Metabolismo da galactose, frutose, ácido glicurónico e aminoaçúcares; Rui Fontes
Metabolismo da galactose, frutose, ácido glicurónico e aminoaçúcares
1. O UDP-glicurónico é o dador de ácido glicurónico na síntese de glicosaminoglicanos e na formação de
bilirrubina conjugada e de glicurono-conjugados de xenobióticos. As enzimas envolvidas nestes
processos são transférases em que um dos produtos é o UDP e o outro o substrato adicionado de um resíduo
de ácido glicurónico:
UDP-glicurónico + X UDP + glicurónico-X
(1)
Os glicosaminoglicanos são sintetizados intra-celularmente mas existem predominantemente no espaço
extracelular de variados tecidos e órgãos para onde são segregados pelas células componentes desses
mesmos tecidos. São longos polímeros lineares em que a unidade que se repete é um dissacarídeo que difere
nos diferentes tipos de glicosaminoglicanos. Com uma excepção (sulfato de queratano) todos os
glicosaminoglicanos podem conter resíduos de ácido glicurónico que se ligam a resíduos de um
aminoaçucar. Alguns glicosaminoglicanos também contêm outro ácido urónico, o L-idurónico, que resulta
da epimerização do ácido glicurónico.1
A glicurono-conjugação da bilirrubina e dos xenobióticos é um passo que precede a sua excreção (biliar ou
renal).
2. O UDP-glicurónico é sintetizado a partir da glicose. No primeiro passo a glicose é fosforilada formando-se
glicose-6-P (cínase da glicose; equação 2); depois a glicose-6-P sofre isomerização originando glicose-1-P
(fosfoglicomútase; equação 3) que por acção da pirofosforílase do UDP-glicose se converte em UDPglicose (equação 4). A conversão da UDP-glicose em UDP-glicurónico envolve uma desidrogénase
dependente do NAD+ e a transferência de quatro electrões (equação 5). O UTP (uridino-trifosfato), substrato
na reacção 4, pode formar-se por fosforilação do UDP (uridino-difosfato) por acção da cínase dos
nucleosídeos difosfatos (equação 6).
Glicose + ATP Glicose-6-P + ADP
Glicose-6-P Glicose-1-P
Glicose-1-P + UTP UDP-glicose + PPi
UDP-glicose + 2 NAD+ UDP-glicurónico + 2 NADH
UDP + ATP ADP + UTP
(2)
(3)
(4)
(5)
(6)
O somatório das equações 2-5 é a equação 7.
Glicose + ATP + UTP + 2 NAD+ UDP-glicurónico + ADP + PPi + 2 NADH
(7)
O somatório das equações 1, 6 e 7 (equação 8) permite compreender que na formação de um conjugado do
ácido glicurónico (e de uma unidade dissacarídica dos glicosaminoglicanos) se gastam 2 “ligações ricas em
energia” do ATP e se formam 2 NADH (eventualmente usados na síntese de ATP na fosforilação oxidativa).
Glicose + 2 ATP + 2 NAD+ + X glicurónico-X + 2 ADP + PPi + 2 NADH
(8)
3. A frutose que é absorvida no intestino pode resultar da hidrólise da sacarose da dieta mas também da
ingestão directa de frutose sobretudo em alimentos que foram processados industrialmente2. A absorção da
frutose no intestino e o transporte do sangue para as células envolve transportadores de membrana mas
1
Em alguns casos (sulfato de dermatano, heparina e sulfato de heparano), já depois da síntese da cadeia, uma percentagem
maior ou menor dos resíduos de ácido glicurónico sofre epimeração no carbono 5 convertendo-se em resíduos de ácido Lidurónico. Esta epimerização não ocorre nos casos do ácido hialurónico e do sulfato de condroitina. O aminoaçucar pode ser
a N-acetil-glicosamina (ácido hialurónico, sulfato de queratano, heparina e sulfato de heparano) ou a N-acetil-galactosamina
(sulfato de condroitina e sulfato de dermatano). Com excepção do ácido hialurónico todos os glicosaminoglicanos contêm
resíduos de sulfato que, na sua maior parte, estão ligados por ligações sulfoéster a grupos hidroxilo do polímero; no caso da
heparina, o sulfato pode substituir alguns dos grupos acetilo da N-acetil-glicosamina havendo, por isso, resíduos de N-sulfilglicosamina.
