Oxidação dos ácidos gordos; Rui Fontes
Oxidação dos ácidos gordos
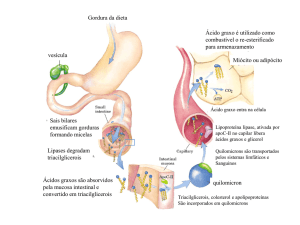
1- Durante o jejum a velocidade de hidrólise dos triacilgliceróis do tecido adiposo excede a
velocidade de síntese sendo o glicerol e os ácidos gordos libertados para o plasma sanguíneo.
Estes ácidos gordos não estão esterificados e por isso dizem-se livres. A maior parte dos ácidos
gordos libertados no tecido adiposo dizem-se de cadeia longa (porque contêm entre 10 e 18
carbonos) e são insolúveis em meio aquoso mas não formam agregados no sangue porque se
ligam (ligação não covalente) à proteína mais abundante no plasma: a albumina. Nos
capilares dos tecidos, os ácidos gordos desligam-se da albumina e penetram nas células. Dentro
das células, por acção catalítica de sintétases de acil-CoA, os ácidos gordos são “activados”,
ou seja, dão origem a acis-CoA: ácido gordo + CoA + ATP → acil-CoA + AMP + PPi.
2- Quer na dieta quer nas reservas lipídicas do organismo a maioria dos ácidos gordos contém
entre 14 e 18 carbonos: são de cadeia longa. O catabolismo dos ácidos gordos ocorre na matriz
mitocondrial e, no caso dos ácidos gordos de cadeia longa, o passo limitante da velocidade do
processo é o transporte de acil-CoA através da membrana mitocondrial interna. Este processo
de transporte é complexo e envolve a acção de uma transférase da membrana mitocondrial
externa (carnitina palmitoil-transférase I: equação 1) que catalisa a transferência do acilo do
acil-CoA para a carnitina, um transportador da membrana mitocondrial interna que é um
antiporter (troca acil-carnitina que entra por carnitina que sai; equação 2) e uma outra
transférase (localizada na membrana interna da mitocôndria mas cujo centro activo está
voltado para a matriz) que reverte o processo catalisado pela primeira transférase permitindo a
formação de acil-CoA na matriz (carnitina palmitoil-transférase II: equação 3). O somatório
das equações 1-3 é a equação 4.
carnitina(fora) + acil-CoA(fora) → acil-carnitina(fora) + CoA(fora)
acil-carnitina(fora) + carnitina(dentro) → acil-carnitina(dentro) + carnitina(fora)
acil-carnitina(dentro) + CoA(dentro) → carnitina(dentro) + acil-CoA(dentro)
acil-CoA (fora da mitocôndria) → acil-CoA (dentro da mitocôndria)
(1)
(2)
(3)
(4)
3- No interior da mitocôndria o acil-CoA vai ser oxidado num processo designado de oxidação
em β: ocorrem ciclos sucessivos em que o carbono β (o carbono 3) do acilo é oxidado; em cada
ciclo liberta-se uma unidade de acetil-CoA (2C) sendo o acil-CoA encurtado em 2 carbonos.
Em cada ciclo ocorrem quatro passos: o primeiro e o terceiro são catalisados por
desidrogénases, o segundo por uma líase (hidrátase) e o último por uma transférase (tiólase).
As equações 5-8 descrevem as acções catalíticas das enzimas envolvidas no processo:
acil-CoA + FAD → ∆2-trans-enoil-CoA1 + FADH2
∆2-trans-enoil-CoA + H2O → L-β-hidroxi-acil-CoA
L-β-hidroxi-acil-CoA + NAD+ → β-ceto-acil-CoA + NADH
β-ceto-acil-CoA + CoA ↔ acetil-CoA + acil-CoA
(5)
(6)
(7)
(8)
O 1º passo é catalisado por uma desidrogénase que tem como grupo prostético o FAD (a
desidrogénase de acil-CoA; equação 5) formando-se um acil-CoA insaturado (com uma dupla
ligação entre os carbonos 2 e 3). No 2º passo, uma hidrátase catalisa a hidratação do acil-CoA
insaturado formando-se um acil-CoA hidroxilado no carbono 3 (equação 6). No 3º passo, uma
outra desidrogénase (neste caso dependente do NAD+, a desidrogénase do β-hidroxi-acil1
∆2-trans-enoil-CoA: enoil é um resíduo de um ácido gordo insaturado; ∆2 significa que a dupla ligação está no carbono
2 (entre o 2 e o 3); trans significa que é o isómero trans e não o cis.
