos rins. E o
efeito diurético é o aumento do volume de urina, a capacidade de excretar água. Todo diurético que nós vamos estudar aumenta tanto a excreção do
sódio como da água, mas nós vamos usar apenas o termo diurético referindo-se tanto a natriurese como a diurese.
Onde diurético age no organismo? Vamos lembrar de alvos protéicos. Existem quatro alvos protéicos: receptor farmacológico, enzima, canal
iônico, proteína transportadora. Os diuréticos podem agir em qualquer um desses alvos protéicos mas a maioria atua em proteínas de transporte.
Aquelas proteínas que estão no néfron fazendo constantemente o transporte de íons da luz do néfron para o vaso sanguíneo. A maioria vai impedir esse
transporte de íons. Porém, há diuréticos que atuam em receptor farmacológico, como a espirolactona; em enzima, como a cetazolamida (que não tem
mais uma aplicação como diurético); em canal iônico, como a amilorida e o triantereno (impedindo a ação da aldosterona sobre os rins). Então a gente
vai ter diuréticos atuando em todos os tipos de alvo protéico que a gente conhece.
No néfron. O néfron é a unidade funcional do rim. (desenho do néfron). Cada parte do néfron tem uma função diferente, de forma que o liquido que é
filtrado aqui no glomérulo é totalmente diferente do liquido que sai aqui. Conforme ele vai passando por determinadas regiões dos túbulos renais, as
características desse liquido (como a pressão osmótica, ou seja, a concentração de água e eletrólitos) vai mudando porque cada região do néfron tem
uma permeabilidade diferente a sais e eletrólitos e também cada região sofre ação de hormônios: aldosterona (retenção de sódio) e hormônio
antidiurético, o ADH ou vasopressina, interferindo com a característica desse liquido que vai caminhar por todo o túbulo renal.
54
No glomérulo, onde ocorre a filtração glomerular. O liquido que chega em contato com o glomérulo corresponde a 20% do fluxo sanguíneo renal.
Esse liquido é lançado nos glomérulos através da força hidrodinâmica e para que um soluto possa atravessar um glomérulo, ele precisa ter certas
características porque ele vai ter que atravessar três camadas de membrana celular: o capilar sanguíneo, a membrana basal e as células epiteliais da
cápsula de Bowman. Só atravessam as moléculas com peso molecular pequeno (< 20 000 daltons). Proteínas não atravessam (albumina = 80 000
daltons). Há muitos íons, muitos eletrólitos que conseguem atravessar as camadas e já entram aqui junto com a água através do processo de filtração.
No túbulo contorcido proximal (TCP). 60 a 70% do sódio que é filtrado já é reabsorvido para o sangue através do TCP. Esse processo é feito por uma
proteína de transporte, uma bomba de sódio-potássio-ATPase. Porém não existe nenhum diurético que bloqueia essa bomba, ou seja, que atua no TCP
inibindo a reabsorção de sódio. 75% do filtrado é reabsorvido em condições isosmóticas. Ácidos e bases orgânicas e muitos medicamentos são
secretados para a luz tubular e sódio, cloro, glicose, bicarbonato são reabsorvidos para o sangue. Então é uma região bastante permeável, tanto aa
água quanto a eletrólitos. Há aqui uma enzima, conhecida como anidrase carbônica. Ela está no interior e na parte interna da membrana das células do
TCP. Promove a reabsorção do bicarbonato de sódio. Sem ela, a gente perderia muito bicarbonato de sódio na urina. (essa figura e todas as figuras
que eu vou mostrar aqui são retiradas do livro de farmacologia básica do Katzung). Então como acontece essa reabsorção de bicarbonato de sódio
para o sangue? A gente tem aqui uma proteína de transporte uma bomba de sódio-hidrogênio, que absorve o sódio pra dentro da célula. Ela troca o
sódio pelo hidrogênio. Quando o hidrogênio é liberado ele vai reagir com o bicarbonato e vai formar o ácido carbônico. A anidrase carbônica que
está na MP da célula desidrata esse ácido carbônico em água + CO2. Esse CO2 entra por difusão passiva na célula do TCP e é re-hidratado pela
anidrase carbônica e é transformado em acido carbônico outra vez. Então é a anidrase carbônica que faz desidratação na luz e re-hidratação no
interior da célula. Esse acido carbônico se ioniza em íon hidrogênio e bicarbonato. O bicarbonato é reabsorvido para o vaso sanguíneo por um
mecanismo de transporte e junto com o sódio ele vai se transformar em bicarbonato de sódio. Então, se não fosse a anidrase carbônica fazendo
desidratação e re-hidratação do ácido carbônico a gente não teria a reabsorção de bicarbonato de sódio. O que aconteceria se a gente usasse um
fármaco que inibisse essa enzima, como é o caso da cetazolamida? Acumulo de bicarbonato de sódio na luz do néfron porque ele não será reabsorvido
(acidose é conseqüência). O que acontece se eu aumentar a quantidade de soluto no liquido? A pressão osmótica vai aumentar. Eu aumentei a
osmolalidade desse liquido, aumentei a quantidade de soluto. O qq vai acontecer se eu tiver uma membrana separando dois líquidos, um com uma
pressão alta e outro com uma pressão normal? Eu vou precisar equilibrar a pressão aqui dentro com o vaso sanguíneo. Então o qq o vaso sanguíneo vai
fazer? Vai diluir esse liquido fornecendo água para a luz e eu vou ter o efeito diurético. Todo diurético que inibe reabsorção de íons vai fazer o efeito
diurético porque quando acumula soluto na luz do néfron, aumenta a pressão e para equilibrar essa pressão com o vaso vai drenar água do vaso para
a luz do néfron. Mas a diurese será alcalina, porque está acumulando bicarbonato de sódio; e se usar por tempo prolongado vai promover uma
acidose metabólica. Por causa desse efeito da acidose, e pelo efeito diurético limitado, a cetazolamida (inibidora da anidrase carbônica) não é
utilizada como diurético hoje. Ela é utilizada para tratar pressão intra-ocular (glaucoma), e também para acelerar a excreção de substancias ácidas
que estão em grande quantidade no sangue, porque ela alcaliniza urina, vai ionizar essas substancias e eu posso tratar uma intoxicação por uma
substancia acida. Resumo do mecanismo de ação da cetazolamida: inibiu a enzima, não vou reabsorver bicarbonato de sódio, ele vai se acumular na luz
da urina, vou aumentar pressão aqui, vou drenar água e ter um efeito diurético.
Na alça de Henle. Ela é dividida em duas partes, o ramo descendente e o ramo ascendente. Eles são totalmente diferentes em relação a
permeabilidade a água e aos eletrólitos. O ramo descendente é altamente permeável a água, então ela vai sendo reabsorvida pro sangue
constantemente conforme o liquido vai passando aqui. E o ramo ascendente, especialmente a parte espessa (ramo ascendente espesso), promove
reabsorção de sódio, potássio e cloro. Então é altamente permeável a íons. A gente chama este ramo ascendente de ramo diluidor da urina porque
promove a reabsorção desses eletrólitos. Essa reabsorção acontece através de uma proteína chamada proteína dissódio-potássio-2-cloros-ATPase. Se
não fosse essa proteína nós não teríamos uma boa reabsorção de sódio no organismo porque ela faz MUITA reabsorção de sais. Como? Ela é uma
proteína que transporta o sódio, o potássio e 2 cloros ao mesmo tempo pra dentro da célula. A gente chama de transportador eletroneutro. Ele coloca
dois íons de carga positiva e dois íons de carga negativa. Então eletricamente ele é neutro. Só que quando ele promove essa reabsorção do sódio, o
sódio sai e vai pro vaso pela bomba de sódio-potássio-ATPase, o cloro também sai por essa bomba de cloro-potássio e volta pro vaso e o potássio
esta sendo reabsorvido aqui, entra na célula aqui e só sai um. Concorda que ta entrando dois íons de carga positiva pra dentro da célula e só um ta
indo pro sangue? Então o interior da célula não vai ficar positivo? Pra equilibrar isso o potássio tem que abrir um canal e sair por aqui. Porque que isso
é importante? Quando essa bomba funciona, ela cria um potencial positivo na luz do túbulo renal porque ela faz com que carga positiva saia pra
equilibrar a positividade no interior da célula. E se eu começar a tirar carga positiva do interior e colocar na luz, eu vou aumentar a positividade da luz
e o qq eu vou ter que fazer pra equilibrar isso? Tirar íons de carga positiva. E o qq sai? O magnésio e o cálcio sai pelo espaço entre as células. Então
vejam, quando essa bomba funciona adequadamente eu promovo reabsorção de sódio, potássio, cloro, magnésio e cálcio. Olha, saem dois cloros. Não
está representado aqui, mas eu não aumento a negatividade, ele acaba saindo na mesma proporção que entra. O que fica acumulado é o potássio.
