Carlos Capela
AMINOÁCIDOS
AMINOÁCIDOS ......................................................... 1
A.
INTRODUÇÃO ......................................................... 4
1.
DEFINIÇÃO .................................................................... 4
1.a.
Aminoácidos proteicos ............................................................4
1.A.I.
1.A.II.
2.
AMINOÁCIDOS PROTEICOS COMUNS ............................................... 4
AMINOÁCIDOS PROTEICOS DERIVADOS ......................................... 5
ESTRUTURA QUÍMICA GERAL ..................................... 6
2.a.
Isómeros ópticos ......................................................................6
2.A.I.
3.
CONFIGURAÇÃO DE FISHER ............................................................ 7
CLASSIFICAÇÃO DOS AMINOÁCIDOS ........................... 9
3.a.
Segundo a Polaridade dos seus radicais “R”.........................9
3.A.I.
3.A.II.
3.A.III.
AMINOÁCIDOS COM RADICAL “R” APOLAR .................................. 9
AMINOÁCIDOS COM RADICAL “R” POLAR NÃO-CARREGADO ..... 9
AMINOÁCIDOS COM RADICAL “R” POLAR CARREGADO ............ 10
3.a.iii.1.
3.a.iii.2.
3.b.
“R” Carregado Positivamente ........................................................... 10
“R” Carregado Negativamente.......................................................... 10
Segundo a Natureza dos seus radicais “R”..........................11
3.B.I.
ÁCIDOS MONOAMINO E MONOCARBOXÍLICOS ............................ 11
3.b.i.1.
3.b.i.2.
3.b.i.3.
Aminoácidos Alifáticos Neutros ........................................................ 11
Aminoácidos Aromáticos ................................................................... 12
Aminoácidos Sulfurados.................................................................... 13
3.b.i.5.
3.b.i.6.
Aminoácidos com um resíduo Amida................................................ 15
3.B.II.
3.B.III.
α-Iminoácido..................................................................................... 14
ÁCIDOS MONOAMINO E DICARBOXÍLICO ..................................... 16
ÁCIDOS DIAMINO E MONOCARBOXÍLICO ..................................... 17
4. AMINOÁCIDOS EM SOLUÇÃO AQUOSA:
PROPRIEDADES QUÍMICAS E ELÉCTRICAS......................... 18
Aminoácidos página 1 de 70
Carlos Capela
B.
METABOLISMO DOS AMINOÁCIDOS ................... 19
1.
METABOLISMO GERAL .............................................. 19
1.a.
1.b.
1.c.
Descarboxilação ....................................................................19
Transaminação......................................................................21
Desaminação .........................................................................23
1.C.I.
1.C.II.
1.d.
1.e.
1.f.
Desamidação..........................................................................26
Transdesaminação ................................................................26
Transferência de corpos em C1 .............................................27
1.F.I.
2.
A DESAMINAÇÃO OXIDANTE .......................................................... 23
DESAMINAÇÃO NÃO OXIDANTE ..................................................... 24
A TRANSMETILAÇÃO ..................................................................... 28
METABOLISMO ESPECIAL .......................................... 29
2.a.
Biossíntese dos aminoácidos – Anabolismo .........................29
2.A.I.
2.A.II.
2.A.III.
INTRODUÇÃO ................................................................................. 29
BIOSSÍNTESE DO ÁCIDO GLUTÂMICO ........................................... 33
BIOSSÍNTESE DA GLUTAMINA ....................................................... 34
2.a.iii.1.
2.A.IV.
2.A.V.
2.A.VI.
2.A.VII.
2.A.VIII.
2.A.IX.
2.b.
Regulação do equilíbrio ácido-base................................................... 35
BIOSSÍNTESE DA ALANINA E ÁCIDO ASPÁRTICO .......................... 36
ASPARAGINA .................................................................................. 36
SERINA E GLICINA ......................................................................... 37
ARGININA, PROLINA E ORNITINA ................................................. 39
METIONINA, CISTEÍNA E CISTINA................................................. 41
FENILALANINA E TIROSINA........................................................... 44
Degradação dos Aminoácidos - Catabolismo ......................45
2.B.I.
2.B.II.
2.B.III.
MECANISMO GERAL DE DEGRADAÇÃO DE AMINOÁCIDOS:......... 45
O PAPEL DO ÁCIDO GLUTÂMICO.................................................. 46
ELIMINAÇÃO DA AMÓNIA: ............................................................ 47
2.b.iii.1.
2.b.iii.2.
2.B.IV.
Transporte de Amónia ao Fígado:..................................................... 47
Ciclo da Ureia ou de Krebs-Henseleit: .............................................. 48
O DESTINO DOS ESQUELETOS CARBONADOS:.............................. 52
2.b.iv.1.
2.b.iv.2.
2.b.iv.3.
2.b.iv.4.
2.b.iv.5.
2.b.iv.6.
2.b.iv.7.
2.b.iv.8.
2.b.iv.9.
2.b.iv.10.
2.b.iv.11.
2.b.iv.12.
Glicina e Serina.................................................................................. 52
Alanina ............................................................................................... 53
Cisteína ............................................................................................... 53
Ácido Aspártico e Asparagina ........................................................... 53
Ácido Glutâmico, Glutamina, Prolina, Arginina e Histidina........... 54
Valina, Isoleucina e Leucina............................................................. 55
Lisina .................................................................................................. 55
Metionina ........................................................................................... 56
Treonina ............................................................................................. 56
Fenilalanina e Tirosina ..................................................................... 56
Triptofano........................................................................................... 57
Em resumo.......................................................................................... 57
Aminoácidos página 2 de 70
Carlos Capela
C.
DERIVADOS DE AMINOÁCIDOS ........................... 59
1.
CREATINA ................................................................... 59
2.
GLUTATIÃO ................................................................. 61
2.a.
Ciclo do γ-Glutamilo ou de Meister .....................................63
3.
NEUROTRANSMISSORES DERIVADOS DA TIROSINA .. 65
4.
NEUROTRANSMISSORES DERIVADOS DO TRIPTOFANO.
..................................................................................... 66
4.a.
4.b.
D.
A Serotonina ..........................................................................66
A Melatonina .........................................................................67
AMINOACIDÚRIAS................................................ 68
1.
A FENILCETONÚRIA ................................................... 68
2.
O ALBINISMO .............................................................. 69
3.
HOMOCISTINÚRIA....................................................... 69
4.
A CISTINÚRIA.............................................................. 70
5.
CISTINOSE ................................................................... 70
Aminoácidos página 3 de 70
Carlos Capela
A. INTRODUÇÃO
1.DEFINIÇÃO
Os aminoácidos são ácidos orgânicos de série saturada, cíclicos ou heterocíclicos
nos quais um dos hidrogénios do átomo de carbono α está substituído por um grupo
funcional amina.
Os aminoácidos podem ser:
•
Proteicos: quando fazem parte da estrutura das proteínas;
•
Não proteicos: quando existem nas células e tecidos, livres ou combinados, mas
nunca nas proteínas. Alguns exemplos: β-Alanina, ornitina e citrulina.
1.a. Aminoácidos proteicos
São as unidades fundamentais das Proteínas. Distinguem-se em comuns e
derivados.
1.a.i. AMINOÁCIDOS PROTEICOS COMUNS
São os aminoácidos para os quais existe pelo menos um codão específico no código
genético. Existem vinte aminoácidos comuns, dos quais o homem pode sintetizar 11.
Os restantes cuja síntese de novo é impossível são denominados essenciais porque
devem ser obtidos na alimentação.
Aminoácidos página 4 de 70
Carlos Capela
1.a.ii. AMINOÁCIDOS PROTEICOS DERIVADOS
São geralmente formados por uma reacção catalisada por uma enzima, a partir de
um aminoácido comum, após a sua incorporação na estrutura de uma proteína. É o
exemplo da cistina que resulta da oxidação das cadeias laterais de 2 cisteínas, unidas
por uma ligação covalente dissulfeto.
Aminoácidos página 5 de 70
Carlos Capela
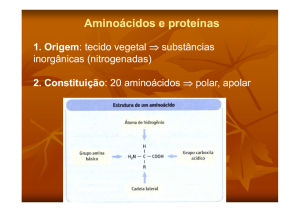
2.ESTRUTURA QUÍMICA GERAL
Os 20 aminoácidos possuem características estruturais em comum, tais como:
•
A presença de um carbono central α, quase sempre assimétrico (à excepção da
glicina);
•
Ligados a este carbono central, um grupo Carboxilo, um grupo Amina e um
átomo de hidrogénio;
•
O quarto ligante é um radical chamado genericamente de “R”, responsável pela
diferenciação entre os 20 AA. É a cadeia lateral dos AA. É o radical “R” quem
define uma série de características dos AA, tais como polaridade e grau de
ionização em solução aquosa.
2.a. Isómeros ópticos
A quiralidade ou assimetria do carbono α (ligação a 4 grupos diferentes) confere
aos aminoácidos actividade óptica. Só o aminoácido Glicina, que têm 2 H no carbono
α, não possui quiralidade.