2
A razão do uso crescente da frutose é o facto deste monossacarídeo ser muito doce, cerca de duas vezes mais doce que a
glicose, por exemplo.
Página 1 de 6
Metabolismo da galactose, frutose, ácido glicurónico e aminoaçúcares; Rui Fontes
ocorre sempre a favor do gradiente: não há transporte activo de frutose. No pólo apical dos enterócitos e das
células tubulares renais o transportador para a frutose é o GLUT5; no pólo basal destas mesmas células e nos
hepatócitos é o GLUT2 [1]. Na criação do gradiente que possibilita a sua absorção e a sua entrada para as
células estão envolvidas enzimas que promovem a sua metabolização. Após a ingestão de frutose, a sua
concentração no plasma sobe para valores que podem atingir 0,5 mM (10% do da glicemia) mas desce
rapidamente para valores que são mais de 100 vezes inferiores aos da glicemia [2]. As enzimas envolvidas
na metabolização específica da frutose (cínase da frutose (equação 9) e aldólase B (equação 10)) são mais
abundantes no fígado3 sendo neste órgão que a frutose absorvida é maioritariamente captada (GLUT2) e
metabolizada [3, 4]. O primeiro passo no seu metabolismo é catalisado pela cínase da frutose que promove a
sua fosforilação no carbono 1. A frutose-1-fosfato formada sofre a acção da aldólase B que catalisa a sua
cisão formando-se como produtos a dihidroxiacetona-fosfato (um intermediário da glicólise e da
gliconeogénese) e o gliceraldeído (equação 10). A conversão do gliceraldeído no correspondente
intermediário fosforilado envolve a acção da cínase das trioses (equação 11). A equação 12 é o somatório
das reacções referidas acima e mostra que a frutose é, no fígado, convertida em dihidroxiacetona-P +
gliceraldeído-3-P.
Frutose + ATP Frutose-1-P + ADP
Frutose-1-P dihidroxiacetona-P + gliceraldeído
Gliceraldeído + ATP gliceraldeído-3-P + ADP
Frutose + 2 ATP dihidroxiacetona-P + gliceraldeído-3-P + 2 ADP
(9)
(10)
(11)
(12)
As trioses fosfato formadas (dihidroxiacetona-fosfato e o gliceraldeído-3-fosfato) são inter-convertíveis
(isomérase das trioses-fosfato; ver equação 13) e são intermediários da glicólise e gliconeogénese podendo
ser convertidas em glicose (via aldólase, fosfátase da frutose-1,6-bisfosfato, isomérase das hexoses-fosfato e
glicose-6-fosfátase)4. Outros destinos possíveis são a formação de glicogénio, lactato e palmitato
(lipogénese), sofrer oxidação completa com produção de CO2 (via glicólise, desidrogénase do piruvato, ciclo
de Krebs e fosforilação oxidativa) ou originar glicerol-3-P (via acção da desidrogénase do glicerol-3P: ver
equação 14) que é um dos precursores na síntese de triacilgliceróis.
Dihidroxiacetona-P gliceraldeído-3-P
dihidroxiacetona-fosfato + NADH glicerol-3-P + NAD+
(13)
(14)
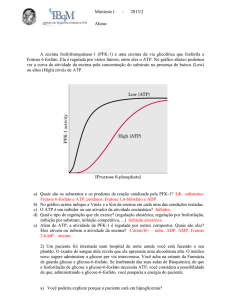
Embora as concentrações intracelulares de glicose possam inibir (inibição competitiva) a actividade de
fosforilação da frutose no carbono 6 pelas diversas hexocínases dos tecidos (incluindo a hexocínase IV, a
hexocínase hepática; ver equação 15) admite-se que uma parte menor do metabolismo da frutose possa
ocorrer via conversão da frutose em frutose-6-fosfato (intermediário da glicólise e gliconeogénese).