Para que o interior da célula não fique positivo ela vai liberar potássio. Então o liquido da luz vai ficar positivo. E para equilibrar isso ele vai fazer com
que o magnésio e o cálcio sejam reabsorvidos.
Quando eu inibo essa proteína (diuréticos mais potentes, os de alça, a furosemida) eu não vou promover só a perda de sódio e cloro, que é o que eu
quero. Eu vou promover perda de potássio, de magnésio e de cálcio (eu não quero). Então inibir esta proteína de transporte significa acumular todos
esses eletrólitos na urina, aumentar MUITO a pressão osmótica aqui na luz, drenando MUITA água nessa região pra conseguir equilibrar essa pressão
osmótica. Por isso que a gente diz que furosemida (Lasics) e seus congêneres são os mais potentes de todos os diuréticos. Eles conseguem eliminar de
15 a 20% do sódio que entra na luz tubular (normalmente eliminamos 5%). Só que vai eliminar outros íons também, provocando reações adversas:
hipopotassemia (câimbras), hipocalcemia e hipomagnesemia, tendo um desequilíbrio eletrolítico pelo uso excessivo de diuréticos de alça. Utilizado pra
casos de hipertensão grave, que não responde a diuréticos menos potentes e também outras aplicações.
Na porção inicial do túbulo contorcido distal (TCD). Aqui também eu tenho uma reabsorção moderada de sódio e cloro através de uma proteína de
transporte. O sódio é reabsorvido junto com o íon cloreto através de um transportador também eletroneutro chamado de bomba de sódio-cloro. Esse
55
transportador também é alvo dos diuréticos. Os diuréticos mais utilizados são inibidores desse transportador, os diuréticos tiazidicos (em segundo lugar
a furosemida, porque ela é muito potente e causa muito mais desequilíbrio eletrolítico). Da mesma forma, eles vão acumular cloreto de sódio no TCD
aumentando pressão osmótica e drenando água. Esse transportador reabsorve sódio e cloro da luz do néfron para o vaso sanguíneo. E os diuréticos
tiazidicos atuam nele. Eles são diuréticos de ação moderada.
Aqui nessa região é também regulada a excreção do cálcio. Aqui também a gente tem a ação de um hormônio produzido pelas glândulas
paratireóides, o paratormônio. A função dele é promover a reabsorção do cálcio para a corrente sanguínea. Ele faz isso atuando em receptores que
estão nas células do TCD e promovendo a síntese desses canais de cálcio. Promovendo a síntese, eu promovo a reabsorção de cálcio para a corrente
sanguínea. Então por isso é que nessa região é regulada a excreção do cálcio, porque ele é reabsorvido e só será excretada a parte que ficará na luz
do néfron.
Nos túbulos coletores. Tem baixa permeabilidade tanto a água quanto aos eletrólitos, mas tem reabsorção de sais de água nessa região. Essa
reabsorção ocorre pela ação de dois hormônios: de sal, aldosterona; e de água, o hormônio antidiurético (ADH). Na célula principal, age a aldosterona
em receptores intracelulares e temos a ação do ADH que vai atuar num receptor de membrana celular. A aldosterona vai atravessar a membrana, se
ligar no seu receptor, vai alterar a síntese protéica dessa célula (é um receptor nuclear, interage com DNA), estimulando a síntese desses canais de
sódio, e promovendo a reabsorção do sódio pra dentro da célula. Só que para equilibrar essa carga positiva que vai ficar a mais dentro da célula, o
potássio vai ter que sair. Então a aldosterona funciona fazendo exatamente isso: estimulando a síntese desses canais de sódio, estimulando a atividade
dessa proteína que transporta o sódio pro vaso e ao mesmo tempo estimulando a excreção do potássio. O hormônio antidiurético atua em receptores
da membrana da célula, estimula a síntese desses canais de água e dessa forma a água é reabsorvida para dentro da célula e por difusão simples
ela vai pro vaso sanguíneo. O ADH e a aldosterona funcionam juntos pra gente não ter uma perda excessiva nem de sal nem de água. Existem algumas
patologias em que a pessoa pode ter deficiência na produção de aldosterona e ai ela perde muito sal pelo organismo e tem uma diurese grande e
existem pessoas que tem também deficiência na produção do hormônio ADH ou na ação do ADH nos seus receptores. E essa pessoa também não
consegue reabsorver água, e conseqüentemente ela também vai ter uma diurese muito aumentada. O álcool é antagonista do ADH e a gente acaba
fazendo muito mais xixi quando a gente toma um copo de cerveja do que se eu tomasse o mesmo volume de água. Aldosterona promove a reabsorção
de sódio e aumenta a excreção de potássio por uma ação tríplice: estimula o permutador sódio-hidrogênio, e através de sua ação nos receptores que
estão dentro da célula ativa canais de sódio, aumenta numero de bombas de sódio basolaterais. Então ela sempre vai trabalhar para reabsorver o
sódio e excretar o potássio. Tem um hormônio que vai interferir com as ações da aldosterona nesse receptor e tem algum diurético que vai interferir com
as suas ações ao abrir os canais de sódio. Então existem os diuréticos poupadores de potássio que podem ou ser antagonistas do receptor da
aldosterona, que é o caso da espironolactona (nome comercial Espirolactona), ou inibidores dos canais de sódio abertos pela aldosterona, que da
mesma forma vai antagonizar o efeito da aldosterona, que é o caso da amilorida ou triantereno (como se fossem uma rolha, bloqueiam o canal e não
deixam o sódio entrar). Esses diuréticos promovem pequena diurese porque a qtde de sódio que vai ser perdida é bem menor do que com um diurético
de alça. O grande motivo de usar esse diurético não é pelo seu efeito diurético, mas sim para impedir a perda de potássio que o diuréticos depletores
de potássio causam. Então eles são utilizados muito mais associados a diuréticos de alça ou tiazidicos para evitar a hipocalemia ou a hipopotassemia.
Em relação ao ADH, a gente não tem nenhum antidiurético utilizado na prática que vai impedir a sua ação pra fazer um efeito diurético. A gente tem o
álcool mas a gente nunca vai prescrever álcool para o paciente como diurético.
(Ver desenho do katsung que ela deixou no portal. Mostra a ação dos diuréticos inibidores da anidrase carbônica, dos diuréticos de alça inibindo a
proteina, dos diuréticos tiazidicos inibindo o transportador eletroneutro, e os diuréticos antagonistas da aldosterona inibindo a reabsorção de sódio.)
A furosemida é contra-indicada para pacientes com osteoporose. Já os diuréticos tiazidicos, por mecanismos desconhecidos, favorecem a reabsorção
desse cálcio. Então para o paciente que tem osteoporose é indicado utilizar um tiazidico e não utilizar um diurético de alça.
