Conteúdo
Leis de Kepler
1
Leis da dinâmica de Newton
3
Campo Eléctrico
5
Constante de Avogadro
10
Efeito fotoeléctrico
12
Electrólise
13
Reagente limitante
15
Modelo atómico de Bohr
17
Modelo atómico de Rutherford
19
Fórmula de estrutura
21
Aminoácido
23
Fórmula estereoquímica
25
Período de semi-desintegração
26
Ligação iónica
28
Auto-ionização da água
30
Modelo atómico de Thomson
31
pH
32
Campo Eléctrico
33
Massa
38
Referências
Fontes e Editores da Página
40
Fontes, Licenças e Editores da Imagem
41
Licenças das páginas
Licença
42
Leis de Kepler
1
Leis de Kepler
Referência : de Araújo, M. (2010), WikiCiências, 1(12):0200
Autor: Mariana de Araújo
Editor: Joaquim Agostinho Moreira
[1]
As leis de Kepler constituem uma base para a descrição do movimento dos planetas em torno do Sol. Foram
descobertas originalmente por Johannes Kepler pela análise dos dados observacionais de Tycho Brahe, relativos à
posição de alguns planetas do Sistema Solar. Posteriormente, Isaac Newton mostrou que as leis de Kepler podem ser
deduzidas a partir das leis da Mecânica e da Lei da Gravitação Universal, para um sistema de dois corpos sujeitos a
uma força central em que um deles, o astro director, tem uma massa muito superior à do outro, o astro dirigido.
Lei das órbitas
A órbita do astro dirigido em torno do astro director é uma elipse, da qual o astro director ocupa um dos focos. Em
geral num sistema de dois corpos estes orbitam em torno do seu centro de massa. No entanto, quando um dos corpos
tem uma massa muito maior que o outro, o centro de massa do sistema praticamente coincide com o centro do corpo
de maior massa, pelo que se pode considerar que este está parado, e que o outro orbita em torno dele. A lei das
órbitas aplica-se a estes sistemas, como é o caso do Sistema Solar ou de satélites que orbitam em torno de um
planeta.
Lei das áreas
O vector de posição de um corpo em
relação ao astro director varre áreas
iguais em intervalos de tempo iguais.
Esta lei é uma consequência da
conservação do momento angular do
astro dirigido que se encontra sob a
acção de uma força central que aponta
sempre para o centro do astro director.
Como a área varrida por unidade de
tempo é constante e o corpo não está
sempre à mesma distância do astro
director, a sua velocidade varia, sendo
máxima quando a distância entre os
dois corpos é mínima, e mínima
quando a distância é máxima.
Lei dos Períodos
Ilustração da lei das áreas. Como o intervalo de tempo decorrido entre A e A' é igual ao
intervalo entre B e B', as áreas A1 e A2 são iguais.
A razão entre o cubo do semi-eixo maior da órbita de um planeta e o quadrado do respectivo período é uma
constante:
A constante K é chamada constante de Kepler e é igual para todos os corpos que orbitam em torno do mesmo astro.
Leis de Kepler
2
Planeta
Período
(anos)
Distância média ao
Sol (UA)
Constante de
Kepler
Erro relativo (%)
Mercúrio
0,24085
0,387
1,001
0,08
Vénus
0,61520
0,723
1,001
0,1
Terra
1,00000
1,000
1,000
-
Marte
1,88071
1,524
0,999
0,07
Júpiter
11,85654
5,203
0,9981
0,2
Saturno
29,44750
9,537
0,9997
0,03
Úrano
84,01697
19,191
0,9987
0,1
Neptuno
164,79124
30,069
0,9989
0,1
|+Tabela 1: Cálculo da constante de Kepler para órbitas em torno do Sol. O
erro é relativo a K = 1 ano2UA-3 para a Terra.
Tabela 2: Cálculo da constante de Kepler para órbitas em torno de Júpiter.
Satélite
Período (anos) Distância média a Júpiter (UA) Constante de Kepler
Io
4,843E-03
2,82E-03
1,04E+03
Europa
9,722E-03
4,49E-03
1,05E+03
Ganymede
1,959E-02
7,15E-03
1,05E+03
Callisto
4,569E-02
1,26E-02
1,05E+03
Referências
1. Kepler, J., New Astronomy, Cambridge University Press, 1993.
2. Feymnan, R., Leighton, R. & Sands, M., The Feynman Lectures on Physics, Vol,. 1, Addison-Wesley Publishing,
1963.
3. Feynman, R., Goodstein, J. & Goodstein, D., A lição esquecida de Feynman, Gradiva, 1997.
4. Copernicus, N., Kepler, J., Galilei, G., Newton, I., Einstein, A. & Hawking, S., On the Shoulders of Giants,
Running Press, 2002.
Criada em 20 de Abril de 2010
Revista em 22 de Novembro de 2010
Aceite pelo editor em 06 de Dezembro de 2010
Leis da dinâmica de Newton
3
Leis da dinâmica de Newton
Referência : de Araújo, M. (2010), WikiCiências, 1(12):0201
Autor: Mariana de Araújo
Editor: Joaquim Agostinho Moreira
[1]
As leis de Newton são um conjunto de três leis que relacionam as forças exercidas sobre um corpo com o seu
movimento, e são suficientes para descrever completamente e de forma determinista a dinâmica de qualquer sistema
clássico, conhecidas as forças que sobre ele actuam, e as posições e velocidades de cada partícula num instante .
Foram enunciadas por Sir Isaac Newton no seu livro Philosophiae Naturalis Principia Mathematica em 1687.[1]
Primeira Lei (Lei da inércia): Um corpo em repouso ou em movimento rectilíneo uniforme
permanecerá nesse estado, se a resultante das forças que nele actuam for nula.
Segunda Lei (Lei fundamental da dinâmica): A taxa de variação temporal da quantidade de
movimento de um corpo é igual à força resultante nele exercida, e tem a direcção dessa força.
Terceira Lei (Lei da acção-reacção): Para cada acção existe uma reacção igual e oposta; i.e, as forças
resultantes da interacção entre dois corpos são iguais e simétricas, cada uma delas aplicada a um dos
corpos.
Os sistemas físicos governados por estas leis são usualmente chamados sistemas clássicos. Estas leis, na sua
formulação original, falham no limite quântico, e situações de altas velocidades e de altas energias, em que é
necessário aplicar a Mecânica Quântica e Relatividade Geral.
É de notar também que a terceira lei, na formulação aqui apresentada, implica que a perturbação que origina as
forças se propagou a uma velocidade infinita. Uma formulação mais geral e correcta não impõe a simetria das forças.
No entanto, na generalidade dos casos clássicos (exceptuando a electrodinâmica), esta lei pode ser assim utilizada,
uma vez que as velocidades dos corpos envolvidos são muito inferiores à velocidade de propagação da interacção,
podendo-se desprezar o intervalo de tempo de propagação e considerar, para todos os efeitos práticos, como
instantânea.
Primeira Lei ou lei da inércia
Um corpo em repouso ou em movimento rectilíneo uniforme permanecerá nesse estado, se a resultante das forças
que nele actuam for nula.
Esta lei é utilizada na definição de um referencial inercial. Apesar de poder aparentar ser um corolário da segunda
lei, na verdade ela define os referenciais em que a segunda lei é válida.
Segunda Lei
A taxa de variação temporal da quantidade de movimento de um corpo é igual à força resultante nele exercida, e tem
a direcção dessa força.
Em notação vectorial, sendo que a força resultante a soma vectorial de todas as forças que actuam no corpo:
Nos casos em que a massa do corpo não varia, esta lei toma a forma mais conhecida:
Traduz também a conservação do momento linear do corpo no caso da resultante das forças ser nula:
Leis da dinâmica de Newton
4
Considere-se agora um sistema formado por N corpos. De um modo geral, estes corpos interactuam entre si e com os
corpos exteriores ao sistema. As interacções entre os corpos do sistema satisfazem a terceira lei de Newton, pelo que
a sua resultante é nula. Contudo, a resultante das forças com origem na interacção do sistema com a vizinhança, pode
não ser nula. A aplicação da segunda lei de Newton ao sistema de N corpos conduz à equação:
sendo
a força resultante das interacções externas sobre o corpo i, e
a sua quantidade de movimento.