Isómeros de aminoácidos rodam a luz polarizada em direcções diferentes. O
isómero que faz rodar o plano de luz polarizada no sentido dos ponteiros do relógio é
designado por dextrogiro (+). Se o isómero faz rodar o plano de luz polarizada no
sentido contrário ao dos ponteiros do relógio é designado por levrogiro (–). Este dado
só é observado experimentalmente, através de um polarimetro.
A aldotriose gliceraldeído é usada como referência para todos os aminoácidos.
Um aminoácido é designado por D ou L dependendo do arranjo dos átomos rodeando o
carbono α. Mas, alguns dos aminoácidos possuem mais do que um carbono quiral, o
que influencia a rotação da luz polarizada. Aminoácidos de cadeia longa possuem
Aminoácidos página 6 de 70
Carlos Capela
grupos adicionais entre o carbono α e o carbono terminal, o que pode promover
designações opostas D/L e +/–.
Mas vejamos como se classifica um isómero em D ou L.
2.a.i. CONFIGURAÇÃO DE FISHER
Quando utilizamos a configuração de Fisher para demonstrar a direcção no espaço
do arranjo dos aminoácidos, o carbono α está no centro, o traço horizontal representa
ligações acima do plano do papel e o traço vertical representa ligações para trás do
plano do papel. A configuração absoluta dos aminoácidos é determinado pela
estereoquimica do átomo de carbono α. Com base na posição do grupo NH2,
convencionou-se que, se este se projectar à esquerda, o aminoácido tem uma
configuração absoluta L. A estrutura simétrica têm uma configuração D.
Moléculas que não são sobreponíveis com a sua imagem especular são conhecidas
como enantiómeros. Ex: D-Alanina e L-Alanina.
Os aminoácidos proteicos encontram-se todos na forma L.
Ao aumentar o número de carbonos quirais, o número de isómeros possíveis
aumenta. Se n é o número de carbonos quirais, o número possível de isómeros é 2n.
Nos aminoácidos com 2 carbonos assimétricos, como a treonina, a configuração no
segundo átomo de carbono na forma que não ocorre nas proteínas, define a forma alo,
que também pode ser D ou L.
Aminoácidos página 7 de 70
Carlos Capela
As soluções de aminoácidos sintetizados em laboratório por vezes são opticamente
inactivas. Isto significa que existe uma mistura equimolar de isómeros L e D, portanto
de formas que apresentam actividade óptica complementar e portanto se anulam. Estas
misturas são designadas por misturas racémicas.
Aminoácidos página 8 de 70
Carlos Capela
3.CLASSIFICAÇÃO DOS AMINOÁCIDOS
O radical “R” permite a classificação dos aminoácidos segundo a sua polaridade e
natureza.
3.a. Segundo a Polaridade dos seus radicais “R”
A polaridade do radical “R” permite a classificação dos aminoácidos em 3 classes:
3.a.i. AMINOÁCIDOS COM RADICAL “R” APOLAR
Possuem radical “R” geralmente formado exclusivamente por carbono e hidrogénio
– grupos alquilo.
São hidrofóbicos e em número de 8:
•
Alanina
•
Valina
•
Leucina
•
Isoleucina
•
Prolina – Forma anel imina
•
Fenilalanina
•
Triptofano
•
Metionina – “R” contém enxofre
3.a.ii. AMINOÁCIDOS
COM
RADICAL
“R”
POLAR
NÃO-
CARREGADO
São aminoácidos com radicais “R” contendo hidroxilos, sulfidrilos e grupos amida.
São hidrofílicos e em número de 7:
•
Glicina – o mais simples dos AA
•
Serina – “R” com função álcool
•
Treonina – “R” com função álcool
•
Cisteína – possui um radical sulfidrilo
•
Tirosina – “R” com grupo fenol
Aminoácidos página 9 de 70
Carlos Capela
•
Asparagina – “R” com função amida
•
Glutamina – “R” com função amida
3.a.iii. AMINOÁCIDOS COM RADICAL “R” POLAR CARREGADO
2 Subclasses:
3.a.iii.1. “R” Carregado Positivamente
São AA diamino e monocarboxílicos, em número de 3:
•
Lisina
•
Arginina
•
Histidina
3.a.iii.2. “R” Carregado Negativamente
São AA monoamino e dicarboxílicos, em número de 2:
•
Ácido Aspártico
•
Ácido Glutâmico
Aminoácidos página 10 de 70
Carlos Capela
3.b. Segundo a Natureza dos seus radicais “R”
3.b.i. ÁCIDOS MONOAMINO E MONOCARBOXÍLICOS
•
Aminoácidos Alifáticos Neutros
•
Aminoácidos Aromáticos
•
Aminoácidos Sulfurados
•
Aminoácidos Álcoois
•
α-iminoácido
•
Aminoácidos com resíduo amida
3.b.i.1. Aminoácidos Alifáticos Neutros
Representação
Aminoácidos Alifáticos
Glicina (Gly)1
Alanina (Ala)
Valina (Val)
Leucina (Leu)
Isoleucina (Ile)
[G]2
[A]
[V]
[L]
[I]
1
Abreviação 3 letras
2
Abreviação 1 letra
Aminoácidos página 11 de 70
Carlos Capela
3.b.i.2. Aminoácidos Aromáticos
Representação
Aminoácidos Aromáticos
Fenilalanina (Phe)
Tirosina (Tyr)
Triptofano (Trp)
[F]
[Y]
[W]
Aminoácidos página 12 de 70
Carlos Capela
3.b.i.3. Aminoácidos Sulfurados
Representação
Aminoácidos Sulfurados
Cisteína (Cys)
Metionina (Met)
[C]
[M]
Aminoácidos página 13 de 70
Carlos Capela
3.b.i.4.
Aminoácidos Álcoois
Representação
Aminoácidos Álcoois
Serina (Ser)
Treonina (Thr)
[S]
[T]
3.b.i.5. α-Iminoácido
Representação
α-Iminoácido
Prolina (Pro)
[P]
Aminoácidos página 14 de 70
Carlos Capela
3.b.i.6. Aminoácidos com um resíduo Amida
Representação
Aminoácidos Amidados
Asparagina (Asn)
Glutamina (Gln)
[N]
[Q]
Aminoácidos página 15 de 70
Carlos Capela
3.b.ii. ÁCIDOS MONOAMINO E DICARBOXÍLICO
Representação
Aminoácidos Ácidos
Ácido Aspártico (Asp)
Ácido Glutâmico (Glu)
[D]
[E]
Aminoácidos página 16 de 70
Carlos Capela
3.b.iii. ÁCIDOS DIAMINO E MONOCARBOXÍLICO
Representação
Aminoácidos Básicos
Histidina (His)
Lisina (Lys)
Arginina (Arg)
[H]
[K]
[R]
Aminoácidos página 17 de 70
Carlos Capela
4.AMINOÁCIDOS
EM
PROPRIEDADES
SOLUÇÃO AQUOSA:
QUÍMICAS
E
ELÉCTRICAS
Os aminoácidos são substâncias Anfotéricas, pois, em solução aquosa, comportamse ou como um ácido ou como uma base, formando também iões dipolares, a saber:
•
O grupo carboxilo ioniza-se em solução aquosa libertando protões, e adquirindo
carga negativa.
•
O grupo amina ioniza-se em solução aquosa aceitando protões e adquirindo
carga positiva.
Este comportamento depende do pH do meio aquoso em que o aminoácido se
encontra.
Em meio ácido, os AA tendem a aceitar protões, comportando-se como base e
adquirindo carga positiva – ionizam os seus radicais amina.
Em meio básico, os AA tendem a doar protões, comportando-se como ácidos e
adquirindo carga negativa – ionizam o seu radical carboxilo.
O valor de pH onde as cargas eléctricas do aminoácido se igualam e se anulam
chama-se Ponto Isoeléctrico, ou pH Isoeléctrico (pHi). No ponto isoeléctrico não há
migração, em meio eléctrico nem para o eléctrodo positivo (anôdo) nem para o
eléctrodo negativo (cátodo). A esse pH a
forma predominante é a de ião dipolar,
mas as formas catiónica e aniónica,
embora em baixa concentração estão
presentes equivalentemente, ou seja,
[COO-] = [NH3+].
Quando o pH da solução é inferior
ao pHi de um dado aminoácido, este
encontra-se
em
meio
ácido,
fica
carregado positivamente e migra para o
cátodo.
Quando o pH da solução é superior ao pHi de um dado aminoácido, este encontrase em meio básico, fica carregado negativamente e migra para o ânodo.
Aminoácidos página 18 de 70
Carlos Capela
B. METABOLISMO DOS
AMINOÁCIDOS
1.METABOLISMO GERAL
1.a. Descarboxilação
A descarboxilação dos aminoácidos é catalisada pelas descarboxilases, enzimas
dependentes do fosfato de piridoxal (forma activa da vitamina B6), que é um cofactor.