Detalhes dos fármacos. Então os diuréticos podem exercer as suas ações por ter uma ação direta sobre a célula do néfron, e todos os que eu desenhei
aqui fazem isso. Ou eles podem modificar indiretamente conteúdo dos filtrado, vcs vão ver ainda. Então eles podem ter ação em três lugares: na alça
de Henle, no inicio do túbulo distal ou nos túbulos e tubos coletores. Então os diuréticos importantes são esses três aqui: diuréticos de alça, tiazidicos e
antagonistas da aldosterona. A cetazolamida não é usada como diurético.
Detalhes dos diuréticos de alça: são os mais poderosos, significa que 15 a 20% do sódio existente no filtrado acaba sendo eliminado com esse
diurético. A furosemida é o representante principal, o protótipo de todos eles. Eles vão inibir essa proteína de transporte, uma bomba de sódiopotassio-2cloros na alça ascendente espessa. Impede a reabsorção desses íons, da água, aumentando torrencialmente o volume urinário. Ele está na
RENAME.
Então
ele
é
uma
arma
terapêutica
muito
importante
pra
casos
de
hipertensão
resistente.
- Estrutura: derivados das sulfas (medicação antibiótica, bacteriostática, o bactrin, sulfametroxazol-dimetropina, com ação muito restrita hoje em dia).
Eles perceberam que pacientes que utilizavam sulfas tinham o efeito diurético e através dessa estrutura eles criaram derivados mais seletivos para essa
proteína, a furosemida. Ela tem o radical sulfonamida. A bumetanida, outro derivado de sulfa, também tem esse radical. Só o acido etacrínico que não
tem esse radical sulfonamida, ele tem o grupo metileno que também é importante pra inibir essa bomba. Por causa de a maioria deles ser derivado de
sulfa eles tem uma reação adversa semelhante a da sulfa: reação de hipersensibilidade.
- Efeitos indesejados: hipocalemia (perda de potássio), hipocalcemia (perda de cálcio), alcalose metabólica por aumentar a excreção de íons
hidrogênio (com o uso prolongado). Outro efeito, que não está relacionado com as suas ações sobre o rim, é a ototoxicidade (perda auditiva com o uso
crônico, devendo ser interrompido o uso do medicamento porque ela é reversível). Outros efeitos: excesso de ácido úrico no sangue (para exercer a sua
ação, a furosemida tem que atravessar a parede do néfron e bloquear a parede de dentro penetrando na célula por uma proteína que transporta a
furosemida pra dentro; essa proteína é a mesma que transporta acido úrico então eles vão competir por ela e a furosemida entra e o acido úrico fica;
esse excesso é mais prejudicial pra quem já tem excesso, gota úrica), perda de magnésio e reações alérgicas por ser derivado das sulfas.
56
- Indicações clinicas: qualquer doença causada por excesso de sal e água no organismo (caso de edema pulmonar agudo, insuficiência cardíaca crônica,
síndrome nefrótica, insuficiência renal, hipertensão resistente, hipercalcemia).
- Interações medicamentosas: interfere na excreção renal de varias substancias: acido úrico, lítio (carbonato de lítio: usado pra transtorno bipolar,
principalmente na fase maníaca, ou para mania/ o lítio tem uma janela terapêutica muito estreita, se eu usar um medicamento que diminua sua
excreção eu vou aumentar seus níveis no sangue e ultrapassar essa janela, se tornando tóxico com a furosemida), aminoglicosideo (antibiótico, bastante
tóxico também; são ototóxicos, se eu associar pode ser irreversível), salicilatos (aumenta a toxicidade), bloqueadores neuromusculares (reduz o efeito
por acelerar a sua eliminação). Em resumo, a furosemida interfere bastante com excreção renal de fármacos. Se impedir a excreção vai aumentar a
toxicidade, se aumentar a excreção vai diminuir o efeito. (é claro que vcs não vão ter que guardar essas coisas, não se assustem, mas é bom a gente
criar esse raciocínio para analisar um caso que colocar para vcs). Geralmente é interação farmacocinética. Que tipo de interação é a furosemida com o
aminoglicosideo? São dois fármacos ototóxicos, interação farmacodinâmica por adição. E furosemida com lítio? Interação farmacocinética com efeito de
potencialização, aumentou a toxicidade do lítio.
Diuréticos tiazidicos. A principal é a Hidroclorotiazida (RENAME, SUS), há também a clortalidona (não é uma tiazida, mas atua na mesma PT de
transporte). Ação diurética moderada; inibem o cotransporte de sódio e cloro; se fixam no lugar do cloro impedindo a reabsorção de cloro e de sódio;
provocando excreção aumentada de sódio e de água. Acabam aumentando a reabsorção do cálcio, favorecendo a ação do paratormônio (indicado
para pacientes com osteoporose). Para que a hidroclorotiazida tenha uma função adequada como diurético, é preciso que haja uma produção
adequada de prostaglandinas renais (PGE e PGI2). Se eu fizer o uso de um AINE que vai inibir a ação dessas prostaglandinas, eu posso ter perda da
eficácia anti-hipertensiva. Os AINEs interferem só com os diuréticos tiazidicos, como interferem também com os IECA, com os beta-bloqueadores; eles
podem diminuir a eficácia anti-hipertensiva de todos eles por reduzir a produção dessas prostaglandinas vasodilatadoras.
- Estrutura química: também são derivados de sulfa, com o grupamento sulfonamida, podendo causar também reações alérgicas.
- Efeitos indesejados/ reações adversas: hipocalemia, excreção reduzida de ácido úrico (também são contra-indicados para pacientes com Gota),
excreção aumentada de magnésio e pode ocorrer alcalose. Os efeitos são parecidos com os dos diuréticos de alça, a diferença é que eles são menos
acentuados. Os diuréticos tiazidicos podem causar hiperglicemia por ações fora do rim, não relacionadas as ações na célula do néfron. Eles podem
reduzir a liberação de insulina e aumentar a glicogenólise e também reduzir a síntese da insulina. Por causa disso, no uso em pacientes diabéticos deve
ser feito um controle para verificar se ela não está interferindo no tratamento. E o paciente que não é diabético, se for usar por muito tempo,é bom
fazer testes de glicemia constantes para ver se ela não está causando o aumento da glicemia. E eles podem causar também hiponatremia (perda
excessiva de sódio), assim como a furosemida.
- Indicações: hipertensão leve a moderada, insuficiência cardíaca, edema grave resistente, diabetes insipidus nefrogenica. Os tiazidicos podem causar
diabetes melitus; a diabetes insipidus é completamente diferente e ocorre porque a pessoa não responde ao ADH (ou os sues receptores não são
sensíveis ao hormônio, ou não ocorre uma síntese adequada do hormônio ADH pela hipófise anterior). A conseqüência será uma diurese excessiva e a
falta do ADH promovendo essa diurese excessiva pode levar a essa diabetes insipidus nefrogenica. Em pacientes com essa doença, os diuréticos
tiazidicos provocam efeito paradoxal (efeito contrario do esperado), ou seja, diminuem o volume de urina. Então, os diuréticos tiazidicos sao indicação
em diabetes insipidus e contra indicação em diabetes melitus. Então efeitos não relacionados a ação no rim são a hipergicemia, podem aumentar
também os níveis de colesterol plasmático no uso prolongado, podem causar impotência masculina, mas isso é reversível com a retirada, e como são
derivados de sulfa podem causar reações de hipersensibilidade. Esses efeitos adversos que eu mostrei não são comuns, mas podem acontecer.
Os diuréticos antagonistas da aldosterona. Há duas formas de impedir a ação da aldosterona, ou eu posso bloquear o seu receptor e não deixar ela
se ligar, ou eu posso bloquear o canal de sódio que ela promove a síntese quando ela se liga no receptor. Quem bloqueia o receptor é a espirolactona,
ela é antagonista competitivo do receptor de aldosterona. E quem bloqueia os canais de sódio ativados pela aldosterona é o triantereno é a amilorida.