Utilizando a definição de quantidade de movimento do centro de massa, é imediato verificar que:
,
isto quer dizer que o movimento global de translação do sistema, sob a acção das forças externas, pode ser descrito
pelo movimento do centro de massa. No entanto, podem actuar no corpo forças que, apesar de terem resultante nula,
provocam movimento de rotação do corpo, não havendo movimento do seu centro de massa.
Consideremos o caso simples de um binário de forças, como ilustrado na figura. Os ponto A e B têm a mesma
massa, estão rigidamente ligados pelo segmento entre eles, e o sistema está inicialmente em repouso num plano. Se
aplicarmos duas forças e , de igual módulo e sentidos opostos, nos pontos A e B respectivamente, o centro de
massa permanecerá fixo, mas os pontos A e B irão descrever um círculo em torno dele.
Binário de forças
Leis da dinâmica de Newton
5
Referências
1. Newton, I., Philosophiae Naturalis Principia Mathematica (“Mathematical Principles of Natural Philosophy”), [1],
London, 1687.
2. Halliday, D., Resnick, R., & Walker, J., Fundamentals of Physics, J. Wiley & Sons, 2001.
3. Feymnan, R., Leighton, R. & Sands, M., The Feynman Lectures on Physics, Vol,. 1, Addison-Wesley Publishing,
1963.
4. Alonso, M. & Finn, E., Física, Addison Wesley, 1999.
Criada em 03 de Novembro de 2010
Revista em 06 de Dezembro de 2010
Aceite pelo editor em 06 de Dezembro de 2010
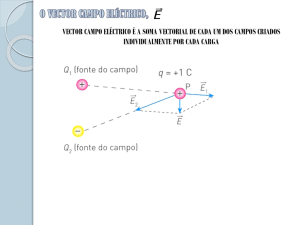
Campo Eléctrico
Referência : Ferreira, M. (2010), WikiCiências, 1(12):0202
Autor: Miguel Ferreira
Editor: Joaquim Agostinho Moreira
[1]
Da experiência sabe-se que uma carga eléctrica cria um campo eléctrico no espaço vizinho.
Quando um corpo electricamente carregado é colocado na região do espaço onde há campo eléctrico, esse corpo fica
sujeito à acção de uma força eléctrica. Por definição, o campo eléctrico num dado ponto do espaço é igual à força
eléctrica que actua na unidade de carga positiva que se coloca nesse ponto:
.
O campo eléctrico pode ser criado por cargas ou por campos magnéticos variaveis no tempo.
No que se segue, apenas referiremos o campo eléctrico criado por cargas em repouso. Por simplicidade, iniciaremos
a discussão com uma única carga pontual, Q, colocada na origem de um referencial, e que cria campo eléctrico. Para
caracterizar o campo produzido pela carga Q, utiliza-se uma carga de prova, positiva, de valor muito pequeno, q. A
força eléctrica que actua na carga de prova é descrita pela lei de Coulomb, que pode ser formulada matematicamente
do seguinte modo:
.
Utilizando a definição operacional de campo eléctrico enunciada anteriormente, o campo eléctrico criado por uma
carga pontual Q, num ponto na posição , é
,
sendo o versor da direcção definida pelo vector
.
Da expressão anterior podemos concluir que o campo eléctrico criado por uma carga pontual é radial, decai com o
quadrado da distância entre o ponto considerado e a carga criado de campo. O cmpo é centrípeto se a carga criadora
for positiva, e centrífugo se a craga for negativa.
A força de Coulomb obedece ao princípio da sobreposição; isto é, a força que várias cargas exercem sobre uma carga
q é igual à soma vectorial das forças individuais que cada carga do conjunto exerce sobre q. De acordo com a
definição operacional, o campo eléctrico obedece ao princípio da sobreposição. Matematicamente, o campo criado
por um conjunto de n cargas é dado por:
Campo Eléctrico
6
No sistema internacional, as unidades de campo eléctrico são newton por coulomb (N/C) ou volt por metro (V/m).
O conceito de campo eléctrico é mais do que uma abordagem matemática
diferente do fenómeno da interacção entre cargas eléctricas. De facto, o
campo eléctrico é uma entidade física real responsável por mediar a
interacção entre as cargas. Quando as cargas que dão origem ao campo
eléctrico se movem, os seus movimentos são transmitidos aos corpos
carregados que se encontram nas vizinhanças sob a forma de perturbações no
campo eléctrico. Estas perturbações difundem-se ao longo do campo à
velocidade da luz e vão alterar as características das forças provocadas pelo
campo nos corpos carregados que se encontram nas vizinhanças.
Dependência do módulo do campo
eléctrico criado por uma carga pontual na
distância à fonte.
Linhas de Campo
Uma maneira útil de representar graficamente o campo eléctrico é através de
linhas imaginárias, paralelas ao vector campo eléctrico em todos os pontos.
Estas linhas têm o nome de linhas de campo. A representação de um campo
eléctrico por linhas de campo permite visualizar a direcção e sentido do
campo eléctrico em cada ponto do espaço, e permite comparar a intensidade
do campo eléctrico em duas regiões do espaço distintas. Ao representar-se um
campo eléctrico através das linhas de campo, a sua densidade espacial deve
ser proporcional à intensidade do campo eléctrico: em zonas onde o campo
eléctrico é mais intenso, as linhas devem estar mais próximas umas das
outras. Para além disso, as linhas nunca se podem cruzar porque nesse caso
haveria uma ambiguidade na determinação do vector campo eléctrico nesse
ponto [ver figura].
Campo eléctrico criado por uma carga
positiva.
Campo eléctrico criado por uma carga
negativa.
Campo Eléctrico
Carga pontual positiva. As linhas
estendem-se até ao infinito e têm a
mesma direcção e sentido do vector
campo eléctrico em todos os pontos
do espaço.
7
Duas carga pontuais positivas. As
linhas de campo não existem onde o
campo é nulo.
Cargas pontuais de sinais opostos. As
linhas de campo começam na carga
positiva e terminam na negativa.
está bem definido: é o único vector
tangente à linha de campo naquele ponto.
Não é possível definir o vector
porque no ponto em questão as linhas de
campo se cruzam.
Campo Eléctrico em Condutores
Um material condutor caracteriza-se por ter cargas eléctrics que se podem mover sob a acção de um campo eléctrico
aplicado, dando origem a uma corrente eléctrica. Diz-se que um condutor está em equilíbrio electroestático quando
não há movimento organizado de carga, mesmo na presença de um campo eléctrico externo.
No interior de materiais condutores em equilíbrio electrostático, o campo eléctrico é nulo. De facto, enquanto o
campo eléctrico não for nulo no interior do condutor, haverá movimentos organizados de carga no sentido de o
anular. Por exemplo, quando um condutor é colocado num campo eléctrico externo, as cargas livres tendem a
reorganizar-se de maneira a anular o campo eléctrico no interior do condutor criando um outro campo eléctrico de
intensidade igual e sentido oposto ao campo eléctrico externo. De acordo com o princípio da sobreposição, na região
interna do condutor os campos somam-se vectorialmente e anulam-se.
Uma vez que o campo eléctrico no interior do candutor é nulo, o seu volume e superfície encontram-se ao mesmo
potencial eléctrico.
O campo eléctrico na superfície de um material condutor carregado e num regime electrostático, é perpendicular a
essa superfície. A condição de se considerar uma situação electrostática é fundamental para se compreender a razão
pela qual o campo só pode ser perpendicular. Se o campo não fosse perpendicular, isto é, se fosse possível decompor
o campo numa componente paralela à superfície do condutor, haveria um movimento de cargas, o que contradiz a
condição de equilíbrio electrostático.
É possível provar [ver Leitura Recomendada],que o excesso de carga eléctrica num condutor em equilíbrio
electrostático se encobtra distribuída na sua superfície externa. Quer isto dizer que não há carga livre no interior do
condutor.