A descarboxilação consiste na remoção do grupo carboxilo dos aminoácidos
transformando-os em aminas.
Aminoácidos página 19 de 70
Carlos Capela
Aminas formadas por descarboxilação de aminoácidos e seu papel
Aminoácido
Amina
Papel
Ácido Aspártico
α-alanina
Composição das proteínas
β-alanina
CoA
Cisteína
Mercaptoetilamina
CoA
Ácido Glutâmico
GABA (ácido γ-aminobutírico)
Mediador do SNC
Histidina
Histamina
Acção Hipotensora
DOPA
Dopamina
Percursor da adrenalina
Serina
Etanolamina
Fosfatidos
5-Hidroxitriptofana
Serotonina
Acções hormonais
Tirosina
Tiamina
Contracção do útero e
enxaquecas
Ornitina
Putrescina
Lisina
Cadaverina
Ácido cisteico
Taurina
Ácido Cisteico-
Hipotaurina
Putrefacções
sulfínico
As aminas obtidas por descarboxilação são depois metabolizadas por intermédio
das monoaminooxidases (MAO) que se encontram no fígado, rim e mucosa intestinal,
formando-se um aldeído.
Aminoácidos página 20 de 70
Carlos Capela
1.b. Transaminação
É a reacção fundamental do metabolismo dos aminoácidos podendo fazer-se em
todos excepto na prolina, lisina e treonina.
Esta reacção consiste na transferência de um grupo amina de um α-aminoácido para
um α-cetoácido formando-se a partir do primeiro um outro α-cetoácido e a partir do
segundo um novo aminoácido, na presença de enzimas designadas aminotransferases
ou transaminases.
As aminotransferases são específicas e quase todas utilizam o ácido glutâmico
como dador de NH3, e o ácido α-cetoglutárico, como par aminoácido-cetoácido. O
ácido glutâmico é assim o principal dador de grupos amina.
Aminoácidos página 21 de 70
Carlos Capela
Exemplos de transaminações:
1. Ácido glutâmico + ácido oxaloacético ⇔ ácido α-cetoglutárico + ácido
aspártico
2. Ácido glutâmico + ácido pirúvico ⇔ ácido α-cetoglutárico + Alanina
As reacções das transaminases têm a seguinte utilidade metabólica:
•
Degradação de aminoácidos;
•
Biossíntese de aminoácidos não essenciais graças à reversibilidade da reacção;
•
Conversão dos aminoácidos em glúcidos mediante a transformação posterior
dos α-cetoácidos (ácido pirúvico, ácido α-cetoglutárico e ácido oxaloacético);
•
Síntese de compostos azotados (como a ureia);
•
Participação, associadas às desaminações, nas reacções de transdesaminação.
Em suma, a transaminação é reversível e permite degradar e sintetizar aminoácidos
e interliga o metabolismo dos aminoácidos e hidratos de carbono via ácido
oxaloacético, ácido α-cetoglutárico e ácido pirúvico.
Aminoácidos página 22 de 70
Carlos Capela
1.c. Desaminação
Pode ocorrer por um processo geral – desaminação oxidante – ou por processos
particulares, específicos de determinados aminoácidos.
1.c.i. A DESAMINAÇÃO OXIDANTE
Este processo decorre em várias etapas. Numa primeira fase ocorre uma
desidrogenação do aminoácido a iminoácido. Os equivalentes redutores retirados ao
substrato vão reduzir a coenzima. O iminoácido hidrolisa-se em α-cetoácidos e
amoníaco.
A coenzima reduzida é geralmente reoxidada pelo oxigénio molecular com a
formação de H2O, na cadeia transportadora de electrões. No entanto, outros aceitadores
de electrões podem permitir a reoxidação da coenzima.
Algumas desaminases importantes:
1. D-aminoácido-oxidase (cujo coenzima é o FAD);
2. L-aminoácido-oxidase (cujo coenzima é o FMN);
Aminoácidos página 23 de 70
Carlos Capela
3. Glicola oxidase que forma o ácido glioxilico;
4. Glutamato desidrogenase – muito importante pois origina o ácido αcetoglutárico, intermediário do ciclo de Krebs. Esta enzima que catalisa esta
reacção reversível, utiliza o NADPH na síntese de ácido glutâmico (aminação
redutora) e o NAD+ na desaminação oxidante.
A interconversão entre o ácido glutâmico e o ácido α-cetoglutárico por
desaminação oxidante constitui um importante ponto de contacto entre os
metabolismos glucídico e proteico.
1.c.ii. DESAMINAÇÃO NÃO OXIDANTE
Este processo ocorre em aminoácidos álcoois como é o caso da serina e treonina.
Formam-se α-cetoácidos a partir de uma desidratação prévia seguida de uma
hidratação. Por outras palavras, o oxigénio do α-cetoácido já pertencia ao aminoácido,
sendo proveniente do grupo OH, o que não acontecia na desaminação oxidativa em que
este elemento provinha da água.
Aminoácidos página 24 de 70
Carlos Capela
No caso da cisteína, o mecanismo reaccional é semelhante. A diferença consiste na
perda prévia de H2S (dessulfuração).
Tanto a 1ª como a 2ª reacção estão representadas de forma simplificada, pois em
todas elas intervêm o grupo prostético das enzimas, o fosfato de piridoxal, que forma
intermediariamente uma base de schiff com cada um dos aminoácidos.
Aminoácidos página 25 de 70
Carlos Capela
1.d. Desamidação
Os ácidos com um grupo amida, como é o caso da asparagina e glutamina, sofrem
um processo semelhante ao da desaminação não oxidante.
As enzimas asparaginase e glutaminase hidrolisam os respectivos aminoácidos,
transformando-os em ácido aspártico e ácido glutâmico pela libertação do radical
amida.
Os aminoácidos formados serão por sua vez transaminados e transformados no αcetoácido respectivo.
1.e. Transdesaminação
O contraste entre a fraca actividade das desaminases (excepto a glutamato
desidrogenase) e a grande actividade das transaminases promove a reacção de
transdesaminação.
Assim considera-se que a maior parte dos aminoácidos são degradados por
desaminação
oxidante
indirecta
graças
ao
sistema
transaminase/glutamato
desidrogenase. Daqui resulta a transdesaminação (junção das transaminase com a
glutamato desidrogenase).
Aminoácidos página 26 de 70
Carlos Capela
1.f. Transferência de corpos em C1
No decorrer do metabolismo dos aminoácidos forma-se vários corpos com o
carbono1 das moléculas:
•
Metilo CH3;
•
Metileno CH2;
•
Metenilo CH;
•
Formilo O=CH;
•
Forminino CHNH;
•
Anidrido carbónico CO2;
O CO2 é utilizado por meio da biotina, enquanto que outros são utilizados pelo
ácido tetrahidrofólico (THF). Este é um cofactor em muitas reacções de aminoácidos e
um transportador de carbono que facilita a interconversão de grupos.
Um exemplo de transferência em C1 é a conversão da serina em glicina pela serina
hidroximetiltransferase. Os corpos em C1 transportados pelo THF são depois utilizados
em reacções anabólicas, como por exemplo, a síntese de purinas, pirimidinas e na
remetilação da homocisteína.
Aminoácidos página 27 de 70
Carlos Capela
1.f.i. A TRANSMETILAÇÃO3
O principal dador de metilo é a metionina, que é assim classificada em aminoácido
essencial. A S-Adenosil-Metionina é o principal dador de grupos CH3, pois embora o
THF possa transportar um grupo metilo no seu azoto 5 ou 10, o seu potencial de
transferência não é suficientemente elevado.
3
Ver ciclo de metilo activado
Aminoácidos página 28 de 70
Carlos Capela
2.METABOLISMO ESPECIAL
2.a. Biossíntese dos aminoácidos – Anabolismo
2.a.i. INTRODUÇÃO
O nitrogénio é um gás que ocorre na atmosfera na proporção aproximada de 79%.
Apesar disso, não é utilizado de forma directa pelos seres vivos, com excepção de
alguns microorganismos. O seu aproveitamento pela generalidade dos seres vivos está
na dependência da sua fixação e posterior nitrificação.
A fixação do N2 pode ser feita através de radiação ou da biofixação, sendo este
último processo o mais importante. A biofixação é realizada por bactérias,
cianobactérias e fungos que podem viver livres no solo ou associados a plantas. Esses
organismos são os únicos que conseguem transformar o N2 atmosférico numa forma
utilizável pelos seres vivos: o amoníaco (NH3) que em solução aquosa origina a amónia
(NH4+). Os biofixadores que vivem associados a plantas são mais eficientes nesse
processo que os de vida livre. Isto porque a planta fornece um habitat apropriado,
geralmente nos nódulos das raízes, que protege esses microorganismos contra um
excesso de O2 (o qual inibe a fixação do nitrogénio) e fornece energia para a realização
do processo. Em troca, a planta recebe um elevado suprimento de nitrogénio sob a
forma assimilável.