Página 1 de 6
Oxidação dos ácidos gordos; Rui Fontes
CoA; equação 7) catalisa a formação de um acil-CoA com um grupo cetónico no carbono 3: um
β-ceto-acil-CoA. As acções catalíticas da desidrogénase de acil-CoA e da desidrogénase do βhidroxi-acil-CoA implicam, respectivamente, a redução do FAD e do NAD+. Por último, o
derivado oxidado no carbono β (β-ceto-acil-CoA) formado pela acção catalítica da
desidrogénase do β-hidroxi-acil-CoA sofre tiólise (o CoA funciona como aceitador numa
reacção de transferência de acilo; equação 8) formando-se um acetil-CoA e um acil-CoA em
que o resíduo acilo está encurtado em dois carbonos relativamente ao acil-CoA donde se partiu.
O acil-CoA gerado pode depois voltar a ser oxidado pelas mesmas enzimas ocorrendo vários
ciclos de “encurtamento”. Quando se forma o derivado β-ceto-acil-CoA com 4 carbonos (o
acetoacetil-CoA) a acção da tiólase gera duas unidades de acetil-CoA. De facto, para cada um
dos passos do processo existem diferentes isoenzimas que se diferenciam funcionalmente por
terem maior ou menor actividade dependendo do tamanho da cadeia do resíduo acilo dos acisCoA que se vão formando sucessivamente. Quer quando estas isoenzimas estão localizadas na
membrana interna da mitocôndria quer quando estão na matriz os centros activos estão sempre
voltados para a matriz.
4- Ao contrário do que pode acontecer com a oxidação da glicose (que pode ser anaeróbia), a
oxidação dos ácidos gordos só pode ocorrer na presença de O2 que, por acção catalítica dos
complexos da cadeia respiratória, oxida o FADH2 e o NADH regenerando o FAD e o NAD+
indispensáveis ao processo. Os electrões do FADH2 da desidrogénase de acil-CoA são
transferidos para a coenzima Q (ubiquinona) através da acção sequenciada de duas oxiredútases
que (tal como a desidrogénase de acil-CoA) também têm FAD como grupo prostético: essas
enzimas são a flavoproteína de transferência de electrões (ETF; electron transfer
flavoprotein) e a oxiredútase da ETF-ubiquinona. Assim, a oxidação dos acis-CoA a ∆2trans-enoil-CoA só pode prosseguir em novos ciclos catalíticos porque o FADH2 formado é
reoxidado transferindo dois electrões para a ubiquinona (Q) que se reduz a ubiquinol (QH2).
Por sua vez, a reoxidação do ubiquinol implica a subsequente acção catalítica dos complexos
III e IV da cadeia respiratória. A reoxidação do NADH que se forma aquando da acção
catalítica da desidrogénase do β-hidroxi-acil-CoA depende da actividade dos complexos I, III e
IV da cadeia respiratória. As unidades de acetil-CoA geradas durante a oxidação em β são
oxidadas a CO2 pelas enzimas do ciclo de Krebs que é, tal como a oxidação em β, um processo
estritamente aeróbio.