Campo Eléctrico
8
1. Condutor em equilíbrio electroestático; 2. Liga-se um campo eléctrico externo
reorganizam-se e criam um campo
e passa a haver campo no interior do condutor; 3. As cargas
; 4. O movimento de cargas pára quando o campo eléctrico que criam tem o mesmo valor que o campo
eléctrico exterior e o anula no interior do condutor.
Blindagem Electroestática
Um material condutor é capaz de isolar uma dada região do espaço da influência do campo
eléctrico, isto é, havendo uma cavidade no interior de um
condutor, um campo eléctrico exterior não consegue penetrar no
condutor e exercer a sua influência no interior dessa cavidade. Da
mesma maneira, se existe uma carga livre dentro da cavidade de
um condutor, o campo a que dá origem não consegue penetrar o
condutor. Contudo, para anular o efeito da carga interior, o
condutor tem que reorganizar a sua carga livre. Esta reorganização
conduz ao aparecimento de um campo eléctrico nas vizinhanças do
condutor. É importante salientar que este campo não se deve à
carga no interior da cavidade, mas às cargas livres do condutor
após a reorganização.
O campo eléctrico numa cavidade de um condutor é
independente do campo eléctrico no seu exterior.
Campo Eléctrico
9
Descontinuidade do Campo Eléctrico numa superfície electricamente
carregada
Considere-se uma superfície electricamente carregada. O vector campo eléctrico num ponto dessa superfície pode ser
decomposto numa componente perpendicular e numa componente paralela à superfície condutora. É possível
mostrar [ver Leitura Recomendada] que a componente perpendicular à
superfície condutora é descontínua, enquanto que a componente paralela é contínua. Isto
quer dizer que se se medir o campo eléctrico nos dois lados da uma superfície carregada,
o valor da componente normal é diferente, e a sua diferença é uma constante que se
relaciona com a densidade superficial de carga da superfície. De facto, é possível mostrar
que
, em que é a densidade superficial de carga.
Exemplos de Campos Eléctricos
1. Campo eléctrico criado por uma esfera maciça de raio R, carregada uniformemente
com carga :
, para
Decomposição do vector
campo eléctrico em duas
componentes:
é
perpendicular à superfície e
,
é paralela.
2. Campo eléctrico criado por uma superfície esférica de raio R
uniformemente carregada, com carga , fora da esfera:
,
3. Campo eléctrico produzido por um filamento rectilíneo de comprimento
ilimitado, com densidade linear de carga constante :
Campo eléctrico criado por uma esfera
maciça carregada com carga Q.
4. Campo eléctrico criado por um plano infinito com densidade superficial de
carga :
, em que
perpendicular ao plano
é o versor que aponta na direcção
Campo eléctrico criado por um filamento
muito comprido.
Campo Eléctrico
Leitura Recomendada
1. Alonso, M. e Finn, E. J., Física, Addison Wesley, 1999
2. Purcell, E. M., Electricity and Magnetism, McGraw Hill, 1985
3. Brito, L., Fiolhais, M. e Providência, C., Campo Electromagnético, McGraw Hill, 1999
Criada em 03 de Outubro de 2010
Revista em 06 de Dezembro de 2010
Aceite pelo editor em 06 de Dezembro de 2010
Constante de Avogadro
Referência : Manuel Ferreira Fernandes, R. (2010), WikiCiências, 1(12):0203
Autor: Ricardo Manuel Ferreira Fernandes
[1]
Editor: Jorge Gonçalves
A constante de Avogadro é uma constante física fundamental, representada pelo símbolo NA (ou L), que corresponde
ao número de entidades elementares (átomos, moléculas, iões, radicais, electrões, fotões, etc.) existentes numa mole
da substância considerada. Esta constante tem o valor de 6,022 141 79 (30) x 1023, correspondente ao número de
átomos existentes em exactamente 0,012 kg de carbono-12† e apresenta as dimensões de mol-1.
A constante de Avogadro foi
introduzida, em 1909, pelo físico
francês Jean Perrin em homenagem ao
químico e físico italiano Amedeo
Avogadro (figura 1) que, em 1811,
publicou que: volumes iguais de gases
nas
mesmas
condições
de
temperatura e pressão contêm o
mesmo número de moléculas.[1]
No início do século XX utilizava-se o
termo molécula-grama para designar a
massa de uma determinada substância
Figura 1 - Selo italiano lançado no âmbito das comemorações do centésimo aniversário da
que no estado gasoso ocupava o
morte de Avogadro ocorrida em 1856.
mesmo volume que dois gramas de
hidrogénio (em iguais condições de
pressão e temperatura). Assim, com base no conceito de molécula-grama, Perrin propôs que a afirmação de
Avogadro era equivalente a dizer que quaisquer duas moléculas-grama contêm sempre o mesmo numero N de
moléculas, logo, N é uma constante universal invariável, à qual seria apropriado que se designasse por constante de
Avogadro.[2]
Jean Perrin, que foi laureado com o prémio Nobel da Física em 1926, calculou a constante de Avogadro a partir do
movimento browniano de partículas coloidais. Porém, não foi o primeiro cientista a determinar o seu valor. Em
1865, o cientista austríaco Johann Josef Loschmidt com base na teoria cinética dos gases determinou a densidade
numérica de partículas existentes num determinado volume de gás, actualmente conhecida como constante de
Loschmidt, que é proporcional à constante de Avogadro. É por causa de Loschmidt que, por vezes, se utiliza o
símbolo L para representar a constante de Avogadro.
10
Constante de Avogadro
11
Desde as primeiras estimativas até à actualidade, diferentes métodos foram utilizados para determinar a constante de
Avogadro. Assim, esta constante pode ser determinada a partir da teoria cinética dos gases, a partir do movimento
browniano, por métodos electroquímicos, a partir da teoria do corpo negro da radiação, pela contagem das partículas
alfa resultantes de decaimento radioactivo ou a partir densidade cristalina obtida por raio-X. Em 1965, através da
utilização da interferometria de raio-X, em conjunto com o uso de um cristal perfeito de silício (Si), conseguiu-se um
progresso assinalável na exactidão do valor da constante de Avogadro. Note-se que os valores da constante de
Avogadro determinados pelas diferentes técnicas são concordantes entre si, o que confirma o postulado da existência
da átomos e moléculas proposto há cerca de dois séculos.[3,4]
Como já foi referido, a constante de Avogadro, quando foi proposta por Perrin, referia-se à quantidade de entidades
elementares existentes numa molécula-grama - unidade daquela época para especificar a quantidade de um composto
ou de um elemento químico. No entanto, em 1971, na 14ª Conferência de Pesos e Medidas introduziu-se quantidade
de substância como grandeza fundamental do Sistema Internacional, tendo-se adoptado a mole como unidade
(representada pelo símbolo mol). Assim, por proposta da IUPAC, da IUPAP e da ISO, a mole foi definida como a
quantidade de substância de um sistema que contém tantas entidades elementares como o número de átomos
existentes em 0,012 kg de carbono-12; em que a natureza das entidades elementares (átomos, moléculas, electrões,
protões, etc.) tem de ser especificada.[5] A introdução da mole como unidade de quantidade de substância conduziu a
que a constante de Avogadro passasse a ser formalmente definida como o número de átomos existentes em
exactamente 0,012 kg de Carbono-12.
O número de entidades elementares existentes numa pequena porção de substância é de tal modo elevado, que seria
um processo praticamente infinito determinar o número de entidades elementares aí existentes. Assim, a constante de
Avogadro permite fazer a transição entre o microscópico e o macroscópico, isto é, conhecendo a massa de uma
substância e a massa molar, é possível a partir da constante de Avogadro determinar o número N de entidades
elementares aí existentes. Considere-se, como exemplo, 30,35642 g de sódio.
O sódio tem uma massa molar, M, igual a 22,98976928 g∙mol-1. Logo, o quociente entre a massa de sódio, m, e a
respectiva massa molar, M, é o número de moles, que multiplicado pela constante de Avogadro permite determinar o
número de átomos existentes:
átomos de sódio (Na)
Verifica-se assim que 30,35642 g de sódio contêm 7.95183 x 1023 átomos.