Aminoácidos página 29 de 70
Carlos Capela
A amónia produzida pelos biofixadores associados é incorporada directamente aos
aminoácidos da planta onde vivem. Já a amónia produzida pelos biofixadores de vida
livre é transformada em nitrito e depois em nitrato, pela acção das bactérias
nitrificantes (Nitrosomonas e Nitrobacter). Essas bactérias são autotroficas
quimiossinteticas, que utilizam a energia da nitrificação para a síntese das suas
substâncias orgânicas
O nitrato pode ser absorvido pelos vegetais e o nitrogénio nele contido é utilizado
na síntese de aminoácidos, proteínas e ácidos nucleicos. Essas substâncias são
transferidas directa ou indirectamente para os animais, ao longo das cadeias
alimentares. Os animais, portanto, só conseguem captar o nitrogénio indispensável para
a síntese das suas proteínas e ácidos nucleicos ingerindo directamente plantas ou,
indirectamente, alimentando-se de outros animais da cadeia alimentar.
O nitrogénio deixa o corpo dos organismos por dois processos: excreção de
produtos nitrogenados e/ou decomposição dos organismos mortos.
As excreções nitrogenadas: ureia e ácido úrico são transformados em amónia por
bactérias e fungos decompositores. Estes organismos também degradam as substâncias
nitrogenadas contidas no corpo dos organismos mortos, transformando-as em amónia.
A amónia pode retornar ao ciclo sendo transformada em nitrito e nitrato pelas
bactérias nitrificantes, ou em nitrogénio (N2), por bactérias desnitrificantes. O N2 volta
Aminoácidos página 30 de 70
Carlos Capela
para a atmosfera, podendo entrar novamente na fase biológica do ciclo através dos
processos de fixação.
A síntese dos aminoácidos compreende, de um modo geral, as seguintes etapas:
1. Formação do amoníaco
2. Incorporação do NH3 num composto orgânico:
a. Aminação redutora do ácido α-cetoglutárico a ácido glutâmico;
b. Formação de uma amida como a glutamina ou asparagina;
c. Aminação do ácido fumárico e oxaloacético a ácido aspártico;
d. Formação do Carbamil-fosfato.
3. Síntese do esqueleto carbonado dos aminoácidos, isto é, formação dos αcetoácidos correspondentes.
Aminoácidos página 31 de 70
Carlos Capela
4. Transferência do grupo amina (NH3) do composto orgânico para o ácido αcetónico ou α-cetoácido.
As plantas e outros organismos fotossintéticos sintetizam os 20 aminoácidos
comuns das proteínas.
Nos mamíferos, um certo número de aminoácidos não pode ser formados através
dos mecanismos referidos anteriormente, quer porque os α-cetoácidos correspondentes
não se encontram presentes e não podem ser sintetizados, quer porque não podem
sofrer aminação ou transaminação.
Estes aminoácidos são, no Homem em número de 9. São designados aminoácidos
essenciais ou indispensáveis, devendo ser fornecidos pela alimentação.
Aminoácidos essenciais para
Aminoácidos não essenciais
o organismo humano adulto
Fenilalanina
Alanina
Histidina
Arginina
Isoleucina
Ácido aspártico
Leucina
Asparagina
Lisina
Ácido glutâmico
Metionina
Glutamina
Treonina
Cisteína
Triptofano
Glicina
Valina
Prolina
Serina
Tirosina
No homem, a arginina sintetizada pelo ciclo da ureia é suficiente para as necessidades do adulto, mas
não para uma criança. A arginina e a histidina são essenciais à criança durante o crescimento. A cisteína
e a tirosina são essenciais quando a metionina e a fenilalanina, respectivamente, são insuficientes ou
estão ausentes na dieta. A tirosina só é essencial na fenilcetonúria.
O esqueleto carbonado dos diferentes aminoácidos resulta de intermediários da
glicólise, da via das pentoses fosfato e do ciclo de Krebs.
Aminoácidos página 32 de 70
Carlos Capela
2.a.ii. BIOSSÍNTESE DO ÁCIDO GLUTÂMICO
O ácido glutâmico resulta de uma aminação redutora do α-cetoácido ácido αcetoglutárico.
No fígado, a amónia é incorporada como grupo amino pela glutamato
desidrogenase. Esta reacção é reversível. O ácido glutâmico serve como um dador de
grupos amina da maioria dos aminoácidos.
O NADPH é usado como coenzima na reacção de síntese, enquanto o NAD+ é
utilizado na reacção de degradação. A formação de NADH durante a reacção de
desaminação oxidante é um bónus bem-vindo, uma vez que pode ser reoxidado pela
cadeia respiratória, com a formação de ATP.
Como se pode verificar, a reacção é reversível, mas como a amónia é tóxica para o
organismo, esta reacção dá-se na direcção da formação do ácido glutâmico ao nível do
intestino, enquanto no fígado é enfatizado a sua degradação para síntese dos
aminoácidos.
A glutamato desidrogenase é regulada alostericamente por nucleótidos púricos.
Quando há necessidade de oxidação de aminoácidos para energia, a actividade é
aumentada no sentido da degradação do ácido glutâmico por ADP e GDP, que são
indicadores de um baixo nível de energia. GTP e ATP são indicativos de um alto nível
de energia, e por isso são activadores na direcção da síntese de ácido glutâmico.
Aminoácidos página 33 de 70
Carlos Capela
2.a.iii. BIOSSÍNTESE DA GLUTAMINA
A amónia livre é tóxica e, é preferencialmente, transportada no sangue na forma de
grupos amino ou amida. 50% dos aminoácidos circulantes são glutamina, um
transportador de amónia. O grupo amida da glutamina é um importante dador de
nitrogénio para as várias classes de moléculas, incluindo bases púricas e pirimídicas, e
o grupo amina da citosina.
Ácido glutâmico e amónia são substratos para a glutamina sintetase. ATP é
necessário para tornar a reacção energeticamente favorável.
A remoção do grupo amida é catalisada pela glutaminase.
A reacção segue predominantemente para a direita no fígado e nos rins, e o NH3 é secretado na urina
directamente ou sob a forma de ureia. A reacção segue predominantemente para a esquerda, por exemplo
no cérebro, removendo o NH3, que é tóxico para as células nervosas.
O principal destino da glutamina e da alanina no sangue é o fígado. Neste órgão, a
amónia é libertada pela alanina aminotransferase, pela glutaminase e pela glutamato
desidrogenase. A glutamato desidrogenase, como já vimos, não liberta apenas amónia,
mas também produz NADH e ácido α-cetoglutárico, um intermediário glucogénico.
Sob condições de necessidade de energia, esses produtos são muito benéficos. Alguma
glutamina e alanina têm como destino o rim. Neste, a amónia é libertada pelas mesmas
enzimas activas no fígado e excretada.
Aminoácidos página 34 de 70
Carlos Capela
2.a.iii.1. Regulação do equilíbrio ácido-base
A maioria da amónia, que se encontra
na urina, é formada pela desaminação da
glutamina, pela actividade da glutaminase
da mitocôndria e da membrana do lúmen,
que é dependente e independente de ATP,
respectivamente. A amónia é relevante no
rim no combate à acidose.
A
amónia
difunde
nos
fluidos
tubulares e é neutralizada pelos H+. O
NH4+ é eliminado por troca com o sódio e
daí
resulta
reabsorção
de
sódio
e
bicarbonato para o plasma. Assim é
mantida a neutralidade eléctrica e a
reserva alcalina.
Por outro lado, quando ocorre acidose,
o organismo desvia glutamina do fígado
para o rim, para conservar o bicarbonato,
uma vez que a formação de ureia, o
principal mecanismo de excreção de NH4+,
necessita de bicarbonato. Para evitar o uso
e a excreção deste anião como ureia,
durante a acidose, a captação de glutamina
pelo fígado é suprimida e mais glutamina é
transportada ao rim, para ser excretada.
Aminoácidos página 35 de 70
Carlos Capela
2.a.iv. BIOSSÍNTESE DA ALANINA E ÁCIDO ASPÁRTICO
Estes aminoácidos são sintetizados numa única reacção, respectivamente a partir do
ácido pirúvico e do ácido oxaloacético, na presença da Alanina aminotransferase e da
Aspartato aminotransferase.
Como já vimos, o ácido aspártico é um percursor do nucleótidos púricos e
pirimídicos, e participa na ureogénese. Por descarboxilação, que pode sofrer quer no
carboxilo α quer no carboxilo β origina a α-alanina ou a β-alanina. Alguma β-alanina é
formada a partir do propionil-CoA e durante a degradação das pirimidinas. Encontra-se
na forma livre no plasma e tecidos ou combinada na carnosina, anserina e ácido
pantoténico, componente da coenzima A.
Por outro lado, o ácido aspártico pode entrar no ciclo de Krebs e na neoglicogénese,
através da perda do seu grupo amina permitindo a formação de outros aminoácidos,
dando origem ao ácido oxaloacético que é o ponto de partida destes ciclos.