Não importa o mecanismo de ação o resultado será o mesmo: perda de sódio e retenção de potássio. A ação diurética deles é limitada, porem eles são
muito utilizados na terapia e são vendidos até associados com diuréticos que são depletores de potássio. Então e gente tem já no mercado associação
de um diurético tiazidico com um diurético poupador de potássio (sua principal importância é devido a essa capacidade de poupar potássio). Qual que
é o prejuízo de perder potássio? Câimbras musculares e depois as arritmias cardíacas, que, dependendo do nível de perda, pode ser muito grave,
podendo levar até a uma parada cardíaca. Então é muito importante os níveis de potássio no sangue, por isso que esse efeito colateral já é prevenido
com a associação. Interação entre um diurético poupador de potássio com um diurético depletor de potássio é uma interação dinâmica de antagonismo
fisiológico (enquanto um depleta potássio, o outro poupa potássio por ações completamente independentes).
Faramcologia
04/06/09
Hipertensivo que interfere com o sistema renina angiotensina não pode associar com diuréticos bloqueadores de potássio.
Inibidores da eca podem aumentar níveis plasmáticas de digoxina e lítio,digoxina é um cardiotônico e o lítio usado em no tratamento da mania e
transtorno bipolar
57
Antagonistas de receptores da angiotensina:
Posso inibir os efeitos da angiotensina 2 de duas formas: inibindo sua síntese ou bloqueando a ação de seus receptores.
A angiotensina tem dois receptores: receptores AT1 e AT2.
Os antagonistas dos receptores da angiotensina bloqueiam somente o receptor AT1. O receptor AT1 esta presente nos vasos, promovendo
vasoconstrição; no rim, quando a angiotensina estimula receptor AT1 renal diminui a liberação de renina (mecanismo para a angiotensina 2 regular sua
própria liberação, quando muito concentrada no sangue estimula AT1 no rim para menor liberação de renina, e assim produz menos angiotensina 2); na
supra-renal, quando a angiotensina 2 se liga aumenta liberação de aldosterona.
Receptor AT2 esta presente em vários locais, nos vasos, promovendo vasodilatação; da mesma forma que adrenalina e noradrenalina que tem
receptores alfa e beta, o alfa 1 fazendo vasoconstrição e beta 2 faz vasodilatação, a angiotensina 2 é a mesma coisa. Quando os dois receptores
estão livres se liga nos dois sendo o resultado é uma vasoconstrição, embora tenha em algumas vezes vasodilatação.
Quando eu tomo um antagonista dos receptores da angiotensina 2, vou somente inibir a ação do receptor AT1, impedindo o efeito da
angiotensina 2 nos vasos, tendo menor vasoconstrição, dessa forma ocorrendo vasodilatação; vou inibir na supra-renal diminuindo a liberação de
aldosterona, porem vou inibir também seu efeito inibitório no rim na diminuição da liberação de renina, assim aumentando a liberação de renina e
consequentemente de mais angiotensina 2. Assim quando eu tomo antagonista AT1 eu aumento a síntese de angiotensina 2, embora seus receptores AT1
estejam bloqueados. Com sua síntese aumentada a angiotensina 2 atua mais no receptor AT2 que está livre , fazendo vasodilatação. O efeito
hipotensor do antagonista AT1 não é só por bloquear AT1, mas também porque aumenta a atividade da angiotensina 2 no receptor AT2, isso que da
seu efeito vasodilatador.
Os fármacos que fazem parte dessa classe é a família TAN, o protótipo é o losartan. Mas a partir da estrutura dele foram criados vários. Não há
nenhuma diferença significativa entre eles, podem ter alguma diferença apenas na farmacocinética. São contra indicados na gravidez, da mesma forma
que captopril e os inibidores da ECA. Efeitos adversos: hipotensão, e raramente pode ocorrer oliguria( diminuição da produção de urina), azotenia
(aumento da uréia no sangue) e insuficiência renal aguda.
A vantagem em relação aos inibidores da ECA é que esses causam menos tosse, quase não há relatos de tosse. O que significa que provavelmente os
níveis de bradicinina sejam os responsáveis pela tosse porque esses não interferem nos níveis de bradicinina.
Comparando os inibidores da ECA com os antagonistas do receptor da angiotensina: o nível de eficácia hipertensiva é a mesma, ambos melhoram a
sobrevida do paciente no tratamento de insuficiência cardíaca e ambos têm menor eficácia em pacientes negros e idosos. A grande diferença entre eles
é a tosse que pode ser menor; e o preço: os antagonistas de AT1 são bem mais caros que os inibidores da ECA.
Fármacos que podem ser utilizados no tratamento da hipertensão:
Simpaticolíticos: beta bloqueadores, alfa bloqueadores, e anti-hipertensivos de ação central.
Inibidores do sistema renina angiotensina: inibidores da ECA e os antagonistas da angiotensina 2.
Diuréticos: tiazidicos, diuréticos de alça, e os poupadores de potássio.
Vasodilatadores: não são muito utilizados, mais comum no tratamento com pacientes idosos, gestantes e antianginosos.
BENZODIAZEPINICOS
Fármacos ansiolíticos, hipnóticos e sedativos. Atuam no SNC. Dos fármacos psicoativos esses são os mais prescritos.
Diferença entre um fármaco psicoativo e psicotrópico: os psicoativos atuam no SNC alterando humor, a parte cognitiva, mas não tem
propriedade reforçadora, não induzem a auto administração, a dependência. Já os psicotrópicos atuam no SNC afetando humor e cognição, porém
possuem propriedades reforçadoras, causam dependência.
Efeito sedativo: droga que diminui a atividade, modera a excitação e acalma.
Fármaco hipnótico: produz sonolência, e facilita o início e a manutenção do sono, porém a pessoa pode ser acordada facilmente.
Ansiolítico: droga utilizada para atenuar os sintomas da ansiedade patológica.
Ansiedade: são reações normais ao medo, para se defender em situações perigosas. Em uma situação de medo ocorrem reflexos autônomos como:
tremor, sudorese, taquicardia, reações de luta, de fuga. O medo é uma emoção negativa. Essas reações ao medo podem ser patológicas, classificada
como ansiedade quando ocorrem de maneira antecipada sem nenhum estímulo para a pessoa sentir medo, sentindo os reflexos autônomos o tempo
todo, como tremor, sudorese, inquietação, taquicardia. Nesses casos é necessário tratamento.
58
Classificação dos distúrbios da ansiedade:
Ansiedade generalizada: pessoa agitada o tempo todo, ansiosa; Levando a situações que prejudicam qualidade de vida, pode comer demais
ou muito pouco, dificuldade de concentração.
Ataque de pânico: ataques de medo opressivo associado com sistema autônomo marcante. Pessoa sente sem nenhum motivo, às vezes em
lugares em que passa, por exemplo, indo para o trabalho ocorre sudorese excessiva, taquicardia, dor no peito, tremor, asfixia, sensação de
morte.
Distúrbios de estresse pós-traumático: ansiedade gerada por lembranças de coisas estressantes que aconteceram no passado, como um
assalto.
Distúrbio obsessivo compulsivo: comportamentos com rituais compulsivos dominado por ansiedade irracional.
Existem três classes de fármacos para tratar esses distúrbios da ansiedade: fármacos ansiolíticos, alguns antidepressivos, principalmente os inibidores
seletivos de recaptação da serotonina como a fluoxetina e sua família, e algumas vezes fármacos antipsicóticos. Os mais utilizados na prescrição são os
antidepressivos, e depois os ansiolíticos benzodiazepínicos.