Para entender melhor a magnitude do valor numérico da constante de Avogadro NA = 6,022 x 1023 imagine-se que se
tinha como desafio contar o número de partículas existentes numa mole durante o intervalo de tempo correspondente
à idade do planeta terra, ou seja, cerca de 4,5 mil milhões de anos (4,5 x 109 anos). Logo, para superar o desafio era
necessário contar 4,2 milhões de partículas por segundo durante os 4,5 mil milhões de anos!
†
Os átomos de carbono-12 devem estar no seu estado fundamental e não estarem quimicamente ligados entre si.
Referências
1. Essay on a Manner of Determining the Relative Masses of the Elementary Molecules of Bodies, and the
Proportions in Which They Enter into These Compounds [2], consultado em 12/06/2010.
2. Brownian Motion and Molecular Reality [3], consultado em 12/06/2010.
3. S. Ramaseasha, Resonance 11 (2006) 79-87, DOI:10.1007/BF02835688 [4].
4. P. Becker, H. Friedrich, K. Fujii,W. Giardini, G. Mana, A. Picard. H. Pohl, H. Riemann, S. Valkiers, Meas. Sci.
Technol. 20 (2009) DOI: 10.1088/0957-0233/20/9/092002 [5], consultado em 12/06/2010).
5. International Bureau of Weights and Measures, 8th ed. (2006) 114–115 [6], consultado em 12/06/2010.
Constante de Avogadro
Criada em 10 de Dezembro de 2010
Revista em 10 de Dezembro de 2010
Aceite pelo editor em 10 de Dezembro de 2010
Efeito fotoeléctrico
Referência : Manuel Ferreira Fernandes, R. (2010), WikiCiências, 1(12):0204
Autor: Ricardo Ferreira Fernandes
[1]
Editor: Jorge Gonçalves
O efeito fotoeléctrico é um fenómeno no qual são emitidos electrões de um material, geralmente metálico, quando
iluminado com radiação de frequência conveniente.
O efeito fotoeléctrico foi observado pela primeira vez, em 1839, por Alexandre Bequerel, através de um eléctrodo
colocado numa solução condutora exposta à luz. Em 1887, Heirich Hertz, observou que eléctrodos irradiados com
luz ultravioleta originavam faíscas eléctricas com mais facilidade. Entre 1888 e 1981, Aleksandr Stoletev estudou
detalhadamente o efeito fotoeléctrico, tendo estabelecido a proporção directa entre a intensidade de radiação
electromagnética que actuava na superfície metálica e a fotocorrente provocada por essa radiação.[1] Em 1902,
Philipp Eduard von Lenard observou que a energia cinética dos electrões ejectados aumentava com a frequência da
luz incidente, o que não estava de acordo com as leis da Física da época, que previam que a energia cinética dos
electrões deveria ser proporcional à intensidade da radiação.
Em 1905, Einstein, baseando-se na teoria do corpo negro de Max Planck, resolveu este aparente paradoxo ao propor
que a luz deveria ser composta por quanta (unidades discretas de energia, actualmente denominados por fotões) e não
por ondas contínuas e que a energia de cada quantum de luz deveria ser igual à frequência multiplicada por uma
constante, mais tarde denominada por constante de Planck. Usando esta hipótese, Einstein foi capaz de explicar o
fenómeno observado de que a energia cinética máxima, Ecin, dos electrões ejectados varia com a frequência,ν, da
radiação incidente através de:
em que h é constante de Plank, ν a frequência da radiação incidente, w0 a chamada função de trabalho, que equivale
à energia mínima necessária para remover um electrão da superfície de um dado material, c a velocidade da luz e λ o
comprimento de onda da radiação incidente.
Pela explicação do efeito fotoeléctrico foi atribuído a Albert Einstein o Nobel da Física em 1921.[2]
Na actualidade, o efeito fotoeléctrico está na base de inúmeras aplicações práticas, sendo as fotocélulas, aparelhos
fotocondutores e células solares exemplo disso.[3] Uma das aplicações mais usadas no quotidiano são as fotocélulas
que actuam como sensores para abrir automaticamente portas ou sistemas semelhantes, usados por exemplo, quando
se entra num edifício, ou para evitar que as mesmas se fechem quando existe algum obstáculo (caso dos elevadores).
12
Efeito fotoeléctrico
Referências
1. Wikipedia (en): Stoletov's law [1], consultado em 06/01/2010
2. The Nobel Prize in Physics, 1921 - Albert Einstein [2], consultado em 06/01/2010
3. Photoelectric Effect - Applications [3], consultado em 06/01/2010
Criada em 23 de Janeiro de 2010
Revista em 10 de Dezembro de 2010
Aceite pelo editor em 10 de Dezembro de 2010
Electrólise
Referência : Manuel Ferreira Fernandes, R. (2010), WikiCiências, 1(12):0205
Autor: Ricardo Ferreira Fernandes
[1]
Editor: Jorge Gonçalves
Etimologicamente, electrólise significa "decomposição pela electricidade". A electrólise é, assim, um processo que
utiliza corrente eléctrica para promover uma reacção química não espontânea. Para isso, um gerador de corrente
eléctrica contínua é ligado aos eléctrodos de uma célula electrolítica forçando os electrões a participar em reacções
provocadas de oxidação num dos eléctrodos (o ânodo) e de redução no outro eléctrodo (o cátodo).
No ano de 1800, os cientistas ingleses William Nicholson (1753-1815) e Anthony Carlisle (1768-1840), quando
tentavam reproduzir as experiências de Allesandro Volta (1745-1827), com o objectivo de analisar as cargas
eléctricas usando um electroscópio previamente desenvolvido por Nicholson, verificaram que ao inserirem os dois
fios condutores metálicos provenientes da pilha de Volta num recipiente com água, se libertavam bolhas gasosas nas
superfícies dos fios condutores (hidrogénio e oxigénio).[1] Posteriormente, em 1807, o químico inglês Sir Humphry
Davy (1778-1840) fez passar uma corrente eléctrica através de hidróxido de potássio e hidróxido de sódio fundidos,
isolando os elementos potássio e sódio, respectivamente. Davy prosseguiu os seus estudos com metais
alcalino-terrosos, tendo isolado de forma semelhante o magnésio, o cálcio, o estrôncio e o bário. Em 1834, Michael
Faraday (1791-1867) introduziu, por sugestão do polímato Rev. William Whewell (1794-1866), o termo electrólise
que deriva do grego electro + lysis e significa decomposição por acção da electricidade.[2]
13
Electrólise
No quotidiano, a electrólise é um processo
muito usado na preparação e purificação de
metais, como por exemplo, na obtenção do
alumínio a partir do mineral bauxite, ou na
refinação do cobre na etapa final da
extracção.
A electrólise é também utilizada para a
obtenção industrial de algumas substâncias
(compostas e elementares), como por
exemplo, o clorato de potássio, o
di-hidrogénio, o dicloro, o hidróxido de
sódio e clorato de sódio.
A electrólise também está presente nos
processos
de
electrodeposição,
Figura 1 - Representação esquemática de uma célula electrólitica utilizada para um
nomeadamente
no
processo
de
processo de galvanoplastia.
galvanoplastia, no qual se pretende o
revestimento de uma superfície condutora
através da deposição, por acção de uma corrente eléctrica, de iões de um dado metal. A superfície que vai receber o
revestimento metálico é ligada ao pólo negativo de uma fonte de alimentação comportando-se como um cátodo. O
metal que vai fornecer o revestimento é ligado ao pólo positivo e comporta-se como ânodo. Quando a fonte de
alimentação é ligada, a acção da corrente eléctrica que flui no circuito provoca a redução (no cátodo) do catião em
solução e a oxidação do metal (no ânodo) (figura 1).