5- O palmitato (CH3-(CH2)14-COOH) pode ser usado como exemplo do acoplamento existente
entre a oxidação dos ácidos gordos e a síntese de ATP. O palmitato contém 16 carbonos e
porque não contém duplas ligações diz-se saturado. A oxidação completa de um mole de
palmitato pode ser descrita pelo somatório (equação 15) das equações que exprimem a
activação do palmitato (equação 9), a hidrólise do PPi (equação 10), a oxidação do palmitilCoA a 8 unidades de acetil-CoA (equação 11), a oxidação do acetil-CoA no ciclo de Krebs
(equação 12), a oxidação do FADH2 e do NADH pelo O2 (que é indissociável da síntese de
ATP; equação 13) e a reacção de conversão do AMP em ADP (cínase do adenilato) (equação
14):
palmitato + CoA + ATP → palmitil-CoA + AMP + PPi
(9)
PPi + H2O → 2 Pi
(10)
(11)
palmitil-CoA + 7 FAD + 7 NAD+ + 7 CoA + 7 H2O → 8 acetil-CoA + 7 FADH2 + 7 NADH
8 acetil-CoA + 16 H2O + 8 ADP + 8 Pi + 24 NAD+ + 8 FAD →
8 ATP + 24 NADH + 8 FADH2 + 16 CO2 + 8 CoA(12)
Página 2 de 6
Oxidação dos ácidos gordos; Rui Fontes
31 NADH + 15 FADH2 + 23 O2 + 100 ADP + 100 Pi →
31 NAD+ + 15 FAD + 46 H2O + 100 ATP + 100 H2O (13)2
ATP + AMP → 2 ADP
(14)
⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯⎯
(15)
CH3-(CH2)14-COOH + 23 O2 + 106 ADP + 106 Pi → 16 CO2 + 16 H2O + 106 ATP + 106 H2O
A equação 11 mostra que na formação de 8 unidades de acetil-CoA (2C) a partir de um ácido gordo
com 16 carbonos ocorrem 7 “ciclos de encurtamento”. A equação 15 (somatório das equações 914) mostra claramente que as enzimas envolvidas no processo de oxidação do palmitato permitem
o acoplamento de um processo exergónico (a oxidação do palmitato) com um processo
endergónico (a síntese de ATP) e que a energia libertada no primeiro faz com que o segundo possa
ocorrer.
6- A velocidade da oxidação dos
nutrientes depende da velocidade de
hidrólise do ATP. Nos músculos
esqueléticos o aumento da velocidade
de hidrólise do ATP provoca (via
cínase do adenilato: 2 ADP → AMP +
ATP) aumento na concentração de
AMP que em última análise vai
provocar aumento na velocidade da
oxidação em β. O passo regulador da
velocidade da oxidação em β é o
transporte dos acis-CoA para dentro da
mitocôndria sendo que a enzima
“marca passo” do processo é a
carnitina-palmitoil-transférase
I.
Esta enzima é inibida pelo malonilCoA que se forma no citoplasma por acção catalítica da carboxílase de acetil-CoA (acetilCoA + CO2 + ATP → malonil-CoA + ADP + Pi). A carboxílase de acetil-CoA é inibida
quando fosforilada por acção da cínase activada pelo AMP (AMPK). A AMPK é activada
por fosforilação catalisada por outra cínase de proteínas e a AMPK é melhor substrato dessa
cínase quando está ligada ao AMP; por outro lado o AMP é activador alostérico da AMPK
fosforilada. Quando a actividade de contracção muscular aumenta, aumenta a concentração de
AMP que activa a AMPK que catalisa a forforilação e consequente inactivação da carboxílase
de acetil-CoA. Esta inactivação provoca diminuição na concentração citoplasmática de
malonil-CoA. A diminuição da concentração de malonil-CoA “desinibe” a carnitina-palmitoiltransférase I. A activação (=desinibição) desta enzima permite a entrada dos acis-CoA para a
mitocôndria e, consequentemente, a oxidação em β fica estimulada [1].
7- Para uma determinada velocidade de hidrólise de ATP existe uma velocidade de oxidação de
nutrientes (glicose e ácidos gordos) que permite manter a velocidade de síntese de ATP igual à
velocidade de hidrólise. Embora o aumento do trabalho muscular provoque aumentos quer na
velocidade de oxidação da glicose quer na dos ácidos gordos, independentemente da
velocidade com que um órgão está a hidrolisar ATP existe uma relação inversa entre a
velocidade de oxidação de glicídeos (glicose plasmática + glicogénio intracelular) e a
2
Na equação 13, para o cálculo do número de ATPs formados admitimos que à oxidação de um mole de NADH
corresponde a formação de 2,5 moles de ATP e que à oxidação de um mole de FADH2 correspondem 1,5 moles de
ATP.