Esses fármacos também são hipnóticos, utilizados no tratamento da insônia. Porem a insônia não é uma doença, é uma queixa de uma doença primária,
geralmente paciente tem insônia por um fator primário que leva a isso, devido a depressão, ansiedade, por estar passando por um momento
estressante. Assim deve ser tratado a causa primaria da insônia. Esses fármacos utilizados pra tratar a insônia devem ser utilizados inicialmente, para
não tratar o paciente que não dorme há uma semana, apenas para recuperar o sono do paciente, para que não haja dependência.
BENZODIAZEPINICOS
Efeito ansiolítico e hipnótico.
Zolpidem: não é estruturalmente um benzodiazepínico, mas atua de forma semelhante e possui menos atividade ansiolítica e mais hipnótica.
Buspirona: agonista parcial dos receptores da serotonina, é um ansiolítico eficaz, porem não hipnótico.
Beta-bloqueadores: não tratam a ansiedade do paciente, mas diminui os sintomas de ansiedade, o tremor, a taquicardia e a sudorese.
Barbitúricos: primeiros ansiolíticos e hipnóticos utilizados, porem hoje não são muito utilizados porque possuem janela terapêutica estreita, possuem
perfil de segurança pequeno, pode causar depressão respiratória. Utilizados hoje em pré anestesia e no tratamento da epilepsia.
Anti histamínicos efeito hipnótico pode ser utilizado para crianças.
BENZODIAZEPINICOS
Até 1960 eram usados apenas os barbitúricos, para acalmar, anestesiar. Porém em doses um pouco maior que a dose terapêutica pode
ocorrer depressão respiratória. Era alto o índice de suicídios com os barbitúricos.
No final da década de 50 ocorreu um erro de síntese química, sintetizaram uma estrutura benzodiazepínica, anel aromático ligado a sete
elementos. Essa molécula foi testada para vários efeitos (teste hipocrático de ??), e verificaram excelente efeito sedativo, hipnótico. O primeiro fármaco
que foi lançado no mercado a partir dessa estrutura foi o clordiazepóxido, foi uma revolução no tratamento da ansiedade e da insônia, pois eles não
causam morte por depressão respiratória em altas doses. Hoje em dia é a classe mais prescrita no tratamento da ansiedade e da insônia.
Estrutura do benzodiazepínico: anel aromático com uma diazepina, estrutura de sete elementos ligada ao anel. Essa estrutura que se liga ao GABA A.
A diferença entre as classes de benzodiazepínicos são os radicais ligados, isso muda a farmacocinética dos benzodiazepinicos, assim quando há
mudança nos radicais a diferença entre eles está no tempo de meia vida. Dessa forma são classificados em ação ultra-curta, curta, ação intermediaria e
ação longa.
Mecanismo de ação: atuam no receptor do GABA, que é um receptor ligado a canal iônico. O GABA é um aminoácido inibitório, produzido no SNC,
tem dois receptores GABA A e GABA B. Os benzodiazepínicos se ligam somente no receptor GABA A, que é um receptor ionotrópico (ligado a canal
iônico), não tem nenhuma ação no GABA B que é um receptor metabotrópico. Todos eles então são agonistas do GABA A.
O receptor GABA A tem um canal iônico no seu interior, quando o aminoácido inibitório GABA se liga em um sítio específico de ação ele vai promover
alteração conformacional dessa proteína, dessa forma vai abrir o canal e íons de carga negativa vão penetrar no neurônio, hiperpolarizando essa
célula e o neurônio hiperpolarizado vai diminuir sua excitação, por isso é um aminoácido inibitório.
Vários fármacos atuam nesse receptor, mas em um sitio de ligação diferente do GABA. Benzodiazepínicos, barbitúricos, álcool, todos eles tem ação no
receptor GABA A, mas cada um em um sitio de ligação diferente do GABA, por isso que na verdade eles não são agonistas e sim moduladores do
GABA. Os benzodiazepínicos quando se ligam sozinho no receptor GABA A não tem efeito nenhum, porém quando o aminoácido inibitório se liga ao
mesmo tempo tenho uma abertura muito maior do canal iônico, entrando mais íons de carga negativa, e assim deprime muito mais os neurônios do SNC
e assim tenho o efeito depressor que todos eles causam. Para esse mesmo sitio de ligação onde se ligam os benzodiazepínicos existem fármacos com
59
ação agonista (benzodiazepínicos), antagonista (flumazenil - usado em casos de intoxicação por benzodiazepinicos) e agonista inverso (beta-carbonilas
- promovem ação inversa do receptor, ao se ligar ao sitio de ligação fecha o canal, pode produzir efeito ansiogenico).
Isso mostra que na verdade muitos receptores não estão funcionalmente silenciosos quando não tem ligação de um ligante, porque se ele não tivesse
nenhuma atividade sem a ação do GABA significa que não estaria entrando nenhum íon Cl sem a ação do GABA. Mas isso não é verdade, existe já
certa abertura do canal quando nenhum ligante está se ligando, é possível saber isso porque quando um agonista inverso se liga esse canal se fecha,
impedindo a entrada de íon Cl, e assim havia a entrada de íon anteriormente, o que significa que ele tem uma atividade intrínseca constitutiva.
Classificação dos benzodiazepínicos:
Ação ultra-rápida: zolpidem, que estruturalmente não é um benzodiazepínico porem atua no mesmo receptor da mesma forma.
Ação curta: tempo de meia vida menor que 6 horas; triazolam e midazolam
Ação intermediaria: tempo de meia vida de 6 a 24 horas; estazolam
Ação longa: tempo de meia vida acima de 24 horas; diazepam
A diferença na pratica entre benzodiazepínicos de ação ultra-rapida, curta, intermediaria e longa de maneira geral é que os de ação ultra-rapida e
curta como hipnóticos, pois quero que dure apenas uma noite e os de ação intermediaria e longa como ansiolíticos, porque a duração deve ser maior,
durante o dia inteiro.
DIAZEPAM
Diazepam lançado em 1963, após o clordiazepóxido. Sendo o mais prescrito desde então. Sua concentração máxima no sangue atingida
entre 30 e 90 minutos. Tempo de meia vida longo de 20 a 80 horas, pois seus metabólitos ainda estão ativos. E o efeito depende muito de função
hepática. Se a função hepática estiver diminuída o tempo de meia vida vai aumentar ainda mais.
Sofre reações de fase I, reações de oxidação pelas enzimas microssomais hepáticas e seus metabolitos são ativos, continuam exercendo
efeito ansiolítico e hipnótico. Metabólitos nordazepam, hidrodiazepam, oxidiazepam continuam exercendo efeito. Excretado principalmente pela urina,
e é metabolizado pelas enzimas do citocromo P450.
Se for metabolizado no fígado e associado a fármacos indutores ou inibidores enzimáticos podem ocorrer problemas de interação
medicamentosa.
Fármacos que podem prolongar seu efeito e aumentar a toxicidade são os inibidores enzimáticos: eritromicina, ritonavir, itraconazol,
cimetidina, todos esses fármacos podem fazer seu efeito depressor aumentado.
Anti-histamínico associado a benzodiazepínico tem efeito aditivo sobre a depressão. Álcool associado ao benzodiazepinico tem efeito
sinérgico, podendo levar a uma depressão cardio-respiratória.
Representantes de tempo de meia vida curta a ultra-curto mais utilizado como hipnóticos: Flurazepam (Dalmadorm), Triazolam (Halcion),
Midazolam (Dormonid), Flunitrazepam (Rohipnol), Bromazepam (Somalium), Lorazepam (Mesmerin), Nitrazepam (Nitrapan), Estazolam (Noctal). Esses
fármacos possuem efeitos adversos mais importantes que os de meia vida longa que é o alto risco de abuso, porque o paciente vai fica sem suas ações
no SNC de forma mais rápida, sentindo necessidade de tomar outra vez já que seu tempo de meia vida é mais curta. E ainda podem causar a síndrome
de abstinência, o que significa que o paciente já está dependente fisicamente do medicamento. Os benzodiazepínicos no terceiro dia de uso já podem
apresentar sinais de tolerância, ou seja, é necessária uma dose maior do medicamento para obter o mesmo efeito.