Referências
1. RSC: Enterprise and electrolysis [1], consultado em 02/03/2010.
2. Online Etymology Dictionary: electrolysis [2], consultado em 02/03/2010.
Criada em 27 de Março de 2010
Revista em 14 de Dezembro de 2010
Aceite pelo editor em 14 de Dezembro de 2010
14
Reagente limitante
15
Reagente limitante
Referência : Manuel Ferreira Fernandes, R. (2010), WikiCiências, 1(12):0206
Autor: Ricardo Ferreira Fernandes
[1]
Editor: Jorge Gonçalves
O reagente limitante é o reagente que numa reacção química se encontra em defeito, de acordo com as proporções
estabelecidas pela equação química correspondente (proporções estequiométricas). Deste modo, a quantidade
máxima possível de produto de reacção é determinada pela quantidade existente de reagente limitante, que é
completamente consumido se as reacções químicas forem completas. Os reagentes que no decurso de uma reacção
química completa não se gastam na totalidade são designados por reagentes em excesso ou excedentários. Porém,
quando os reagentes estão em proporções estequiométricas, todos os reagentes são reagentes limitantes.
Considere-se o exemplo da equação química seguinte que representa a reacção entre o trióxido de diferro (Fe2O3) e o
alumínio (Al):
Fe2O3(s) + 2 Al(s)
2 Fe(s) + Al2O3(s)
Através da equação verifica-se que uma mole de trióxido de diferro (Fe2O3) reage com duas moles de alumínio (Al),
originando duas moles de ferro (Fe) e uma mole de trióxido de dialumínio (Al2O3). Supondo que se colocam 30,0 g
de Fe2O3 na presença de 12,0 g de Al em condições propícias à reacção, qual é o reagente limitante?
O primeiro passo consiste em converter a massa dos reagentes em quantidade de substância. Atendendo aos valores
de massa molar de cada um dos reagentes tem-se:
De seguida, para identificar o reagente limitante podem utilizar-se três métodos:
Método 1) Verificar qual dos reagentes se encontra em menor quantidade relativa
O reagente limitante é o que, numa reacção química, se encontra em menor quantidade relativa, ou seja, é aquele que
apresenta menor quociente entre a respectiva quantidade de substância e o respectivo coeficiente estequiométrico na
equação química que descreve a reacção. Algebricamente, o reagente limitante corresponde ao valor mínimo
obtido através da equação (1) para os diferentes reagentes.
Assim, para a reacção acima referida em que a proporção estequiométrica (Fe2O3:Al) é 1:2, virá:
O menor dos dois quocientes é correspondente a Fe2O3 (0,188), logo este é o reagente limitante. Esta forma de
identificar o reagente limitante é particularmente útil quando há mais do que dois reagentes.
Método 2) Determinar o reagente que se encontra em defeito
Reagente limitante
Através das proporções estequiométricas, determina-se qual dos reagentes se encontra em defeito. Deste modo,
atendendo a que uma mole de Fe2O3 reage com duas moles de Al, qual o número de moles de Al necessário para
reagir com 0,188 moles de Fe2O3?
Verifica-se que são necessárias 0,376 moles de Al para reagir com 0,188 mol de Fe2O3.
Assim, uma vez que existem 0,455 mol de Al disponíveis para reagir, ou seja, uma quantidade superior à necessária
para reagir com 0,188 mol de Fe2O3, o Al encontra-se em excesso. Logo, Fe2O3 é o reagente limitante.
Método 3) Calcular qual dos reagentes origina uma maior quantidade de produto de
reacção
As proporções estequiométricas entre os reagentes e os produtos de reacção estabelecidas pela equação química,
permitem calcular a quantidade máxima de produto que teoricamente se pode formar. Assim, o reagente limitante é
aquele que, de acordo com a quantidade existente e com a proporção estequiométrica, produz uma menor quantidade
de produto. Note-se que para calcular a quantidade de produto de reacção formado a partir dos regentes basta utilizar
um dos produtos de reacção obtidos, uma vez que os resultados são independentes quer se opte por um ou por outro.
No caso da reacção acima optou-se por calcular a quantidade formada de Fe.
De seguida, de acordo com a relação estequiométrica da equação, calcula-se a quantidade de produto de reacção que
é formada a partir da quantidade de substância de cada reagente.
Quantidade de substância de Fe formada a partir de 0,188 mol de Fe2O3:
Quantidade de substância de Fe formada a partir de 0,445 mol de Al:
Verifica-se que é o reagente limitante é Fe2O3, uma vez que origina uma menor quantidade de produto de reacção,
neste caso Fe.
Referências
General Chemistry Glossary: limiting reactant [1], consultado em 08/04/2010.
Source Book: Stoichiometry [2], consultado em 08/04/2010.
UCDavis ChemWiki: Limiting Reagents [3], consultado em 08/04/2010.
Criada em 10 de Dezembro de 2010
Revista em 14 de Dezembro de 2010
Aceite pelo editor em 14 de Dezembro de 2010
16
Modelo atómico de Bohr
Modelo atómico de Bohr
Referência : Corrêa, C. (2010), WikiCiências, 1(12):0207
[1]
Autor: Carlos Corrêa
[1]
Editor: Jorge Gonçalves
A teoria atómica de Bohr foi apresentada em dois artigos publicados na revista Philosophical Magazine and Journal
of Science, em Julho e Setembro de 1913, com o título “On the Constitution of Atoms and Molecules", Partes I e II.
Bohr acentua que o seu artigo constitui uma tentativa de aplicação das ideias de Rutherford para uma teoria de
constituição do átomo, propondo-se discutir no primeiro artigo o mecanismo da ligação dos electrões a um núcleo
com carga positiva utilizando a teoria de Planck. É neste primeiro artigo que apresenta o seu modelo atómico e se
explica a posição das riscas do espectro do átomo de hidrogénio
Refere a inadequabilidade da termodinâmica clássica para explicar as propriedades dos átomos com base num
modelo como o de Rutherford, considerando um sistema constituído por um núcleo de pequeníssimas dimensões,
com carga positiva +E, e um electrão descrevendo órbitas elípticas estacionárias à sua volta, tal qual sucede com os
planetas em volta do Sol. Por simplicidade considera a massa do electrão desprezável em relação à massa do núcleo
fixo, a velocidade do electrão pequena em relação à velocidade da luz e que não há qualquer emissão de energia.
A permanência do electrão em óbita (por simplicidade considerada circular, de raio a) exige que a força atractiva ao
núcleo (Ee/a2) seja igual à força centrífuga [ma(2
)2]*, sendo m a massa do electrão. Representando por W a
energia necessária para afastar o electrão a uma distância infinita do núcleo, obten-se o valor da frequência de
rotação e do diâmetro da órbita, 2a:
Contudo, dado que uma carga eléctrica em movimento circular emite radiação electromagnética, as órbitas do
electrão deixariam de ser estacionárias; W aumentaria sucessivamente e o electrão descreveria órbitas cada vez
menores, de maior frequência, acabando por cair no núcleo. Bohr concluiu que "é óbvio que o comportamento deste
sistema é muito diferente do que se verifica nos sistemas atómicos que se encontram na Natureza".
É aqui que surgem as ideias de Planck: "Now the essential point in Planck´s theory of radiation is that the energy
radiation from an atomic system does not take place in the continuous way assumed in the ordinary electrodynamics,
but that it, on the contrary, takes place in distinctly separated emissions, the amount of energy radiated out from an
atomic vibrator of frequency in a single emission being equal to
, where is an entire number, and is a
universal constant".
Na aplicação das ideias de Planck ao modelo atómico, Bohr assume que no processo de ligação do electrão ao núcleo
é emitida radiação de frequência igual a metade da frequência de rotação do electrão em torno do núcleo (
) e a quantidade de energia emitida no processo é
e, considerando as relações
anteriores, obteve as energias, frequências e dimensões das várias órbitas permitidas para o electrão:
Note-se que o valor de W é máximo para = 1, o que corresponde ao estado mais estável do sistema, isto é, ao
estado em que o electrão está mais ligado e que requer maior quantidade de energia para ser removido†.
Para o átomo de hidrogénio, E = e, e substituindo as constantes m, e, e h pelos respectivos valores3, vem 2a = 1,1 ×
10-8 cm, = 6,2 × 1015 s-1 e W/e = 13 V, "valores que são da mesma ordem de grandeza das dimensões do átomo,
das frequências "ópticas" e dos potenciais de ionização" conhecidos na época.