2.a.v. ASPARAGINA
O ácido aspártico pode ser aminado a Asparagina. O grupo amida da asparagina
vem da glutamina, e não da amónia livre como na síntese da glutamina.
Aminoácidos página 36 de 70
Carlos Capela
O ATP é necessário para activar o grupo carboxilo receptor. A enzima responsável
pela síntese é a asparagina sintetase. A molécula de ácido glutâmico regressa ao ciclo
glutamina sintetase para incorporar mais amoníaco.
A enzima responsável pela degradação é a asparaginase.
2.a.vi. SERINA E GLICINA
A serina e a glicina são 2 aminoácidos interconvertíveis. No entanto, a glicina que é
o mais simples, é sintetizada posteriormente.
A Serina resulta do ácido 3-fosfoglicérico, um intermediário da glicólise. Uma
desidrogenase NADH-dependente converte o ácido 3-fosfoglicérico a um α-cetoácido,
o ácido 3-fosfopirúvico, capaz de subsequente transaminação. A actividade de uma
aminotransferase com o ácido glutâmico como dador produz 3-fosfoserina, a qual é
convertida a serina pela fosfoserina fosfatase.
Aminoácidos página 37 de 70
Carlos Capela
A grande via de síntese de glicina é uma reacção catalisada pela serina
hidroximetilltransferase. Esta reacção envolve a transferência do grupo hidroximetil da
serina para o cofactor tetrahidrofolato (THF), produzindo N5,N10-metileno-THF.
Na realidade, a reacção serina-glicina é a principal fonte de unidades
monocarbonadas na forma N5,N10-metileno-THF.
Aminoácidos página 38 de 70
Carlos Capela
A Glicina está envolvida em muitas reacções anabólicas incluindo a síntese de
nucleótidos de purina, heme, glutatião, creatina e serina.
2.a.vii. ARGININA, PROLINA E ORNITINA
A ornitina (percursora da citrulina e Arginina) e a Prolina são ambas sintetizadas a
partir do ácido glutâmico e degradadas por uma via levemente diferente, novamente a
ácido glutâmico. A síntese destes dois aminoácidos, a ornitina e a prolina, começa a
partir do ácido glutâmico, com uma reacção compartilhada que utiliza ATP e NADH, e
forma o γ-semialdeído glutâmico. Este, espontaneamente cicliza-se formando uma base
de Schiff – ácido pirrolidina-5-carboxílico – entre o aldeído e o grupo amina. O ácido
pirrolidina-5-carboxílico é então reduzido pelo NADPH ou NADH, originando a
Prolina.
O γ-semialdeído glutâmico pode submeter-se à transaminação do grupo aldeído,
evitando a ciclização e produzindo a Ornitina.
A produção de Arginina para síntese proteica e não como intermediário do ciclo da
ureia, ocorre no rim, que não possui arginase (e por isso não produz ureia). O principal
local de síntese da citrulina para ser usada como percursora da arginina é a mucosa
intestinal que possui todas as enzimas necessárias para converter o ácido glutâmico, via
ornitina, a citrulina, que é então transportada para o rim a fim de produzir arginina.
Aminoácidos página 39 de 70
Carlos Capela
Aminoácidos página 40 de 70
Carlos Capela
2.a.viii. METIONINA, CISTEÍNA E CISTINA
Existem 3 aminoácidos sulfurados, a Metionina, a Cisteína e a Cistina. Os 2
últimos são facilmente inconvertíveis por oxidação-redução. A cistina é bastante
importante na estabilização da estrutura das proteínas, uma vez que é responsável pela
formação das pontes dissulfeto.
A metionina é um aminoácido essencial, o que não acontece com a cisteína.
Contudo, quando se fornece cisteína, as necessidades de metionina diminuem.
Além da sua importância como aminoácido constituinte das proteínas e iniciador da
biossíntese proteica, a metionina, é sobretudo um fornecedor de grupos metilo. Mas
para actuar como tal, tem de ser activada pelo ATP a S-Adenosil-Metionina (SAM),
que é o composto activado que participa nos processos de transmetilação.
A S-Adenosil-Metionina é, assim, sintetizada pela transferência de um grupo
Adenosilo do ATP para o átomo de enxofre da metionina. É a carga positiva adquirida
pelo enxofre que torna este grupo metilo muito mais reactivo que o ácido N5-metilTHF. Note-se que nesta reacção são hidrolisadas todas as ligações de elevado potencial
energético do ATP (visto que o pirofosfato também será hidrolisado).
A S-Adenosil-Metionina pode ceder o seu metilo a compostos diversos, passando a
S-Adenosil-Homocisteína. Este mecanismo intervém, por exemplo, na síntese da
colina, da creatina, da adrenalina, etc.
A S-Adenosil-Metionina é seguidamente hidrolisada em homocisteína e adenosina.
A homocisteína é agressiva para o endotélio. Esta pode ser eliminada por remetilação
Aminoácidos página 41 de 70
Carlos Capela
(transferência de um grupo metilo) do ácido N5-metil-THF, formando Metionina. Se a
homocisteína for fornecida na alimentação, pode-se formar Metionina.
O conjunto de reacções aqui descrito constitui o Ciclo do Metilo activado.
A homocisteína, formada pela desmetilação da metionina, além de ser um percursor
da metionina no ciclo do metilo activado, pode ser eliminada através da síntese de
cisteína. Esta reacção catalisada pela Cistationina sintetase e Cistationina Liase, tratase de uma transulfuração. No Homem é irreversível, daí não podermos sintetizar
Metionina através da cisteína, e somente cisteína a partir da Metionina.
Aminoácidos página 42 de 70
Carlos Capela
Aminoácidos página 43 de 70
Carlos Capela
2.a.ix. FENILALANINA E TIROSINA
A fenilalanina e tirosina são discutidas juntas, uma vez que a tirosina resulta da
hidroxilação da fenilalanina.
A maior parte da fenilalanina é utilizada na síntese de proteínas e Tirosina nos
indivíduos normais. ¾ da fenilalanina ingerida é metabolizada a tirosina. O
metabolismo da fenilalanina inicia-se pela conversão irreversível a tirosina, catalisada
pela fenilalanina-4-monooxigenase ou fenilalanina-4-hidroxilase, na presença de
oxigénio molecular e tetrahidrobiopterina (THP).
O NADPH é o dador de H+ na hidroxilação e mantém a biopterina na forma de
tetrahidrobiopterina. A dihidropteridina redutase permite a reutilização da coenzima,
cuja função é transferir equivalentes redutores do NADPH para o oxigénio. A
tetrahidrobiopterina é um composto ácido fólico – “like”, mas raramente é utilizada
como transportador de electrões.
Aminoácidos página 44 de 70
Carlos Capela
2.b. Degradação dos Aminoácidos - Catabolismo
É o conjunto complexo de vias de degradação dos aminoácidos para produção de
energia.
Não há uma via catabólica comum a todos os 20 aminoácidos; existem grupos de
aminoácidos que possuem vias de degradação distintas.
Os aminoácidos são obtidos a partir da hidrólise das proteínas, e a sua utilização
como fonte de energia ocorre em 3 situações metabólicas principais:
•
Na dinâmica normal de renovação das proteínas;
•
No jejum prolongado;
•
Na “diabetes mellitus”.
2.b.i. MECANISMO
GERAL
DE
DEGRADAÇÃO
DE
AMINOÁCIDOS:
Apesar de cada grupo de aminoácidos possuir um caminho metabólico próprio de
degradação, há um mecanismo comum de remoção do nitrogénio da molécula, através
da acção das transaminases.
AA ⇒ esqueleto carbonado + amónia
O esqueleto carbonado do AA é dirigido para a oxidação, geralmente como
intermediário do Ciclo de Krebs, e a amónia, por ser muito tóxica, é eliminada do
organismo.
Alguns aminoácidos são desaminados por vias especiais, onde a reacção não é
catalizada por transaminases.
Aminoácidos página 45 de 70
Carlos Capela
2.b.ii. O PAPEL DO ÁCIDO GLUTÂMICO
O ácido glutâmico é um intermediário que funciona como um reservatório
temporário de grupos amina de vários aminoácidos.
Ele é formado pela acção das transaminases, que removem o grupo amina do AA e
o doam para o ácido α-cetoglutárico:
AA + ácido α-Cetoglutárico ⇒ α-Cetoácido + Ácido Glutâmico
Ex: na reacção catalizada pela Alanina-Transaminase (ALT):
Alanina + ácido α-Cetoglutárico ⇒ Ácido Pirúvico + Ácido Glutâmico
O Ácido Glutâmico formado reage com o ácido oxaloacético para formar
novamente o ácido α-Cetoglutárico e o ácido Aspártico, sendo assim consumido e
deslocando para a direita o equilíbrio da reacção acima.
O ácido Aspártico também é utilizado na síntese da ureia, a forma de eliminação do
grupo amina libertado como amónia nas reacções de desaminação:
Ácido Glutâmico + ácido oxaloacético ⇒ ácido α-Cetoglutárico + ácido Aspártico
A reacção é catalizada pela Aspartato-Transaminase (AST), a transaminase mais
activa dos tecidos de mamíferos.