Frutose + ATP frutose-6-P + ADP
(15)
4. Embora a glicose seja o glicídeo com o papel mais importante no metabolismo energético dos mamíferos, o
nutriente dos espermatozóides é a frutose que é sintetizada a partir da glicose nas vesículas seminais. O
processo de síntese de frutose envolve a redução, dependente do NADPH, da glicose a sorbitol (redútase
das aldoses; equação 16) e a oxidação, dependente do NAD+, do sorbitol a frutose (desidrogénase do
sorbitol; ver equação 17). O sorbitol é o polialcool que resulta da redução do grupo aldeído da glicose.5 O
facto de os espermatozóides consumirem frutose e de o líquido seminal conter este açúcar dá aos gâmetas
masculinos uma vantagem competitiva sobre outras células (nomeadamente fungos e bactérias) que povoam
a vagina normal contribuindo para a sua sobrevivência (e para a sobrevivência dos genes neles contidos). A
membrana citoplasmática dos espermatozóides contém GLUT5, o transportador da frutose [1], mas nestas
células, o metabolismo da frutose, ao contrário do que acontece no fígado, envolve a hexocínase (conversão
em frutose-6-P; ver equação 15) [5].
3
Também existem nas células tubulares renais onde seriam importantes para metabolizar a frutose criando o gradiente
necessário para a sua reabsorção.
4
Alguns autores, alargando o conceito clássico de gliconeogénese, incluem na gliconeogénese os processos de formação de
glicose a partir de frutose e galactose.
5
A redútase das aldoses também existe no cristalino do olho e pensa-se que a acumulação de sorbitol neste tecido é, pelo
menos, uma das causas das cataratas dos doentes diabéticos.
Página 2 de 6
Metabolismo da galactose, frutose, ácido glicurónico e aminoaçúcares; Rui Fontes
glicose + NADPH sorbitol + NADP+
sorbitol + NAD+ frutose + NADH
(16)
(17)
5. A intolerância hereditária à frutose é uma doença congénita rara causada por uma alteração no gene da
aldólase B. A aldólase B dos indivíduos com intolerância hereditária à frutose, sendo capaz de catalisar com
eficácia as conversões próprias da glicólise e gliconeogénese (frutose-1,6-bisfosfato gliceraldeído-3-P +
dihidroxiacetona-P) não é capaz de cindir a frutose-1-fosfato (ver equação 10) [4]. Os doentes têm episódios
agudos de vómitos e hipoglicemia quando ingerem frutose (ou sacarose). Na patogenia das crises estão
alterações das concentrações intra-hepatocitárias de metabolitos como a acumulação de frutose-1-P e a
depleção de ATP e Pi. Na maioria dos casos, os doentes (ou os seus pais) aprendem a evitar os alimentos que
contêm frutose e a terapêutica é exactamente essa: restrição na ingestão de frutose e sacarose.6
6. A galactose é ingerida ligada à glicose na lactose que, por acção da lactase intestinal, sofre hidrólise. A
absorção da galactose nos enterócitos (e, a reabsorção nas células tubulares renais) envolve a acção da
SGLT1 (transporte activo secundário dependente do Na+). A galactose absorvida é rapidamente convertida
em glicose (ou glicogénio) e estes processos ocorrem maioritariamente no fígado; as concentrações
plasmáticas de galactose após ingestão de grandes quantidades de galactose ou em jejum são semelhantes às
apontadas acima para o caso da frutose [6, 7]. Tal como no caso da frutose o primeiro passo é o seu
transporte via GLUT2 seguido da sua fosforilação no carbono 1; a enzima envolvida denomina-se cínase da
galactose (ou galactocínase; equação 18). A transférase de uridilato da galactose-1-fosfato (ou uridiltransférase da galactose-1-P; equação 19) catalisa a transferência de um resíduo de uridilato (UMP) entre a
UDP-glicose e a galactose-1-P; um dos produtos formados é a UDP-galactose e o outro a glicose-1-fosfato.
A reacção de isomerização da UDP-galactose em UDP-glicose é catalisada pela UDP-galactose-4epimérase (equação 20).