Bohr refere que a importância da teoria de Planck tinha sido já apontada por Einstein e aplicada a uma série de
fenómenos por Stark, Nernst e Sommerfield. Refere os trabalhos de Nicholson, que publicou vários artigos em que
17
Modelo atómico de Bohr
18
mostrou ser possível explicar as riscas, até então de origem desconhecida, nos espectros luminosos da corona solar e
de nebulosas com base na presença de certos elementos nesses corpos celestes, aplicando a teoria de Planbck. No
entanto, havia sérias objecções aos modelos de Nicholson.
O maior sucesso da teoria de Bohr surgiu ao explicar quantitativamente o espectro de emissão do átomo de
hidrogénio, estudado por vários cientistas, entre os quais Rydberg, que verificou empiricamente que a posição as
riscas (frequência, ) obedecia à relação
onde c é a velocidade da luz, RH é a constante de Rydberg (1,09678 × 107 m-1) e n1 e n2 são números inteiros e
positivos, tais que n1 < n2.
A quantidade de energia emitida na passagem do sistema de um estado correspondente a
correspondente a
é
para um estado
o que permite obter os valores das frequências das riscas do espectro do hidrogénio.
"A concordância é quantitativa e também qualitativa. Para e = 4,7 × 10-10, e/m = 5,31 × 10-17 e h = 6,5 ×10-27
obtém-se
e o valor experimental é 3,29 × 1015 ".
Bohr nota que em descargas através de tubos a baixa pressão não foi possível encontrar mais de 12 riscas na série de
Balmer, enquanto nos espectros de emissão de corpos celestes se observam 33 riscas, que são explicadas pela sua
teoria.
Bohr explica a necessidade de se utilizarem baixas pressões para se obter um grande número de riscas nos espectros
atómicos. De acordo com o diâmetro que calculou para as órbitas (2a), para = 12, o diâmetro do átomo excitado é
igual a 1,6 × 10-6 cm que é a distância média entre dois átomos à pressão de 7 mmHg. Para = 33 o diâmetro do
átomo é de 1,2 × 10-5 cm, que é a distância média entre moléculas à pressão de 0,02 mmHg. Assim, para que a
desexcitação possa ocorrer por emissão de radiação é necessário que os átomos excitados se encontrem
suficientemente afastados uns dos outros, o que exige pressões muito baixas.
Após uma série de outras discussões, o artigo de Bohr termina com uma generalização da hipótese utilizada no seu
modelo:
"Em qualquer sistema molecular constituído por um núcleo de carga positiva e electrões movendo-se em órbitas
circulares, considerando o núcleo em repouso em relação aos electrões, o momento angular de cada electrão em
relação ao centro da sua órbita é igual a h/2 , em que h é a constante de Planck".
Modelo atómico de Bohr
*
Note-se que Bohr representou a frequência de rotação do electrão por que é o símbolo vulgarmente utilizado para
a pulsação,
, sendo f a frequência.
†
É a energia de ionização. Actualmente considera-se a energia W negativa, pois para o electrão localizado a uma
distância infinita, considera-se W = 0.
‡
As unidades são as do Sistema cgs.
Criada em 22 de Abril de 2010
Revista em 06 de Setembro de 2010
Aceite pelo editor em 14 de Dezembro de 2010
Modelo atómico de Rutherford
Referência : Corrêa, C. (2010), WikiCiências, 1(12):0208
[1]
Autor: Carlos Corrêa
[1]
Editor: Jorge Gonçalves
O modelo atómico de Rutherford (E. Rutherford, F.R.S., Universidade de Manchester) foi apresentado numa
comunicação efectuada na Manchester Literary and Philosophical Society em 1911 e publicada na revista
Philosophical Magazine and Journal of Science, em Maio de 1911, com o título “The Scattering of and
Particles by Matter and the Structure of the Atom”.
Era convicção geral que os desvios de partículas (experiências de Geiger e Marsden[1]) e de partículas
(trabalhos de Crowthers[2]) quando feixes destas partículas atravessavam finas lâminas metálicas (por exemplo, de
ouro, com espessura de cerca de 0,00004 cm) resultavam de uma série de sucessivos pequenos desvios.
Thomson havia concluído, baseado nos resultados experimentais de Crowthers com partículas e em cálculos
realizados sobre o seu modelo de “bolo de passas”, que os ângulos de cada desvio deveriam ser pequenos e
resultantes da interacção sucessiva com N electrões. Crowthers, em experiências de deflexão com vários metais,
calculou até o número de electrões, N, que seria responsável pelos sucessivos desvios e que diferia do número de
electrões actualmente conhecido (Al. 27 em vez de 13, Cobre 42 em vez de 29, prata 78 em vez de 47, etc.).
No entanto, alguns dos desvios de partículas ao atravessarem finas láminas de ouro eram mesmo superiores a 90º,
o que era dificil de explicar com base na existência de pequenos desvios sucessivos, pois o cálculo da probabilidade
de ocorrência de desvios sucessivos conduzia a valores extremamente baixos.
Rutherford pensou que era razoável supor que os desvios elevados das partículas se deviam a um único encontro
da partícula com uma zona de intenso campo eléctrico e não a uma série sucessiva de pequenos desvios. Assim,
considerou um modelo, sobre o qual efectuou alguns cálculos, com a seguinte estrutura: “...um átomo que contém
uma carga eléctrica Ne no seu centro rodeada por uma esfera electrificada de carga -Ne (ou +Ne)
uniformemente distribuída numa esfera de raio R, em que e é a unidade fundamental de carga e N a carga central
do átomo.”
Por comodidade, Rutherford considerou a carga central positiva, +Ne, rodeada por carga negativa -Ne. Considerou
que os desvios das partículas , carregadas positivamente, se deviam somente à carga central do átomo.
Aqui surge o verdadeiro modelo de Rutherford: uma zona central - o núcleo - com carga positiva e uma zona
difusa à sua volta, com carga negativa - a nuvem electrónica.
Com base neste modelo, Rutherford determinou as trajectórias hiperbólicas das partículas no seu percurso através
dos átomos que constituíam as folhas de ouro e calculou os ângulos de desvio quando os feixes de partículas
passavam na vizinhança do centro do átomo (à distância p). Para valores de p pequenos, os ângulos de desvio
19
Modelo atómico de Rutherford
podiam alcançar valores tão elevados como 120º ou 150º.
Para lâminas tão finas como 0,0001 cm, deduziu que a probabilidade de uma segunda interacção com outro átomo
era diminuta (da ordem de 0,000001). Geiger, realizando experiências com diferentes lâminas metálicas, concluiu
que o valor de N era aproximadamente proporcional aos seus pesos atómicos.
Tanto para a deflexão de partículas como , a carga central Ne é proporcional ao peso atómico da partícula.
Verificou que a carga positiva do núcleo era aproximadamente igual a 1/2 de Ae, em que A é o peso atómico
(referido ao hidrogénio). Quer dizer que o número de electrões do átomo é cerca de metade do respectivo peso
atómico.
Curiosamente, Rutherford conclui neste célebre artigo “The deductions from the theory so far considered are
independent of the sign of the central charge, and it has not so far been found possible to obtain definitive evidence
to determine whether it is positive or negative.”
No seu segundo artigo, de Março de 1914 (Philosophical Magazine and Journal of Science, Série 6, Volume 27, pag.
488-498), com o título "The Structure of the Atom", Rutherford começa por referir que o artigo vai tratar de alguns
pontos ligados à teoria do "núcleo" do átomo que tinha deliberadamente omitido no artigo anterior (1911).
Escreve "Para explicar os grandes desvios angulares das experiências de dispersão das partículas , supuz que o
átomo consistia num núcleo de pequenas dimensões carregado positivamente no qual se concentrava praticamente
toda a massa do átomo. Considerei o núcleo rodeado de electrões, de modo a tornar o átomo electricamente neutro,
distribuidos a distâncias comparaveis ao que se considera ser o raio do átomo". Note-se que nada adianta sobre o
modo como os electrões se moveriam em torno do núcleo.