Aminoácidos página 46 de 70
Carlos Capela
2.b.iii. ELIMINAÇÃO DA AMÓNIA:
A amónia que se forma no catabolismo das proteínas e aminoácidos será
biotransformada no fígado a ureia, um composto menos tóxico que pode ser levado
pela corrente sanguínea aos rins, onde será filtrada e eliminada.
O processo envolve:
•
O transporte de amónia ao fígado;
•
A biotransformação no Ciclo da Ureia.
2.b.iii.1. Transporte de Amónia ao Fígado:
A amónia é transportada ao fígado de 2 formas principais: como Glutamina e como
Alanina.
2.b.iii.1.a. Transporte de amónia como Glutamina
Ocorre na remoção da amónia de muitos tecidos periféricos (glutamina sintetase)
ATP + NH4+ + ácido Glutâmico ⇒ ADP + Pi + Glutamina + H+
A glutamina, no fígado, é convertida por hidrólise em ácido glutâmico
(glutaminase), libertando a amónia para biotransformação.
Glutamina + H2O ⇒ Ácido Glutâmico + NH4+
2.b.iii.1.b. Transporte de Amónia como Alanina:
A alanina faz o transporte da amónia do músculo ao fígado para biotransformação.
Este transporte depende da formação do ácido glutâmico a partir do ácido αCetoglutárico, e da posterior formação da alanina através da reacção da AlaninaTransaminase, já descrita.
A reacção inversa ocorre quando a alanina chega ao fígado.
Aminoácidos página 47 de 70
Carlos Capela
2.b.iii.2. Ciclo da Ureia ou de Krebs-Henseleit:
O ciclo da ureia é uma via metabólica cíclica também descrita por Krebs, e que
transforma 2 moléculas de amónia e 1 molécula de gás carbónico numa molécula de
ureia.
Possui várias e complexas etapas enzimáticas, e gasta 3 ATPs para cada ureia
sintetizada.
Compostos como o ácido fumárico, o ácido aspártico, o ácido glutâmico e o ácido
α-Cetoglutárico participam no processo, que envolve também 2 enzimas mitocôndriais
e 3 citosólicas.
2.b.iii.2.a. Formação de Carbamil-fosfato
A 1ª etapa, consiste na condensação de CO2, amoníaco e ATP para formar o
Carbamil-fosfato (uma forma activada de azoto), através de uma reacção catalisada
pela Carbamil fosfato sintetase, tendo como coenzima a Biotina. Esta enzima tem
como factor alostérico positivo o ácido-N-acetil-glutâmico e negativo a Arginina.
2.b.iii.2.b. Formação de Citrulina
Na 2ª etapa, o Carbamil-fosfato é transferido para a Ornitina formando a
Citrulina, sendo esta reacção catalisada pela Ornitina Transcarbamilase. A ornitina e
a citrulina são dois aminoácidos não proteicos ou livres.
Aminoácidos página 48 de 70
Carlos Capela
Estas duas primeiras reacções ocorrem no interior da mitocôndria. A citrulina é
então transferida para o citoplasma, onde ocorre o resto do ciclo, através da citrulina
translocase.
2.b.iii.2.c. Formação do Ácido Arginosuccínico
Já no citoplasma, dá-se a 3ª reacção do ciclo, que consiste, na condensação da
citrulina com o ácido aspártico. Esta reacção é catalisada pela arginosuccinato
sintetase, na presença de ATP e Mg2+, e dá formação ao Ácido Arginosuccínico.
Nesta reacção o ATP é hidrolisado a AMP, em vez de ADP (como acontece
normalmente). Como o AMP pode receber um fosfato do ATP, dando origem a 2 ADP,
hidrolisar ATP a AMP é equivalente a hidrolisar 2 ATP a 2 ADP.
2.b.iii.2.d. Cisão do Ácido Argininosuccínico
Na 4ª etapa, o ácido Arginosuccínico cinde-se em Arginina e Ácido Fumárico,
pela acção da Arginosuccinase.
Aminoácidos página 49 de 70
Carlos Capela
O ácido fumárico é convertido em ácido málico e este em ácido oxaloacético pelo
ciclo de Krebs. O ácido oxaloacético reage com uma molécula de NH3 proveniente do
ácido glutâmico e regenera o ácido aspártico que, por sua vez, dá origem ao ácido
arginosuccínico por acção da arginosucinato sintetase.
2.b.iii.2.e. Formação da ureia
A arginina é hidrolisada pela Arginase, enzima que se encontra principalmente no
fígado, dando origem à ureia, regenerando a Ornitina.
O rim não possui esta enzima, mas possui as restantes, daí que seja o principal local
de síntese do aminoácido arginina.
Aminoácidos página 50 de 70
Carlos Capela
Diagrama-resumo do Ciclo da Ureia
Ureia
Arginase
Carbamil-P
L-Ornitina
L-Arginina
Sintetase
Ornitina
Transcarbamilase
Carbamil-P
Citrulina Translocase
Arginosuccinase
Ácido
Fumárico
Arginosuccinato
Sintetase
L-Citrulina
Ácido
Arginosuccínico
L-Ácido
aspártico
Aminoácidos página 51 de 70
Carlos Capela
2.b.iv. O DESTINO DOS ESQUELETOS CARBONADOS:
Os α-Cetoácidos formados pela desaminação dos aminoácidos podem ser
convertido a ácido Pirúvico, Acetil-CoA ou intermediários do Ciclo de Krebs, para
serem oxidados com produção de energia.
Consoante os compostos que originam por degradação, os aminoácidos são
classificados em 2 grupos:
•
Glicogénicos
ou
Glicoformadores:
se
originam
ácido
pirúvico
ou
intermediários do ciclo de Krebs;
•
Cetogénicos ou Cetoformadores: se originam compostos cetónicos.
Glicogénicos
Cetogénicos
Ácido aspártico
Ácido glutâmico
Leucina
Asparagina
Glutamina
Lisina
Alanina
Arginina
Treonina
Prolina
Cisteína
Histidina
Glicina
Metionina
Serina
Valina
Glicogénicos e Cetogénicos
Isoleucina
Fenilalanina
Triptofano
Tirosina
2.b.iv.1. Glicina e Serina
A glicina pode ser convertida reversivelmente em serina pela intervenção de 2
coenzimas: o fosfato de piridoxal e o ácido tetrahidrofólico (THF). A serina é
catabolisada directamente a ácido pirúvico por desaminação não oxidativa, catabolisada
pela serina desidratase, que elimina o NH3 e requer fosfato de piridoxal.
Aminoácidos página 52 de 70
Carlos Capela
2.b.iv.2. Alanina
A sua transaminação origina directamente ácido pirúvico.
2.b.iv.3. Cisteína
A cisteína sofre uma desaminação não oxidativa originando ácido pirúvico com
libertação de NH3.
2.b.iv.4. Ácido Aspártico e Asparagina
O ácido aspártico pode ser transaminado directamente a ácido oxaloacético. O ácido
aspártico pode também ser convertido em ácido fumárico pelo ciclo da ureia.
A asparagina é hidrolisada pela asparaginase a ácido aspártico e NH3.
Aminoácidos página 53 de 70
Carlos Capela
2.b.iv.5. Ácido Glutâmico, Glutamina, Prolina, Arginina e
Histidina
A glutamina origina ácido glutâmico e NH3 por acção da glutaminase.
A prolina e a arginina são convertidas em γ-semialdeido glutâmico, o qual é
oxidado a ácido glutâmico.
A histidina é um aminoácido essencial percursor do ácido glutâmico, por isso, é um
aminoácido glucogénico.
Aminoácidos página 54 de 70
Carlos Capela
O ácido glutâmico através de uma desaminação oxidante, catalisada pela glutamato
desidrogenase, origina ácido α-cetoglutárico.
2.b.iv.6. Valina, Isoleucina e Leucina
O catabolismo da valina, isoleucina e leucina é comum no 1º e 2º passos e envolve a
transaminação aos correspondentes α-cetoácidos, seguida de descarboxilação oxidativa
a ésteres de CoA.
Assim, a leucina é exclusivamente cetogénico (acetil-CoA e ácido acetoacético),
enquanto a Valina é glucogénico (propionil-CoA) e a Isoleucina é simultaneamente,
glucogénico e cetogénico (acetil-CoA e propionil-CoA). O propionil-CoA é
metabolisado a succinil-CoA.
2.b.iv.7. Lisina
A lisina é catabolisada a ácido acetoacético (cetogénico).
A carnitina deriva da lisina. Esta é fundamental no transporte de ácidos gordos de
cadeia longa activados através da membrana interna da mitocôndria, local onde são
oxidados.
Aminoácidos página 55 de 70
Carlos Capela
2.b.iv.8. Metionina
A metionina é catabolisada a cisteína e a ácido α-cetobutírico. Este último dá
origem ao propionil-CoA que entra no ciclo de Krebs como Succinil-CoA.