Galactose + ATP Galactose-1-P + ADP
Galactose-1-P + UDP-glicose UDP-galactose + Glicose-1-P
UDP-galactose UDP-glicose
(18)
(19)
(20)
O somatório das equações 18-20 é a equação 21. Esta equação mostra que a galactose se converte em
glicose-1-fosfato e que não é errado dizer-se que o par UDP-galactose/UDP-glicose (inter-convertíveis por
acção da epimérase) tem um papel “catalítico” (ou pseudo catalítico) neste processo de conversão.
Galactose + ATP Glicose-1-P + ADP
(21)
O acção sequenciada da fosfoglicomútase operando sobre a glicose-1-fosfato formada (equação 3) e da
glicose-6-fosfátase sobre a glicose-6-fosfato (equação 22) permite compreender que se pode formar glicose
a partir da galactose ingerida e que a glicemia aumente quando se ingere galactose.
Glicose-6-P + H2O Glicose + Pi
(22)
A glicose-1-fosfato também pode ser substrato para a síntese de UDP-glicose (equação 4) que é o dador de
unidades de glicose na síntese de glicogénio, de glicoproteínas e de glicolipídeos.
É comum pensar-se que a galactose pode ser convertida em galactose-6-P por acção das hexocínases dos
tecidos. Contudo, esta ideia é errada: a actividade das hexocínases na galactose é, mesmo in vitro,
praticamente nula [8].
7. Para além da lactose, muitos componentes estruturais dos tecidos (como glicoproteínas, glicolipídeos e
proteoglicanos) contêm resíduos de galactose. A UDP-galactose é o dador de galactose aquando da acção de
galactosil-transférases (equação 23) que participam na síntese desses componentes estruturais e, no caso do
6
O deficit de cínase da frutose não é uma verdadeira doença. Os afectados não têm qualquer sintoma e as únicas anomalias
são, aquando da ingestão de frutose, a excessiva excreção renal de frutose (daí a designação clássica de “frutosúria
essencial”), subidas mais marcadas na frutosemia e uma metabolização lenta da frutose (via hexocínase) que não se perde na
urina.
Página 3 de 6
Metabolismo da galactose, frutose, ácido glicurónico e aminoaçúcares; Rui Fontes
tecido mamário em fase de lactação, na de lactose. Nestas acções catalíticas, o carbono anomérico da
galactose que estava ligado ao fosfato da UDP passa a estar ligado a uma proteína, a outro resíduo
glicídico ou a um lipídeo.
UDP-galactose + X UDP + galactose-X
(23)
Embora a UDP-galactose possa ter origem na galactose da dieta (galactose galactose-1-P glicose-1-P
UDP-glicose UDP-galactose; ver equações 18, 19, 4 e 20), a esmagadora maioria das moléculas de
UDP-galactose consumidas nos processos de biossíntese é sintetizada a partir da glicose. Nesta síntese
participam hexocínases (no caso do fígado, a hexocínase IV) que catalisam a formação de glicose-6-P (ver
equação 24), a fosfoglicomútase (equação 3) e a pirofosforílase da UDP-glicose (equação 4). A UDP-glicose
(formada por acção sequenciada destas 3 enzimas: glicose glicose-6-fosfato glicose-1-fosfato UDPglicose) pode, por acção da mesma isomérase que, no fígado, participa na conversão da galactose da dieta
em glicose (epimérase da UDP-galactose; equação 20), levar à formação de UDP-galactose. Assim se
compreende que a galactose (e a lactose) não seja um nutriente essencial (é dispensável na dieta).
Glicose + ATP glicose-6-P + ADP
(24)
8. Na glândula mamária inactiva existe uma galactosil-transférase que está normalmente envolvida na síntese
de glicoproteínas (equação 23). Imediatamente após o parto começa a sintetizar-se na glândula mamária uma
outra proteína (lactalbumina) que se liga à galactosil-transférase. O complexo galactosil-transféraselactalbumina tem actividade catalítica e designa-se como síntase da lactose porque catalisa a formação de
lactose (equação 25): a lactalbumina modifica a actividade da galactosil-transférase no que diz respeito ao
substrato aceitador que passa a ser a glicose.