Concentra-se novamente em experiências de deflexão de Geiger e Marsden[3] realizadas em 1913, que continuam a
substanciar o seu modelo atómico. Rutherford [4] estendeu a sua análise à interacção de partículas com átomos
menores, como hidrogénio e hélio, concluindo que o núcleo do átomo de hidrogénio teria uma só carga positiva e o
núcleo de hélio (partícula ) teria duas.
Previu que, dada a carga e massa do átomo de hidrogénio, a aproximação das partículas levaria os átomos de
hidrogénio a moverem-se com uma velocidade 1,6 vezes maior do que a das partículas , devendo ser possível
detectar a ejecção destes átomos de hidrogénio. A frase de Rutherford "Mr Marsden has kindly made experiments for
me to test whether the presence of such hydrogen atoms can be detected" mostra a sua íntima colaboração com estes
experimentalistas.
Refere a diferença entre o comportamento das partículas e , notando que estas, por terem carga contrária à carga
do núcleo, são aceleradas na sua aproximação ao núcleo, podendo mesmo ser apanhadas numa órbita em espiral,
acabando por cair no núcleo, o que explicava o desaparecimento de partículas na sua passagem através da matéria.
Sobre as dimensões do núcleo escreve: "Para explicar a velocidade adquirida pelos átomos de hidrogénio nas suas
"colisões" com partículas , é possível verificar por cálculo que que os centros dos núcleos de He e de H se devem
aproximar a cerca de 1,7 × 10-13 cm. Isto é uma quantidade muito pequena, um pouco menor do que o valor
geralmente aceite para o diâmetro do electrão, cerca de 2 × 10-13 cm".
Rutherford interroga-se se existirão electrões no núcleo, questão já levantada por Bohr, que também concluiu que as
partículas provinham do núcleo.
Segundo as palavras de Rutherford "é claro na base da teoria do núcleo que as propriedades físicas e químicas dos
elementos dependem inteiramente da carga nuclear, que determina o número e a distribuição dos electrões que o
rodeiam".
A existência de isótopos é claramente prevista, pois "deve ter-se em mente que não é impossível, com base na teoria
do núcleo, que os átomos possam diferir no seu peso atómico mas terem a mesma carga nuclear". O mesmo sucede
com a existência de isóbaros: "Se o núcleo for considerado uma mistura de núcleos de hidrogénio com carga + e
núcleos de hélio com carga 2+, pode conceber-se que a existência de atomos com a mesma carga nuclear mas
diferentes pesos atómicos".
20
Modelo atómico de Rutherford
21
Este segundo artigo termina referindo os trabalhos de Bohr "Bohr chamou a atenção para as dificuldades de
construir átomos baseados na teoria do "núcleo" e mostrou que as posições estáveis dos electrões não podem ser
deduzidas da Mecânica Clássica. Por introdução de conceitos relacionados com o quantum de Planck, Bohr
mostrou que, sob simples suposições, é possivel construir átomos simples e moléculas.(...) Embora haja muitas
opiniões acerca da validade das suposições em Bohr que se baseia, não há nenhuma dúvida de que as suas teoprias
são de grande interesse e importância para todos os físicos, como primeira tentativa de construir simples átomos e
moléculas e explicar os seus espectros."
Referências
1. Geiger e Marsden, Proc. Roy. Soc. A. Ixxxii. p. 495(1909)
2. Crowther, Proc. Roy. Soc. A. Ixxxiv. p. 226(1910)
3. Geiger e Marsden, Phil. Mag. xxv . p.604 (1913)
4. Rutherford e Nuttall, Phil. Mag. xxvi . p.702 (1913)
Criada em 27 de Abril de 2010
Revista em 06 de Setembro de 2010
Aceite pelo editor em 14 de Dezembro de 2010
Fórmula de estrutura
Referência : Manuel Ferreira Fernandes, R. (2010), WikiCiências, 1(12):0209
Autor: Ricardo Ferreira Fernandes
[1]
Editor: Jorge Gonçalves
A fórmula de estrutura consiste na representação gráfica da
sequência e modo de ligação dos átomos entre si (figura 1).
Figura 1 - Fórmulas de estrutura de isómeros
constitucionais: (a) etanol; (b) metoximetano.
Fórmula de estrutura
Figura 2 - Representação do anel do benzeno retirado
de Kekulé's Lehrbuch der organischen Chemie
(1861–1867). Imagem obtida em:Chemical Heritage
[1]
Foundation
22
O químico escocês Archibald Scott Couper (1831–1892) foi um
dos primeiros cientistas a representar a estrutura molecular de um
determinado composto. Couper introduziu, em 1858, as fórmulas
de estrutura, em que os átomos eram representados através dos
respectivos símbolos dos elementos químicos e as ligações entre
os átomos eram representadas através de traços contínuos. No
entanto, Couper, devido aos adiamentos impostos pelo seu
supervisor – o químico francês Charles Adolphe Wurtz
(1817-1884), não publicou imediatamente os seus trabalhos. Como
consequência, o químico alemão August Kekulé von Stradonitz
(1829–1896) publicou um artigo com representações similares
algumas semanas antes que Couper, tendo assim Kekulé ficado
historicamente conhecido pela introdução da estrutura molecular e
da tetravalência do carbono.[1] No decurso dos seus trabalhos
Kekulé ficou igualmente conhecido pela proposta da estrutura
cíclica do benzeno (figura 2).
Posteriormente o químico norte-americano Gilbert Newton Lewis
(1875-1946) publicou, em 1916, o artigo The atome and the
molecule em que introduziu as fórmulas químicas actualmente
designadas por fórmulas ou estruturas de Lewis, nas quais
representa os electrões de valência que participam nas ligações
covalentes entre os átomos e os respectivos electrões não
partilhados existentes na molécula. Assim, utiliza-se uma
linha/traço (–) para representar uma ligação simples ou um par de
electrões, podendo utilizar-se o ponto (•) como símbolo gráfico
para representar um electrão ou dois pontos para um par de
electrões (••). Deste modo, as fórmulas de estruturas indicam a
conectividade e o tipo de ligação que ocorre entre os átomos, em
Figura 3 - Fórmulas de Lewis: (a) molécula de H2
que as ligações simples são caracterizadas pela partilha de um par
(ligação covalente simples); (b) molécula de O2
(ligação covalente dupla); (c) molécula de N2 (ligação
de electrões (figura 3a), as ligações duplas por dois pares de
covalente tripla).
electrões partilhados (figura 3b) e as ligações triplas pela partilha
de três pares de electrões (figura 3c). Os electrões de valência que
não partilhados são colocados de forma adjacente ao átomo ao qual estão associados (figura 3b e 3c).
As fórmulas de estrutura são muito utilizadas em Química Orgânica, devido à enorme variedade de compostos que o
carbono pode originar. Assim, de modo a tornar a representação molecular mais simples utilizam-se formas
abreviadas em que, por exemplo, os compostos representados na figura 1 podem ser escritos ao longo de um texto de
uma forma condensada como CH3-CH2-OH (etanol) e CH3-O-CH3 (metoximetamo).
Fórmula de estrutura
23
Adicionalmente, foram introduzidas fórmulas de estrutura ainda
mais simplificadas, que têm a vantagem de serem fáceis de
desenhar, para representar estruturas complexas de uma forma
clara e concisa. Nestas estruturas os átomos de carbono e de
hidrogénio estão representados de modo implícito na extremidade
das linhas e nos vértices ao longo da estrutura, representando-se
apenas os átomos que não sejam carbono ou hidrogénio
(heteroátomos). Por exemplo, na figura 4 estão representadas as
fórmulas dos isómeros constitucionais etanol e éter dimetílico.
Figura 4 - Isómeros constitucionais: (a) etanol; (b) éter
dimetílico.
Referências
1. August Kekulé and Archibald Scott Couper [2], consultado em 20/08/2010.
IUPAC Gold Book: Structural formula [3], consultado em 20/08/2010.
IUPAC Gold Book: Lewis formula [4], consultado em 20/08/2010.