Página 3 de 6
Oxidação dos ácidos gordos; Rui Fontes
velocidade de oxidação de ácidos gordos. No músculo, a hiperglicemia, a insulina e altos
níveis de glicogénio provocam aumento na velocidade de oxidação dos glicídeos e
diminuição na de oxidação dos ácidos gordos. De forma reversa o aumento da oxidação dos
ácidos gordos durante o jejum também diminui a oxidação da glicose.
8- Após uma refeição que contenha glicídeos o músculo oxida preferencialmente glicídeos. A
insulina (elevada) mobiliza vesículas intra-citoplasmáticas que contêm GLUT4 para a
membrana celular aumentando a velocidade de entrada da glicose e favorecendo a sua
oxidação. Para esta estimulação da oxidação da glicose também contribui a acção estimulante
da insulina na fosfátase da desidrogénase do piruvato cuja acção é desfosforilar a
desidrogénase do piruvato e, consequentemente, acelerar a conversão de piruvato em acetilCoA. A diminuição da velocidade de oxidação de ácidos gordos quando a insulina
plasmática e a glicemia estão elevados (ou/e o glicogénio muscular está elevado) envolve o
aumento da actividade da carboxílase de acetil-CoA, o consequente aumento da concentração
intracelular de malonil-CoA e a consequente inibição da carnitina-palmitoil-transférase I. A
carboxílase de acetil-CoA é activada pela insulina e inibida por fosforilação dependente da
AMPK mas também tem regulação alostérica: é activada pelo citrato e inibida pelos acis-CoA
[2-5]. Crê-se actualmente que a inibição da oxidação de ácidos gordos no músculo quando a
insulina plasmática, a glicemia e os níveis de glicogénio estão elevados seja mediada, pelo
menos parcialmente, pela diminuição da concentração de acis-CoA citoplasmáticos [2-5]. A
insulina diminui a lipólise no tecido adiposo provocando diminuição na concentração
plasmática de ácidos gordos livres; consequentemente, quando a insulina está aumentada a
concentração plasmática dos ácidos gordos livres está diminuída e diminuída a sua entrada para
dentro das células o que diminui a formação de acis-CoA.
9- Pelo contrário, no estado de jejum, a descida da insulina estimula a lipólise no tecido adiposo
com aumento na concentração plasmática dos ácidos gordos livres e aumento dos acis-CoA
citoplasmáticos. Este aumento da concentração dos acis-CoA aumenta a velocidade de
oxidação dos ácidos gordos por pelo menos dois mecanismos: (1) mais acis-CoA significa
mais substrato para a carnitina-palmitoil-transférase I mas (2) também inibição (alostérica) da
carboxílase do acetil-CoA e, consequentemente, inibição da formação de malonil-CoA e
desinibição da carnitina-palmitoil-transférase I. A diminuição da oxidação de glicose quando
um músculo está a oxidar ácidos gordos pode ser um simples consequência do estado
nutricional: no jejum há baixa de insulina. Contudo para além disto o aumento dos ácidos
gordos no plasma por si só também tem um outro efeito que é diminuir a sensibilidade do
tecido muscular à insulina e deste modo diminuir a entrada de glicose para o músculo e a sua
oxidação [6].
10- Quando, em jejum, se faz exercício físico de baixa intensidade os ácidos gordos são os
principais combustíveis do músculo. No entanto, mesmo em jejum, quando a intensidade do
exercício é elevada os combustíveis mais importantes passam a ser a glicose e o glicogénio
intramuscular. Nesta mudança pode ter importância a mobilização (independente da insulina)
de GLUT4 para a membrana sarcoplasmática, a estimulação da cínase da frutose-6-P pelo
AMP e a estimulação da glicogenólise pelo AMP (activação alostérica da fosforílase muscular)
e pelo Ca2+ (via activação da cínase da fosforílase). De forma recíproca este aumento da
oxidação dos glicídeos acompanha-se de diminuição da oxidação de ácidos gordos. Um dos
mecanismos que estará na base desta diminuição é a activação da glicólise anaeróbia (formação
de ácido láctico a partir de glicose) e a consequente descida do pH intracelular que inibe a
carnitina-palmitoil-transférase I e portanto a oxidação dos ácidos gordos [7].