UDP-galactose + glicose lactose + UDP
(25)
9. Estão descritas patologias congénitas raras causadas por alterações nos genes codificadores da galactocínase,
da uridil-transférase da galactose-1-fosfato e da UDP-galactose-4-epimérase que causam deficit de cada uma
destas enzimas [9]. Todas estas alterações causam aumento da galactose no plasma sanguíneo quando se
ingere galactose (ou lactose) e por isso são conhecidas pela designação de galactosemias. No entanto,
porque se conhece há mais tempo (desde 1935) e porque tem consequências mais graves, quando se diz
simplesmente “galactosemia” o mais provável é estar a falar-se do deficit de uridil-transférase da
galactose-1-fosfato. No deficit de uridil-transférase da galactose-1-fosfato ocorre bloqueio da conversão
galactose-1-fosfato glicose-1-fosfato o que leva à acumulação de galactose-1-fosfato e galactose nas
células. Não se sabe a patogenia da maior parte das alterações que podem manifestar-se nestes doentes
(atraso de crescimento, insuficiência hepática, anomalias no sistema nervoso central, alterações renais,
disfunção ovárica, etc.) mas sabe-se porque é que desenvolvem cataratas. Quando se acumula galactose nas
células do cristalino a redútase das aldoses presente nestes tecidos converte a galactose em galactitol, o
polialcool correspondente à galactose (ver equação 26). O galactitol acumula-se nas células do cristalino e
porque tem poder osmótico provoca a acumulação secundária de água que está na origem da opacificação do
cristalino. No caso do deficit de galactocínase acumula-se galactose mas não galactose-1-fosfato e a única
alteração é o desenvolvimento de cataratas que tem a mesma etiologia. O tratamento é, em ambos os casos,
uma dieta restritiva onde a galactose (e lactose) está totalmente proibida. A dieta pode curar e previne o
desenvolvimento das cataratas. Esta dieta também previne o desenvolvimento da insuficiência hepática e a
morte precoce nos casos de deficit de uridil-transférase da galactose-1-fosfato mas outras anomalias não são
prevenidas. De notar que a galactose é nutricionalmente dispensável já que a UDP-galactose pode ser
formada a partir da glicose via acção da UDP-galactose-4-epimérase.7
galactose + NADPH galactitol + NADP+
(26)
10. As oses aminadas são importantes constituintes de glicoproteínas, glicolipídeos e glicosaminoglicanos. Tal
como no caso da glicose, da galactose e do ácido glicurónico o substrato dador das oses aminadas para a
síntese destes compostos são derivados contendo o resíduo UDP. As transférases envolvidas catalisam
7
Os doentes com deficit de UDP-galactose-4-epimérase são muitíssimo raros e a doença é mal compreendida. Nos casos de deficiência
mais grave a restrição absoluta de galactose não é possível porque, para estes doentes, a galactose é nutricionalmente indispensável.
Página 4 de 6
Metabolismo da galactose, frutose, ácido glicurónico e aminoaçúcares; Rui Fontes
reacções análogas às descritas pelas equações 1 e 23. As oses aminadas formam-se a partir da frutose-6fosfato que, por acção de uma transférase, aceita um grupo amida da glutamina gerando a glicosamina-6fosfato (equação 27). A transformação da glicosamina-6-fosfato em UDP-glicosamina ocorre através de
transformações semelhantes às que sofre a glicose-6-fosfato quando se converte em UDP-glicose:
isomerização a glicosamina-1-fosfato e aceitação do resíduo uridilato do UTP (equações 28 e 29). É
frequente que o grupo amina esteja acetilado (ligação amida); a acetilação do grupo amina ocorre através da
transferência do grupo acetilo da acetil-coenzima A para a glicosamina-6-fosfato (equação 30). A formação
da UDP-N-acetil-galactosamina (ou da UDP-galactosamina) ocorre por isomerização da UDP-N-acetilglicosamina (ou da UDP-glicosamina), numa reacção análoga à descrita pela equação 20.