2.b.iv.9. Treonina
A treonina é geralmente catabolisada a ácido pirúvico. Um intermediário desta via
pode sofrer tiólise com a CoA e dar origem a Acetil-CoA e glicina. Outra via
alternativa, converte a treonina em ácido α-cetobutírico e este em propionil-CoA.
2.b.iv.10. Fenilalanina e Tirosina
A degradação da tirosina acompanha a da fenilalanina, uma vez que esta é
hidroxilada a tirosina pela fenilalanina hidroxilase.
A via principal do metabolismo da tirosina é a formação do ácido acetoacético e
ácido fumárico, por isso é simultaneamente, um aminoácido cetogénico e glucogénico.
Aminoácidos página 56 de 70
Carlos Capela
2.b.iv.11. Triptofano
O catabolismo do triptofano tem muitos pontos de ramificação.
O triptofano é simultaneamente, glucogénico e cetogénico, dado que são formados
Alanina e acetoacetil-CoA, respectivamente.
2.b.iv.12. Em resumo
2.b.iv.12.a.
Os aminoácidos que são convertidos a ácido pirúvico são:
•
Alanina
•
Triptofano
•
Cisteína
•
Serina
•
Glicina
•
Treonina
2.b.iv.12.b.
Os aminoácidos que são convertidos em ácido α-Cetoglutárico:
•
Ácido Glutâmico
•
Glutamina
•
Prolina
•
Arginina
•
Histidina
2.b.iv.12.c.
Aminoácidos que são convertidos a ácido Oxaloacético:
•
Ácido Aspártico
•
Asparagina
2.b.iv.12.d.
Aminoácidos que são convertidos a Ácido Fumárico:
•
Tirosina
•
Fenilalanina
Aminoácidos página 57 de 70
Carlos Capela
2.b.iv.12.e.
Aminoácidos que são convertidos a Succinil-CoA:
•
Valina
•
Isoleucina
•
Treonina
2.b.iv.12.f.Aminoácidos que são convertidos a Acetil-CoA
•
Leucina
•
Triptofano
•
Lisina
Aminoácidos página 58 de 70
Carlos Capela
C. DERIVADOS DE AMINOÁCIDOS
1.CREATINA
A Creatina é sintetizada no fígado a partir da metilação do ácido Guanidinoacético,
sendo este processo catalisado pela enzima Guanidinoacetato metiltransferase que
utiliza a S-Adenosil-Metionina como dador do grupo metilo.
O ácido Guanidinoacético é formado no rim a partir dos aminoácidos arginina e
glicina, reacção catalisada pela Glicina amidinotransferase.
Glicina
amidinotransferase
no Rim
Ácido Guanidinoacético
Glicina
Ornitina
Guanidinoacetato
S-Adenosil
metiltransferase
Metionina
no Fígado
S-Adenosil
Arginina
Homocisteina
Creatina
Não enzimática
Creatina
Fosfocinase
Creatinina
Creatina Fosfato
Aminoácidos página 59 de 70
Carlos Capela
A creatina é usada como armazenador de energia. O fosfato do ATP é transferido
para a creatina, gerando creatina fosfato, através da acção da Creatina fosfocinase
(CPK). Esta reacção é reversível, daí que quando é necessário energia (ex: contracção
muscular), a creatina fosfato doa o seu fosfato ao ADP para formar ATP. Tanto a
creatina como a creatina fosfato são encontradas no músculo, cérebro e sangue.
Cerca de 1-2% da creatina fosfato pré-existente é ciclada não enzimaticamente a
creatinina com a perda do fosfato e de uma molécula de água. A creatinina é excretada
na urina, e nova creatina é sintetizada para a substituir. A quantidade de creatina
excretada por um indivíduo é, portanto constante de um dia para outro, e está
relacionada com a massa muscular. Assim o nível de creatinina excretada (Creatinina
clearance rate) é uma medida da função renal.
Aminoácidos página 60 de 70
Carlos Capela
2.GLUTATIÃO
O glutatião, o tripéptido γ-glutamilcisteinilglicina (abreviação GSH), tem várias
funções importantes no organismo:
•
É um redutor;
•
É conjugado com fármacos para torná-los mais solúveis em água;
•
Está envolvido no transporte de aminoácidos através da membrana celular (o
ciclo do γ-Glutamilo);
•
É parte da estrutura de alguns leucotrienos;
•
É um cofactor para algumas reacções enzimáticas;
•
É um auxiliar no rearranjo de pontes dissulfeto das proteínas.
O glutatião é sintetizado a partir de três aminoácidos. É sintetizado pela formação
do dipéptido γ-glutamilcisteína e adição subsequente de glicina. Ambas as reacções
requerem a activação dos grupos carboxilo pelo ATP. A síntese do glutatião é
amplamente regulada pela disponibilidade de cisteína.
Ácido Glutâmico
Cisteína
γ-Glutamilcisteína
Glicina
γ-Glutamilcisteinilglicina
ou Glutatião
Aminoácidos página 61 de 70
Carlos Capela
Como redutor é muito importante na manutenção da estabilidade das membranas
eritrocitárias. O seu grupo sulfidrilo pode ser usado para reduzir os peróxidos formados
durante o transporte de oxigénio (glutatião peroxidase). A forma oxidada do glutatião
consiste em 2 moléculas unidas por uma ponte dissulfeto. Esta forma é reduzida pela
glutatião reductase a 2 moléculas de glutatião à custa de NADPH. Como vemos a via
das pentoses fosfato é muito importante nos eritrócitos, para a produção de NADPH,
necessário à glutatião reductase. De facto, 10% do consumo da glicose, pelos
eritrócitos, é na via da pentoses-fosfato.
NADP+
NADPH + H+
Glutatião Reductase
Glutatião Peroxidase
H2O2
2 Glutatião (GSH)
2 H2 O
Glutatião dissulfeto (GSSG)
A conjugação de fármacos com o glutatião, depois de uma reacção pré-eliminar
catalisada pelo citocromo P450, torna-os mais polares e portanto mais solúveis em água.
Aminoácidos página 62 de 70
Carlos Capela
2.a. Ciclo do γ-Glutamilo ou de Meister
O teor em aminoácidos do “pool” plasmático circulante depende do equilíbrio que
se estabelece entre os processos que adicionam aminoácidos (dieta, degradação das
proteínas endógenas e síntese de novo), e os que retiram aminoácidos (síntese de
proteínas, síntese de compostos nitrogenados) e a perda na urina.
Os aminoácidos do “pool” plasmático são transportados, rapidamente, através da
membrana plasmática para o interior da célula, tendo sido evidenciados pelo menos sete
transportadores diferentes.
Sistemas transportadores dos
aminoácidos na membrana da
célula dos mamíferos
Transportador
Sistema A
Aminoácidos
Ala, Gly, Met, Pro e
Ser
Sistema ASCP
Ala, Cys, Pro e Ser
Sistema L
Phe, Ile, Leu, Met,
Trp, Tyr e Val
Sistema Ly
Arg, His, Lys e Orn
Sistema dos
Asp e Glu
dicarboxílicos
Sistema β
β-alanina e taurina
Sistema N
Asn, Gln e His
Todavia, a especificidade não é absoluta, sendo influenciada por múltiplos factores,
em particular a concentração dos aminoácidos.
O ciclo do γ-glutamilo é um sistema de transporte diferente dos anteriores. Foi
proposto por Alton Meister e opera, fundamentalmente, nas células hepáticas e renais.
Inicia-se com a transpeptização (transferência) de um resíduo do glutatião (γ-Glu-CysGly) para o grupo α-amina do aminoácido candidato ao transporte, catalisada pela γ-
Aminoácidos página 63 de 70
Carlos Capela
glutamiltransferase ou γ-glutamiltranspeptidase da superfície externa da membrana
plasmática.
O γ-Glu-aminoácido (γ-glutamilaminoácido) é transportado para o citosol e,
posteriormente, clivado pela γ-glutamilciclotransferase, que liberta o aminoácido e
cicliza o grupo γ-glutamilo a 5-oxoprolina ou ácido piroglutâmico. A Cys-Gly
(cisteinilglicina) do citosol é hidrolisada por uma dipeptidase a Cys e Gly.
Para regenerar o glutatião, o ácido glutâmico é regenerado a partir da 5-oxoprolina
pela 5-oxoprolinase com gasto de ATP, e assim o glutatião é novamente sintetizado a
partir dos seus 3 componentes. 3 ATPs são usados na regeneração do glutatião, um na
formação do ácido glutâmico a partir da 5-oxoprolina e 2 na formação das ligações
peptídicas.
Apesar de ser energeticamente dispendioso é um ciclo bastante rápido. É graças a
este ciclo que todos os aminoácidos (excepto a prolina) e pequenos péptidos podem ser
transportados para dentro da célula.