Página 4 de 6
Oxidação dos ácidos gordos; Rui Fontes
11- A oxidação dos ácidos gordos de cadeia impar (muito mais raros que os de cadeia par) geram
no processo oxidativo (para além de acetil-CoA) propionil-CoA (CH3CH2CO-SCoA):
quando, após um certo número de ciclos oxidativos que libertam acetil-CoA, a tiólase actua no
pentanoil-CoA liberta-se acetil-CoA e propionil-CoA. O propionil-CoA é carboxilado
(carboxílase do propionil-CoA: propionil-CoA + CO2 + ATP → metil-malonil-CoA + ADP +
Pi) a metil-malonil-CoA que através da acção de isomérases se converte em succinil-CoA, um
intermediário do ciclo de Krebs. A oxidação completa do succinil-CoA a CO2 requer a sua
prévia conversão em acetil-CoA. Uma via pelo qual os intermediários do ciclo de Krebs podem
ser convertidos em acetil-CoA envolve a acção da carboxicínase do fosfoenolpiruvato, da
cínase do piruvato e da desidrogénase do piruvato (succinil-CoA → succinato → fumarato
→ malato → oxalacetato → fosfoenolpiruvato → piruvato → acetil-CoA). Uma outra via
possível envolve intervenção da enzima málica e da desidrogénase do piruvato (succinilCoA → succinato → fumarato → malato → piruvato → acetil-CoA). Nos órgãos que contém
as enzimas da gliconeogénese o succinil-CoA pode converter-se em glicose.
12- Os ácidos gordos insaturados naturais (como o oleico (18:1,9) e o palmitoleico (16:1;9))
contêm, pelo menos, uma dupla ligação e as duplas ligações dos ácidos gordos insaturados
naturais têm sempre uma configuração cis. Quando a dupla ligação se encontra num carbono
impar forma-se, em dado passo do processo oxidativo, o ∆3-cis-enoil-CoA que não é substrato
nem da desidrogénase de acil-CoA nem da hidrátase. A acção da hidrátase só é possível após a
acção de uma isomérase que converte o ∆3-cis-enoil-CoA em ∆2-trans-enoil-CoA. O ∆2trans-enoil-CoA é, por acção sequenciada da hidrátase, da desidrogénase do β-hidroxi-acilCoA e da tiólase, encurtado em dois carbonos. É de notar que um ácido gordo insaturado está
mais oxidado que o seu homólogo com igual números de carbonos e que o número de ATPs
que se pode obter aquando da sua oxidação é menor (cerca de 1,5 ATPs menos).
13- Alguns ácidos gordos naturais (como o linoleico (18:2;9,12) e o α-linolénico (18:3;9,12,15))
contêm duplas ligações em carbonos par. Neste caso, depois de se terem libertado 4 acetisCoA, forma-se o ∆4-cis-enoil-CoA; este é oxidado pela desidrogénase de acil-CoA formandose ∆4-cis-∆2-trans-dienoil-CoA que não é substrato da hidrátase. A acção da hidrátase só é
possível após a acção de uma redútase dependente do NADPH (NADPH + ∆4-cis-∆2-transdienoil-CoA → NADP+ + ∆3-trans-enoil-CoA) e de uma isomérase (∆3-trans-enoil-CoA→∆2trans-enoil-CoA) que, por acção sequenciada, convertem o ∆4-cis-∆2-trans-dienoil-CoA em
∆2-trans-enoil-CoA. O ∆2-trans-enoil-CoA é o substrato da hidrátase e a oxidação em β já
pode prosseguir.