Também podem ser utilizados em pré-anestesia, principalmente o Lorazepam (Lorax), pois é amnésico (perda de memória durante o uso do
medicamento, por isso usado em cirurgias para não lembrar o que aconteceu).
Os fármacos de meia vida longa são mais utilizados como ansiolíticos. Esses fármacos por possuírem tempo de meia vida mais longo
apresentam mais efeitos colaterais como: sedação, diminuição da coordenação motora, amnésia.
Alprazolam (Frontal), Bromazepam (Lexotam), Diazepam (Valium), Clordiazepóxido (Psicosedin), Lorazepam (Lorax). Transtorno do pânico: Alprazolam,
Clonazepam (Rivotril).
ANTICONVULSIVANTES: Não para prevenir, mas para tratar o paciente em convulsão. Diazepam, Clonazepam possuem tempo de meia vida longa,
rápida entrada no SNC.
Mecanismo de ação: atuar em receptor GABA A no SNC.
Efeitos farmacológicos: diminuição da ansiedade e agressão. Teste em animais.
Efeito sedativo, hipnótico. Diminuem tônus muscular e coordenação motora. Os de tempo de meia vida longa têm efeito anticonvulsivante, e também
efeito amnésico.
60
Efeito paradoxal: Triazolam aumentou a irritabilidade em alguns pacientes.
Sintomas da síndrome de abstinência: insônia, ansiedade, disforia, irritabilidade, sudorese noturna, sincopes e tontura. Nesse caso a medicação deve ser
retirada gradualmente.
Diferença entre dependência física e psicológica: na física há tolerância e síndrome de abstinência. Na psicológica o paciente só pensa em obter a
medicação.
REDUÇÃO DO TONUS MUSCULAR E COORDENAÇÃO: Há testes (teste rotarod- ratos sobre uma barra giratória) em animais para testar novos
fármacos benzodiazepínicos com efeito relaxante muscular, devido seu efeito farmacológico sobre o tônus muscular. Pacientes ansiosos geralmente
possuem um aumento do tônus muscular.
EFEITO ANTICONVULSIVANTE: Eficazes contra as convulsões induzidas quimicamente em animais (leptazol, bicuculina), mas não reverte a convulsão da
estricnina (veneno de rato- é antagonista da glicina ).
AMNÉSIA ANTERÓGRADA: Bloqueiam a memória dos eventos experimentados sob sua influência (procedimentos cirúrgicos).
Classificação dos efeitos indesejáveis dos benzodiazepínicos:
Efeitos tóxicos agudos resultantes de doses excessivas: causam sono prolongado, sem depressão séria na respiração ou função cardiovascular.
Depressão respiratória severa em associação com álcool (tratamento- flumazenil).
Efeitos indesejáveis devido seu uso em doses terapêuticas: sonolência, confusão mental, amnésia, coordenação motora prejudicada (
desempenho de dirigir), quanto mais longa duração mais efeitos colaterais.
Tolerância e dependência: ocorre com todos os benzodiazepínicos, tolerância (gradual aumento da dose para produzir o mesmo efeito):
devido alteração no receptor GABA A que está sendo tão estimulado que o neurônio passa a expressar menos receptor, dessensibilizando
receptor GABA A, assim tem que tomar doses maiores para atingir maior numero de receptores para produzir mesmo efeito, isso é uma
tolerância farmacodinâmica. Dependência: interrupção do uso após semanas ou meses pode causar a síndrome de abstinência (nervosismo,
tremor, perda de apetite e convulsões).
Existem novos fármacos que são agonistas dos benzodiazepínicos, mas não são classificados como benzodiazepínicos, pois sua estrutura química é
diferente. Indicados no tratamento da insônia e não causam dependência, mas não são muito utilizados porque não são tão bons ansiolíticos. Assim
sendo mais usado em idosos, pacientes que possuem muita sedação no uso de benzodiazepínicos e em pacientes com historia de dependência.
Ansiolíticos serotoninérgicos têm apenas um representante é a buspirona.
BUSPIRONA
Agonista parcial de receptores 5-HT1A, esse é um auto receptor inibitório que quando a serotonina estimula, diminui a liberação dela
mesma.
Inibem a atividade dos neurônios noradrenérgicos interferindo com as reações de despertar.
Ineficaz nos distúrbios do pânico.
Efeitos colateriais: náuseas, tonteira, cefaléia, agitação.
Vantagens: ausência de interação com álcool, ausência de dependência com uso prolongado e sem amnésia, usada para tratar
ansiedade crônica e persistente, idosos, história de abuso.
Desvantagem: retardo no início da ação (efeitos adaptativos dos receptores).
BARBITURICOS
Até os anos 60 eram o maior grupos de substâncias hipnóticas sedativas prescritas. Atividade depressora do SNC produzindo efeitos semelhantes aos
anestésicos inalatórios. Altas doses podem causar morte por depressão respiratória e cardiovascular. Pentobarbital, fenobarbital, tiopental.
Os barbitúricos causam depressão respiratória e os benzodiazepínicos não apesar de atuarem sobre o mesmo receptor, isso se deve ao fato que
quando o barbitúrico se liga sozinho ele já promove a abertura do canal, não é necessário que o GABA se ligue para a abertura do canal. E o
benzodiazepínico precisa da ligação do GABA para a abertura do canal. Devido a isso que os barbitúricos podem causar mais depressão respiratória,
pois já tem uma função agonista sozinho.
Principais efeitos colaterais:
Tolerância e dependência.
Interações farmacológicas muitos importantes: são indutores das enzimas do citocromo P450, aumentando seu próprio metabolismo
(tolerância farmacocinética) e o metabolismo de muitos fármacos.
Interage com o álcool e outros depressores do SNC potencializando seus efeitos.
61
Farmacologia
10/06/09
Uma das principais causas de incapacidade e morte prematura é a depressão severa. É a 2ª doença crônica mais comum na população
(depois de hipertensão). 10 a 15% da população mundial apresentam algum sintoma depressivo, podendo caracterizar de uma depressão leva a uma
severa. Pelo menos 1 em cada 10 desses pacientes apresenta depressão maior, mas a maioria não é diagnosticada e conseqüentemente
inadequadamente tratada. Estudos apontam que 80% dos suicidas consultaram um clinico geral no mês que precedeu seu suicídio e não foram
diagnosticados com depressão maior.
TIPOS DE DEPRESSÃO
- Unipolar – humor oscila sempre na mesma “vibração” (a pessoa esta sempre triste).
- Bipolar – momentos de depressão alternam-se com episódios de mania (pode ser no mesmo dia). Esses episódios de mania abrangem sentimentos de
exuberância, entusiasmo e autoconfiança excessivos, ações impulsivas, irritabilidade, impaciência, agressão e delírios.
A depressão maior é diagnosticada pelo psiquiatra através de questionários. Esses pacientes podem apresentar insônia ou hipersonia,
sentimentos de desvalorização e culpa excessiva, falta de energia, redução da capacidade de se concentrar e pensar, alteração significativa no
apetite ou peso (alguns pacientes perdem o apetite, outros aumentam seu apetite, alguns ganham peso e outros perdem muito peso), retardo ou
agitação psicomotora e pensamentos recorrentes de morte ou suicídio. Se o paciente apresentar três ou quatro desses sintomas por mais de uma semana
têm-se um dos critérios diagnósticos para a depressão maior.