Frutose-6-P + glutamina glicosamina-6-P + glutamato
Glicosamina-6-P (ou N-acetil-glicosamina-6-P) Glicosamina-1-P (ou N-acetil-glicosamina-1-P)
Glicosamina-1-P (ou N-acetil-glicosamina-1-P) +UTP
UDP-glicosamina (ou UDP-N-acetil-glicosamina) + PPi
Glicosamina-6-P + acetil-CoA N-acetil-glicosamina-6-P + CoA
(27)
(28)
(29)
(30)
1. Douard, V. & Ferraris, R. P. (2008) Regulation of the fructose transporter GLUT5 in health and disease, Am J Physiol Endocrinol
Metab. 295, E227-37.
2. Chong, M. F., Fielding, B. A. & Frayn, K. N. (2007) Mechanisms for the acute effect of fructose on postprandial lipemia, Am J Clin
Nutr. 85, 1511-20.
3. Bais, R., James, H. M., Rofe, A. M. & Conyers, R. A. (1985) The purification and properties of human liver ketohexokinase. A role
for ketohexokinase and fructose-bisphosphate aldolase in the metabolic production of oxalate from xylitol, Biochem J. 230, 53-60.
4. Gitzelmann, R., Steinman, B. & Van der Berghe, G. (1995) Disorders of fructose metabolism in The metabolic and molecular bases of
inherited disease. (Scriver, C., Beaudet, A., Sly, W. & Valle, D., eds) pp. 905-933, McGraw-Hill, New York.
5. Jones, A. R. & Connor, D. E. (2000) Fructose metabolism by mature boar spermatozoa, Reprod Fertil Dev. 12, 355-9.
6. Schadewaldt, P., Hammen, H. W., Loganathan, K., Bodner-Leidecker, A. & Wendel, U. (2000) Analysis of concentration and (13)C
enrichment of D-galactose in human plasma, Clin Chem. 46, 612-9.
7. Sunehag, A., Tigas, S. & Haymond, M. W. (2003) Contribution of plasma galactose and glucose to milk lactose synthesis during
galactose ingestion, J Clin Endocrinol Metab. 88, 225-9.
8. Grossbard, L. & Schimke, R. T. (1966) Multiple hexokinases of rat tissues. Purification and comparison of soluble forms, J Biol Chem.
241, 3546-60.
9. Segal, S. & Berry, G. T. (1995) Disorders of galactose metabolism. in The metabolic and molecular bases of inherited disease.
(Scriver, C., Beaudet, A., Sly, W. & Valle, D., eds) pp. 967-1000, McGraw-Hill, New York.
Página 5 de 6
Metabolismo da galactose, frutose, ácido glicurónico e aminoaçúcares; Rui Fontes
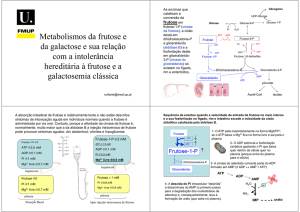
Galactose
Glicogénio
UDP
ATP
2 NAD+
ADP
2 NADH
UDP-Glicurónico
UDP-Glicose
Galactose-1-P
Lançadeiras do
malato ou do
glicerol-3-P
Fosforilação
oxidativa
H2O
X
2 Pi
PPi
UDP
Pi
UTP
Glicurónico-X
UDP-Galactose
UDP-N-acetil-glicosamina
Glicose-1-P
PPi
ATP ADP
Glicose
UTP
UDP-N-acetilgalactosamina
Glicose-6-P
Pi
ATP
N-acetilGlicosamina-1-P
H2O
glutamina glutamato
ADP
ATP
Pi
ADP
CoA
N-acetilGlicosamina-6-P
Glicosamina-6-P
Frutose-6-P
Frutose
Acetil-CoA
ATP
ADP
H2O
Frutose-1,6-bisP
Frutose-1-P
Gliceraldeído-3-P
Di-hidroxi-acetona-P
Gliceraldeído
Pi + ADP +
NAD+
ADP
ATP
QH2
ATP + NADH
Fosfoenolpiruvato
NADH
ADP
Q
ATP
NAD+
Piruvato
Glicerol-3-P
Página 6 de 6