14- Os ácidos gordos ditos de cadeia muito longa (com mais de 18 carbonos) são muito menos
abundantes que os de cadeia longa e são parcialmente oxidados nos peroxissomas. Nestes
organelos não há cadeia respiratória e a enzima que catalisa a oxidação dos acis-CoA a ∆2trans-enoil-CoA é uma oxídase que têm como grupo prostético o FAD: o O2 funciona como
oxidante que se reduz a peróxido de hidrogénio (acil-CoA + O2 → ∆2-trans-enoil-CoA +
H2O2). A catálase catalisa a dismutação do H2O2 formado (2 H2O2 → 2 H2O + O2). Também
existem nos peroxissomas as outras enzimas da oxidação β capazes de formar unidades de
acetil-CoA e de encurtar os acis-CoA num número par de carbonos. Admite-se que o NADH
formado durante a acção da desidrogénase do β-hidroxi-acil-CoA dos peroxissomas seja
oxidado na mitocôndria mas desconhecem-se os mecanismos envolvidos na transferência dos
equivalentes redutores entre os peroxissomas e as mitocôndrias [8]. Nos peroxissomas também
não há ciclo de Krebs: os mecanismos envolvidos no transporte das unidades de acetil-CoA e
dos acis-CoA encurtados em unidades de 2 carbonos para a mitocôndria são ainda controversos
[8].
Página 5 de 6
Oxidação dos ácidos gordos; Rui Fontes
15- Alguns (raros) ácidos gordos da dieta, como o ácido fitânico, contêm um grupo metilo no
carbono β o que impede a acção catalítica da desidrogénase de acil-CoA. A oxidação em β do
ácido fitânico (ou melhor, do derivado “activado” fitanoil-CoA) só é possível após a oxidação
do carbono α seguida de descarboxilação do carbono 1 (o carbono carboxílico). Estas reacções,
que ocorrem nos peroxissomas [8], para além de encurtarem numa unidade o número de
carbonos do ácido fitânico, transformam o carbono que contém o metilo no carbono 2 (era o
carbono β e passou a ser o α). O ácido pristânico, assim originado, depois de activado a
pristanil-CoA já é substrato da desidrogénase de acil-CoA e a oxidação em β já pode
processar-se.
16- Em grau variável, alguns ácidos gordos podem ser oxidados no carbono ω (ómega, o último). O
processo ocorre no retículo endoplasmático do fígado e rim envolvendo uma oxigénase de
função mista contendo o citocromo P450 (que promove a formação de um grupo hidroxilo no
carbono ω) e duas desidrogénases que oxidam, sucessivamente, esse grupo hidroxilo a aldeído
e a carboxilo. Este processo leva à formação de ácidos dicarboxílicos que são maioritariamente
excretados na urina.
1. Hardie, D. G., Hawley, S. A. & Scott, J. W. (2006) AMP-activated protein kinase--development of the energy sensor
concept, J Physiol. 574, 7-15.
2. Rasmussen, B. B., Holmback, U. C., Volpi, E., Morio-Liondore, B., Paddon-Jones, D. & Wolfe, R. R. (2002)
Malonyl coenzyme A and the regulation of functional carnitine palmitoyltransferase-1 activity and fat oxidation in
human skeletal muscle, J Clin Invest. 110, 1687-93.
3. Chien, D., Dean, D., Saha, A. K., Flatt, J. P. & Ruderman, N. B. (2000) Malonyl-CoA content and fatty acid
oxidation in rat muscle and liver in vivo, Am J Physiol Endocrinol Metab. 279, E259-65.
4. Kiens, B. (2006) Skeletal muscle lipid metabolism in exercise and insulin resistance, Physiol Rev. 86, 205-43.
5. Bavenholm, P. N., Pigon, J., Saha, A. K., Ruderman, N. B. & Efendic, S. (2000) Fatty acid oxidation and the
regulation of malonyl-CoA in human muscle, Diabetes. 49, 1078-83.
6. Roden, M. (2004) How free fatty acids inhibit glucose utilization in human skeletal muscle, News Physiol Sci. 19,
92-6.
7. Jeukendrup, A. E. (2002) Regulation of fat metabolism in skeletal muscle, Ann N Y Acad Sci. 967, 217-35.
8. Wanders, R. J. (2004) Peroxisomes, lipid metabolism, and peroxisomal disorders, Mol Genet Metab. 83, 16-27.
Página 6 de 6