Outro critério que pode ser utilizado é o TESTE DE SUPRESSÃO DA DEXAMETASONA. Quando administrados corticóides exógenos a produção
de corticóide endógeno fica diminuída devido a um efeito inibitório no hipotálamo e hipófise. Em pacientes com depressão esse efeito inibitório não
existe, a administração de dexametasona não é capaz de inibir a secreção de CRF e ACTH. Recentemente descobriu-se que existem mecanismos
neuroendócrinos alterados nos pacientes que apresentam depressão, com aumento dos níveis de cortisol sanguíneo.
Sabe-se também que a produção de GH diminui no paciente deprimido e a de prolactina aumenta. A depressão está envolvida também gera
processos neurodegenarativos (Alzheimer, Parkinson, esclerose, corea de Hunginton).
A explicação bioquímica para a depressão é muito antiga. Em 1965 foi postulada a TEORIA MONOAMINÉRGICA DA DEPRESSÃO. Uma
teoria muito antiga que embora falha ainda é a única aceita para explicar a depressão. Ela diz que a depressão resulta de um déficit funcional das
monoaminas no SNC e que a mania resulta de um excesso desses transmissores. Essas monoaminas são principalmente a noradrenalina e a serotonina (as
que são alteradas pelos anti-depressivos).
Para essa teoria o paciente com distúrbio bipolar teria alterações constantes nesses níveis de monoaminas, hora apresentando altos níveis e
hora baixos níveis. O paciente com depressão maior teria esses níveis sempre baixos e o paciente com mania teria constantemente altos níveis de
monoaminas. É uma teoria simplista, porem, todos os fármacos presentes no mercado para tratar depressão trabalham alterando os níveis de
monoaminas e tem uma ótima eficácia antidepressiva.
A teoria nos diz então que pacientes normais, sem sintomas de depressão, teriam a síntese, armazenamento e liberação das monoaminas de
uma forma normal. O que aconteceria então no cérebro de um paciente depressivo seria uma baixa liberação de monoaminas com conseqüente baixa
estimulação de neurônios pós-sinápticos. Essa baixa estimulação resulta na UP-REGULATION, que seria uma super-expressão de receptores pós
sinápticos, porque ele tem pouco neurotransmissor para estimular esses receptores. Sabe-se que existe um componente genético (ainda não definido)
que aumenta a possibilidade de um paciente desenvolver a depressão. Essa teoria não deixa de ser uma teoria porque ela não explica tudo. Existem
evidencias a favor e evidencias contra essa teoria.
(DESENHANDO UMA TRANSMISSÃO MONOAMINÉRGICA NO QUADRO)
De uma forma geral precursores entram na célula e no citoplasma sofrem a ação de uma enzima que o transforma em uma monoamina. Essa
monoamina, para não ser degradada no citoplasma, é coloca dentro de vesículas, as quais vão se fundir à membrana do neurônio quando o nível de
62
Cálcio intracelular aumentar (em resposta ao potencial de ação) liberando esses transmissores na fenda sináptica. Essa monoamina atua nos receptores
pós-sinápticos e após isso acontecem os processos de recaptação.
Quais as evidências que confirmam a teoria monoaminérgica?
1º Sabe-se que quando é usada uma droga que, por qualquer mecanismo de ação, diminua os níveis de monoaminas têm-se efeitos depressivos. Por
exemplo: A α-metil-tirosina inativa a enzima envolvida na síntese de noradrenalina e dopamina (tirosina hidroxilase). Muito pouco desses
neurotransmissores são então sintetizados e liberados quando o paciente está sob o efeito dessa droga, o que resulta em efeitos depressores (efeito
colateral do medicamento). Portanto, a diminuição dos níveis de monoaminas gerando efeitos depressores confirma a teoria monoaminérgica.
2º A reserpina impede o armazenamento de monoaminas (dopamia, serotonina e noradrenalina) nas vesículas. Isso diminui o nível de monoaminas na
fenda sináptica, e sua principal reação adversa é a depressão. Mais uma evidência que confirma a teoria monoaminérgica.
3º Antidepressivos aumentam os níveis de monoaminas, seja por bloquear a recaptação ou por inativar a MAO (enzima que degrada as monoaminas
nos neurônio pré-sinápticos). Esses fármacos (inibidores seletivos de recaptação ou inibidores da MAO) têm efeito antidepressivo porque aumentam os
níveis de monoaminas.
Quais as evidências que contradizem a teoria monoaminérgica?
1º Se administrados triptofano, tirosina ou L-dopa (precurssores da serotonina, noradrenalina e dopamina respectivamente) há um aumento nos níveis
desses transmissores, porém não se tem efeito antidepressivo.
2º A cocaína e as anfetaminas bloqueiam a recaptação desses transmissores e consequentemente aumentam seus níveis, mas não têm efeitos
antidepressivos.
3º O carbonato de lítio melhora a depressão sem aumentar os níveis de monoaminas.
4º Os antidepressivos tricíclicos e inibidores da MAO aumentam imediatamente os níveis de monoaminas, mas o efeito antidepressivo só se percebe de
7 a 21 dias após o início do tratamento. Isso se da provavelmente porque o efeito antidepressivo é causado pela neuroadaptação gerada pelo
aumento constante dos níveis moniaminérgicos, e não pelo aumento imediato das monoaminas. Por isso não podemos pensar que somente o aumento dos
níveis de monoaminas é suficiente para que se tenha um efeito antidepressivo, é necessário que haja a neuroadaptação dos receptores dos neurônios
para que esse efeito seja percebido.
Pesquisadores postularam também que a depressão maior está associada à perda neuronal no hipocampo e no córtex pré-frontal e que as
terapias antidepressivas de diferentes tipos autuam por inibição ou reversão dessa perda por estimularem neurogênese (formação de novos neurônios).
Como a hipótese monoaminérgica se relaciona com a neurodegeneração neuronal e o aumento dos níveis de cortisol para explicar a
depressão?
Quando as monoaminas, em quantidades normais, estimulam seus receptores no SNC, mecanismos de transdução de sinal celular ativam os
genes de resposta benéfica para a neurogênese, e inibem os genes responsáveis pela apoptose. Se o paciente é deprimido as monoaminas estão em
níveis mais baixos (teoria monoaminérgica), e, portanto, não estimulam receptores, isso culmina na não ativação desses genes que promovem a
neurogênese, além dos genes da apoptose continuarem ativos. Altos níveis de cortisol por sua vez estimulam os genes de apoptose, levando à
neurodegeneração. Na Síndrome de Cushing, por exemplo, os pacientes apresentam sintomas depressivos pelos altos níveis de cortisol.
63
ANTIDEPRESSIVOS
INIBIDORES DA MAO
TRANILCIPROMINA – Inibidor irreversível da MAO
MOCLOBEMIDA – Inibidor reversível
ANTIDEPRESSIVOS TRICÍCLICOS (inibem recaptação de serotonina e/ou noradrenalina)
*IMIPRAMINA (Trofanil), *AMITRIPTILINA (Tryptanol), *CLOMIPRAMINA (Anafranil) e NORTRIPTILINA (Pamelor)
(Os com “asterístico” estão na RENAME)
INIBIDORES SELETIVOS DE RECAPTAÇÃO DE SEROTONINA
FLUOXETINA, CITALOPRAM, ESCITALOPRAM, PAROXETINA, SERTRALINA e FLUVOXAMINA.
INIBIDORES SELETIVOS DE RECAPTAÇÃO DE NORADRENALINA
INIBIDORES SELETIVOS DE RECAPTAÇÃO DE SEROTONINA E NORADRENALINA
INIBIDORES SELETIVOS DE RECAPTAÇÃO DE DOPAMINA
ANTIDEPRESSIVOS ATÍPICOS
Porque o efeito antidepressivo dos fármacos só acontece após aproximadamente 20 dias após o início do tratamento?
Porque existe um mecanismo de neuroadaptação cerebral ao uso do antidepressivo. Essa mudança acontece nos receptores pós-sinápticos, com
diminuição da sua expressão (DOWN-REGULATION). Isso acontece para qualquer inibidor seletivo de recaptação ou para qualquer inibidor da MAO.