Aminoácidos página 64 de 70
Carlos Capela
3.NEUROTRANSMISSORES
DERIVADOS DA
TIROSINA
Parte da tirosina é usada como percursora das catecolaminas. O destino metabólico
eventual dos carbonos da tirosina é determinado pelo primeiro passo da via. A síntese
de catecolaminas começa com a tirosina hidroxilase, que, como a fenilalanina e a
triptofano hidroxilase, é dependente de Tetraidrobiopterina.
A tirosina hidroxilase produz diidroxifenilalanina, também conhecido como
DOPA. Na substância cinzenta e em algumas partes do cérebro, esta é a ultima enzima
da via. A medula da supra-renal converte a Dopamina em norepinefrina e epinefrina
(também designada adrenalina). O grupo metil da epinefrina é derivado da S-AdenosilMetionina.
As catecolaminas são metabolizadas pela monoamina oxidase.
Tirosina
Tirosina
Hidroxilase
Tetraidrobiopterina
+ O2
Diidrobiopterina
+ H2O
Epinefrina
S-adenosilHomocisteina
S-adenosilmetionina
Feniletanolamina
N-metiltransferase
Norepinefrina
DOPA
DOPA
Dopamina
Descarboxilase
β-Hidroxilase
3,4-Diidroxifenilalanina
Dopamina
Aminoácidos página 65 de 70
Carlos Capela
4.NEUROTRANSMISSORES
DERIVADOS DO
TRIPTOFANO
O Triptofano é um percursor para a síntese de Serotonina (5-hidroxitriptamina, 5HT) e Melatonina (N-acetil-5-metoxitriptamina).
4.a. A Serotonina
A serotonina (5-hidroxitriptamina) resulta da hidroxilação do triptofano por uma
enzima dependente de tetraidrobiopterina e da descarboxilação por uma enzima que
contêm fosfato de piridoxal. É um neurotransmissor no cérebro e causa a contracção da
musculatura lisa das arteríolas e bronquíolos. É amplamente distribuída no corpo e deve
possuir outros papéis fisiológicos.
Aminoácidos página 66 de 70
Carlos Capela
4.b. A Melatonina
A Melatonina (N-acetil-5-metoxitriptamina) é uma molécula indutora do sono. A
Acetiltransferase necessária para a sua
síntese encontra-se na glândula pineal
e
na
retina.
A
melatonina
está
envolvida na regulação do ritmo
circadiano,
sendo
sintetizada
principalmente durante a noite. Parece
funcionar através da inibição da síntese
e
da
secreção
de
outros
neurotransmissores, como a dopamina
e o GABA.
A libertação de melatonina pela
glândula pineal é um exemplo de um
biorritmo. O sinal interno é fornecido
por um neurotransmissor, neste caso a
norepinefrina
produzida
por
um
neurónio adrenérgico. Neste sistema, o
controlo é exercido pela luz que entra
nos olhos e é transmitida à glândula
pineal através do SNC. O neurónio
adrenérgico que enerva o pinealócito é
inibido pela luz transmitida através dos
olhos. A norepinefrina, libertada como
neurotransmissor no escuro, estimula a formação de cAMP através de um receptor β na
membrana celular do pinealócito, que leva à síntese aumentada de Acetiltransferase. A
actividade aumentada desta enzima causa a conversão de serotonina a Nacetilserotonina e a hidroxiindol-O-metiltransferase (HIOMT) catalisa a conversão da
N-acetilserotonina em melatonina, que é secretada nas horas de escuro, mas não nas
horas de claridade. A melatonina circula até às células que contem receptores, gerando
efeitos sobre funções reprodutivas e outras. Demonstrou-se que a melatonina exerce um
efeito antigonadotrófico.
Aminoácidos página 67 de 70
Carlos Capela
D. AMINOACIDÚRIAS
As Aminoacidúrias são doenças raras do metabolismo dos aminoácidos, quase
sempre associadas ao catabolismo.
As aminoacidúrias primárias são autossómicas recessivas, e são devidas à ausência
ou diminuição da actividade das enzimas. Afectam normalmente um aminoácido.
As
aminoacidúrias
secundárias
estão,
geralmente,
associadas
à
elevada
concentração dos aminoácidos no plasma, ou à deficiência nos mecanismos de
transporte dos aminoácidos, e afectam, simultaneamente vários aminoácidos. A
desnutrição proteica, lesões em órgãos ou disfunção tubular generalizada estão na
origem das aminoacidúrias secundárias.
1.A FENILCETONÚRIA
A fenilcetonúria ou oligofrenia fenilpirúvica é a aminoacidúria mais comum e
estudada.
Resulta
da
deficiência
autossómica
recessiva
da
fenilalanina-4-
monoxigenase/fenilalanina-4-hidroxilase ou, mais raramente, da tetrahidrobiopteridina,
fundamental à conversão de fenilalanina em tirosina.
Na fenilcetonúria clássica, a deficiência da fenilalanina hidroxilase é total em 50%
dos casos, ou a actividade é baixa por diminuição da síntese ou por alteração da enzima.
Devido a esta deficiência, a fenilalanina é metabolizada por uma via alternativa, e os
metabólitos (ácido fenilacético e ácido fenil-láctico) são acumulados nos fluidos
biológicos. Estes metabólitos são inibidores competitivos do transporte dos
aminoácidos na célula e podem alterar o metabolismo das aminas biogéneas. São, ainda
responsáveis, por lesões progressivas e irreversíveis do cérebro nos primeiros meses de
vida, que conduzem a anormalidades neurológicas e a atraso mental. Outra
característica é a cor clara da pele e dos olhos devido à falta de pigmentação, cujo
motivo é a falta de tirosina.
O tratamento convencional é uma dieta pobre em fenilalanina, mas contendo
tirosina (que passa a ser uma aminoácido essencial pois não é formado a partir da
fenilalanina) é extremamente eficaz, por isso o diagnóstico precoce é fundamental. Por
Aminoácidos página 68 de 70
Carlos Capela
isso o teste de triagem de rotina “o teste do pezinho” é obrigatório em muitos países do
mundo. A fenilalanina não deve ser completamente retirada por ser necessária ao
crescimento e desenvolvimento normal do organismo, pois não nos esqueçamos que se
trata de um aminoácido essencial.
A deficiência em tetrahidrobiopteridina, por deficiência, quer da dihidrobiopteridina
sintetase, quer da dihidrobiopteridina redutase, pode ocorrer em 1-3% dos recém
nascidos com fenilcetonúria clássica.
A tetrahidrobiopteridina não é somente coenzima da fenilalanina hidroxilase, mas
também da tirosina hidroxilase e triptofano hidroxilase, ou seja, é necessária à síntese
da dopamina, nor-adrenalina e serotonina (neurotransmissores) e, a deficiência causa
doença neurológica progressiva grave, que não é prevenida pela dieta. Contudo, a
terapêutica com percursores dos neurotransmissores tem relevado algum benefício
clínico.
2.O ALBINISMO
O albinismo deve-se à deficiência da tirosinase necessária à síntese de melanina. A
melanina é responsável pela pigmentação da pele, cabelo e olhos, por isso, os albinos
são muito susceptíveis a queimaduras solares, carcinomas da pele e fotofobia.
3.HOMOCISTINÚRIA
A homocistinúria mais comum é devida à deficiência da cistationina sintetase, que
resulta num aumento de homocisteína e a sua remetilação origina elevados níveis de
metionina. A sua deficiência é hereditária.
O excesso de homocisteína no sangue (hiperhomocisteinémia), pode formar um
tiolactona, a homocisteína tiolactona, um intermediário altamente reactivo no qual o
grupo amino tiolato livre, provoca a oxidação das LDL (lipoproteínas de baixa
densidade), o que faz com elas se agreguem e sejam endocitadas por macrófagos. Os
depósitos excessivos de lípidos na parede das artérias formam ateromas. A
Aminoácidos página 69 de 70
Carlos Capela
homocisteína e a sua lactona, podem ter outros efeitos, incluindo a lipoperoxidação e
agregação plaquetária, que por sua vez levam a fibrose e calcificação das placas
ateroscleróticas.
Cerca de ¼ dos pacientes com aterosclerose que não apresentam nenhum dos outros
factores de risco (hábito de fumar e terapia contraceptiva oral) têm mostrado serem
deficientes na actividade da cistationina sintetase.
4.A CISTINÚRIA
A cistinúria é um defeito no transporte de membrana de cistina e aminoácidos
básicos (lisina, arginina e ornitina), que resulta na sua excreção renal aumentada.
A baixa solubilidade da cistina, resulta em cristais e formação de cálculos, uma
característica desta doença. O tratamento é limitado à remoção dos cálculos, à
prevenção da precipitação pela ingestão de grandes quantidades de água, à
alcalinização da urina para solubilizar a cistina ou à formação de derivados solúveis
pela conjugação com fármacos.
5.CISTINOSE
É mais grave que a cistinúria, pois a cistina acumula-se no lisossomas. A cistina
forma cristais em muitas células, com a perda séria da função dos rins, normalmente
causando insuficiência renal em 10 anos. O defeito encontra-se no transporte da cistina
na membrana dos lisossomas.
Aminoácidos página 70 de 70