O efeito só é percebido quando já aconteceu essa neuroadaptação. No início do tratamento, quando os receptores estão expressos em maior
quantidade, acontecem mais efeitos colaterais. Daí a importância de orientar o paciente de que nos primeiros dias haverá o surgimento de efeitos
colaterais e não melhora do quadro depressivo, para que ele não abandone o tratamento. A fluoxetina agudamente causa muitos efeitos colaterais,
mas isso cessa em algumas semanas, diferente dos antidepressivos tricíclicos que não causam tanto efeito colateral agudamente, mas os efeitos que
aparecem persistem durante todo o tratamento.
INIBIDORES DA MAO
Inibindo a enzima que degrada as monoaminas (monoamiaoxidase) elas são mais armazenadas em vesículas no neurônio pré-sináptico e
consequentemente mais liberadas.
Primeiros criados, mas hoje utilizados como segunda escolha porque tem perfil de interação medicamentosa e reação adversa ruim. Utilizados
para os tipos de depressão resistentes aos outros antidepressivos, em casos de ansiedade e transtorno do pânico e fobia associada.
Existem dois tipos de monoaminaoxidase: MAOA e MAOB, e os inbidores da MAO podem ser seletivos (para a MAOA) ou não-seletivos (inibem
a MAOA e na MAOB). Seletivos para a MAOB não tem efeito antidepressivo, eles agem principalmente inibindo a degradação de dopamina e são
então usados no tratamento do Parkinson.
Os inibidores não seletivos irreversíveis da MAO (representante - tranilcipromina) têm uma interação muito perigosa com alimentos
fermentados, como queijos, cerveja e vinhos. Esses alimentos contêm tiramina, substância que estimula a liberação de monoaminas pelas células nervosas.
A enzima não estando ativa causa um excesso da liberação de noradrenalina podendo levar a um quadro de hipertensão, principalmente em pessoas
que tem histórico de hipertensão. Pode levar à morte. Nos inibidores reversíveis esse efeito é diminuído pela competição pela MAO entre o
medicamento e as monoaminas. O principal motivo então para que esses fármacos sejam de segunda escolha é por eles apresentarem interação
perigosa com alimentos e com outros fármacos. Estão em segundo dos fármacos que mais interagem com outros fármacos (1º - varfariiiiina).
64
Os inibidores reversíveis podem causar boca seca, cefaléia, desconforto gástrico e etc. Os inibidores irreversíveis podem causar agitação,
aumento do apetite, bradcardia, diminuição do desejo sexual, etc.
ANTIDEPRESSIVOS TRICÍCLICOS
A Imipramina foi sintetizada com finalidade antipsicótica. Porém não foi eficaz para esse fim, mas teve sucesso como antidepressivo.
*PROVA – QUAIS OS EFEITOS COLATERAIS DOS ANTIDEPRESSIVOS TRICICLICOS E PORQUE EXISTEM ESSES EFEITOS?
Esses medicamentos não são específicos, inibem a recaptação de serotonina e adrenalina e são ainda antagonistas muscarínicos, antagonistas α1 e
antagonistas H1. Efeitos colaterais:
- Pelo bloqueio dos receptores muscarínicos – boca seca, visão turva e constipação.
- Pelo bloqueio dos receptores α1 – hipotensão.
- Pelo bloqueio H1 – sedação e aumento do apetite.
- Causam ainda hepatotoxicidade, depressão medular (raro), convulsões, fotossensibilidade, efeitos cardiovasculares severos (cardiotoxicidade –
Amitriptilina é o mais cardiotóxico e a Imipramina o menos cardiotóxico) e neuropatia periférica.
Quando administrar então um tricíclico?
Pensar no seu perfil de efeito colateral. Se um paciente depressivo come demais não se deve prescrever os tricíclicos, porque isso vai piorar, se ele tem
hiperssonia também deve-se evitar os tricíclicos porque eles tem efeito sedativo (bloqueio de H1). Se, por outro lado, o paciente deprimido tem insônia
ou falta do apetite, os tricíclicos seriam uma indicação.
A Amitriptilina tem alta ligação as proteínas plasmáticas e é extensamente metabolizada no fígado. Por isso causa interação com outros
medicamentos que se ligam às proteínas plasmáticas e àqueles que são indutores ou inibidores enzimáticos.
INIBIDORES SELETIVOS DE RECAPTAÇÃO DE SEROTONINA
Tem pouco efeito sobre os receptores α1 e H1, não causando então tantos efeitos colaterais comparado com os tricíclicos. Não acontece morte
por altas dosagens como com os tricíclicos, isso porque os inibidores seletivos de recaptação de serotonina têm a janela terapêutica mais larga. Por isso
são os de primeira escolha para o tratamento de depressão.
A Flouxetina age nos dendritos e no terminal axônico. Nos dendritos tem os receptores 5HT14 para a serotonina, um receptor auto-inibitório
que quando ativado diminui a liberação da serotonina. Com as altas concentrações causadas pelos ISRS acontece uma neuroadaptação em forma de
diminuição da expressão desses receptores auto-inibitórios, o que gera perda do efeito inibitório com conseqüente aumento maior ainda a liberação da
serotonina. Nos primeiros dias do tratamento com os ISRS, como ainda não aconteceu a DOWN-REGULATION dos receptores serotoninérgicos póssinápticos e os receptores pré-sinápticos já estão diminuídos acontecem muitos efeitos colaterais. Depois de 1 mês os efeitos colaterais tendem a diminuir
e os efeitos terapêuticos aparecem, devido a neuroadaptação (down-regulation dos receptores pós-sinápticos). Os efeitos colaterais relacionados à
ação aguda da fluoxetina nesses receptores pós-sinápticos hiper-expressos são, de uma forma geral, representados pela agitação, ansiedade, ataque
de pânico, dificuldade de coordenação motora, não controle dos movimentos de mãos e pés, despertar noturno, movimentos oculares rápidos, disfunções
sexuais, náuseas, vômito e alterações do apetite (a maioria das pessoas apresentam falta de apetite, mas algumas apresentam aumento do apetite).
Relembrar a importância da orientação ao paciente sobre esses efeitos.
A fluoxetina se liga altamente as proteínas plasmáticas e é altamente metabolizada no fígado. (Interação medicamentosa).
INIBIDORES SELETIVOS DE RECAPTAÇÃO DE DOPAMINA
Bupropiona – mais utilizada no tratamento de pacientes que querer parar de fumar. Por aumentar os níveis de dopamina gera a sensação de
prazer que o cigarro trás. Só ela não resolve os quadros de depressão.
65
ANTIDEPRESSIVOS ATÍPICOS
Os mais recentemente lançados. Além de inibir a recaptação de serotonina são antagonistas seletivos dos receptores de serotonina. Aqueles
efeitos colaterais causados pelo excesso de serotonina são diminuídos porque os receptores estão bloqueados.
Prazolona – antagonista 5HT2 (bloqueia também H1).
INTERAÇÕES IMPORTANTES COM ANTIDEPRESSIVOS
Dois medicamentos que aumentam os níveis de serotonina administrados concomitantemente podem causar a Síndrome Serotoninérgica –
excesso de serotonina no SNC que causam alterações como ansiedade, confusão, agitação, alucinação, inquietação, tremor, convulsão, hiperreflexia,
incoordenação motora, febre, sudorese, vômito, diarréia e hipertensão e coma. Esses medicamentos são os INIBIDORES DA MAO, INIBIDORES SELETIVOS
DA RECAPTAÇÃO DE SEROTONINA, ANTIDEPRESSIVOS TRICÍCLICOS e o TRIPTOFANO.
Fármacos antidepressivos utilizados concomitantemente que se liguem a proteínas plasmáticas ou que tenham alta metabolização hepática
também podem gerar interação medicamentosa.
66