Criada em 14 de Dezembro de 2010
Revista em 15 de Dezembro de 2010
Aceite pelo editor em 15 de Dezembro de 2010
Aminoácido
Referência : Manuel Ferreira Fernandes, R. (2010), WikiCiências, 1(12):0210
Autor: Ricardo Manuel Ferreira Fernandes
[1]
Editor: Jorge Gonçalves
Um aminoácido é uma substância cuja molécula
contém os grupos funcionais - amino (-NH2) e
carboxilo (-COOH). Num α-aminoácido existe um
substituinte na posição 2 (R), que pode ser uma cadeia
alquílica ou arílica, podendo conter um dos seguintes
grupos: hidroxilo, amino, mercapto, sulfureto,
carboxilo, guanidininilo ou imadazolilo (figura 1).
O primeiro aminoácido natural a ser descoberto, a
asparagina, foi isolado a partir do espargo (Asparagus
officinalis), em 1806, pelos químicos franceses Louis
Figura 1 - Estrutura genérica de um α-aminoácido.
Nicolas Vauquelin (1736-1829) e Pierre Jean Robiquet
(1780-1840). Posteriormente, em 1810, foi descoberto
o aminoácido cistina, que mais tarde, em 1884, se verificou ser um dímero constituído por duas moléculas de
cisteína. À medida que a química orgânica se foi desenvolvendo, novos aminoácidos foram isolados e na actualidade
já se identificaram cerca de 700.[1]
Aminoácido
Os aminoácidos são compostos anfotéricos, uma vez que a sua estrutura apresenta dois grupos funcionais - amino e
carboxilo - que actuam como base e como ácido, respectivamente. Assim, ocorrem transferências de protões dos
grupos ácidos para os grupos básicos, formando espécies designadas por iões dipolares ou zwitteriões (do germânico
zwitt, que significa ambivalente). A elevada polaridade da estrutura zwitteriónica permite que o aminoácido forme
estruturas cristalinas relativamente solúveis em água. Porém, quando aquecidos (473-573 K ou 200-300 ºC), tendem
a decompor-se, antes de atingirem a temperatura de fusão.
Em solução, os aminoácidos, se não têm cadeias laterais ionizáveis, apresentam dois grupos capazes de sofrer
protonação/desprotonação. Assim, a carga do aminoácido varia com o pH da solução. A pH baixo o grupo amina
encontra-se protonado originando um catião (figura 2a). À medida que o pH aumenta o grupo carboxílico é
desprotonado, existindo um pH designado por pH ou ponto isoeléctrico, em que a que a extensão de protonação é
igual à extensão de desprotonação, correspondendo à concentração máxima de aminoácido sob a forma de zwitterião
(figura 2b). Aumentando mais o pH, apenas o grupo ácido se encontra desprotonado, ficando assim o aminoácido
com carga negativa (figura 2c).
Figura 2 - As três formas de um aminoácido de acordo com o pH da solução: (a) forma catiónica; (b) forma zwitteriónica; (c) forma
aniónica.
Os aminoácidos são extremamente importantes a nível bioquímico, uma vez que são a unidade básica de construção
(monómeros) das proteínas, as quais desempenham funções vitais nos organismos como, por exemplo, na respiração
celular e no metabolismo. Apesar do elevado número de aminoácidos identificados até à actualidade, apenas cerca de
duas dezenas de α-aminoácidos entram na constituição das proteínas de todas as espécies, desde os humanos até às
bactérias. No organismo humano, alguns α-aminoácidos são sintetizados pelo próprio organismo, porém, existem 8
α-aminoácidos (fenilalanina, isoleucina, leucina, lisina, metionina, treonina, triptofano e vanilina) que o organismo
não consegue produzir, mas indispensáveis para o seu funcionamento. Assim, estes compostos designados por
aminoácidos essenciais, necessitam obrigatoriamente de ser incluídos na dieta alimentar humana.
Os aminoácidos apresentam diversas aplicações tecnológicas, sendo principalmente utilizados como aditivos
alimentares em rações de animais, uma vez que o componente principal destas é à base de soja ou outras
leguminosas similares, que apresentam baixa percentagem de aminoácidos essenciais. Na indústria alimentar o ácido
glutâmico é utilizado como um aromatizante.[2] Os aminoácidos são igualmente utilizados como precursores na
síntese de alguns medicamentos utilizados, por exemplo, no tratamento da síndrome de Parkinson. Para aumentar a
biodegrabilidade e biocompatibilidade de polímeros e tensioactivos os aminoácidos são também incorporados na sua
síntese.[3]
24
Aminoácido
25
Referências
1. A. Quintas, A. P. Freire, M. J. Halpern, Bioquímica - Organização Molecular da Vida, Lidel: Lisboa, 2008, ISNB:
978-972-757-431-5.
2. S. Garattini, J. Nutrition 130 (2000), 901S-909S Glutamic Acid, Twenty Years Late [1]
3.
F.
Sanda,
T.
Endo,
Macromol.
Chem.
Phys.
200
DOI:10.1002/(SICI)1521-3935(19991201)200:12<2651::AID-MACP2651>3.0.CO;2-P [2]
(1999)
2651–2661,
INTRODUCING AMINO ACIDS [3], consultado em 3/06/2010.
Criada em 28 de Setembro de 2010
Revista em 15 de Dezembro de 2010
Aceite pelo editor em 16 de Dezembro de 2010
Fórmula estereoquímica
AVISO: Não foi possível gerar a página – será produzido texto simples.
As causas potenciais do problema são: (a) um erro do programa responsável pelo PDF (b) sintaxe problemática do
MediaWiki (c) uma tabela demasiado larga
Referência : Manuel Ferreira Fernandes, R. (2010), WikiCiências, 1(12):0211Autor: Ricardo Ferreira
FernandesEditor: Jorge Gonçalves Figura 1 - Fórmulas estereoquímicas de um par de enantiómeros. O arranjo
espacial relativo dos átomos é diferente, porém, os compostos estão um para o outro como a imagem está para o
objecto num espelho plano.A fórmula estereoquímica é uma representação que indica a disposição espacial relativa
dos átomos numa molécula (figura 1).Estas fórmulas foram introduzidas, por volta de 1859, no âmbito da
estereoquímica (área que estuda as propriedades químicas dos compostos em função da disposição espacial dos
átomos que compõem as moléculas) quando o químico francês Louis Pasteur observava cristais de sais do ácido
tartárico. Pasteur verificou que os cristais apresentavam duas formas distintas que eram a imagem uma da outra num
espelho. Quando separadas, estas duas formas de cristais apresentavam actividade optica, isto é, tinham a capacidade
de rodar o plano de polarização da luz polarizada. Posteriormente, em 1872, o químico holandês Jacobus Henricus
van’t Hoff, baseado na tetravalência do carbono enunciada pelo químico alemão August Kekulé, propôs que a
actividade óptica poderia ser explicada a partir dos arranjos espaciais dos substituintes em torno do carbono.Figura 2
- Fórmulas estereoquímicas de isómeros cis-trans: (a) cis-1,2-dicloroeteno; (b) trans-1,2-dicloroeteno.As fórmulas
estereoquímicas permitem compreender a relação entre a disposição espacial dos átomos e as propriedades das
moléculas. Veja-se o exemplo dos isómeros cis-trans do 1,2-dicloroeteno (figura 2), em que os átomos de cloro se
podem encontrar do mesmo lado ou em lados opostos da ligação dupla. Como consequência, as duas moléculas
apresentam diferentes momentos dipolares, o que afecta várias propriedades físicas. A forma cis, de maior
polaridade, será menos volátil. Assim, a temperatura de ebulição é 333 K (60 ºC) para a forma cis e 321 K (48 ºC)
para a forma trans.Figura 3 - Projecção de Fisher da D-glucose.O arranjo espacial relativo dos átomos nas moléculas
pode também ser representado de um modo simplificado através da projecção da molécula no plano da folha de
leitura (plano do papel). As projecções de Fisher, Haworth e Newman são as mais conhecidas.As projecções de
Fisher, assim designadas em homenagem ao químico alemão Hermann Emil Fisher, são particularmente utilizadas
em Química Orgânica na representação de monossacarídeos (glucose, frutose). Nestas projecções, cada carbono e as
quatro ligações que dele partem representam-se por uma cruz, na qual o átomo central (carbono) se encontra no
Fórmula estereoquímica
ponto de intersecção. As linhas horizontais representam as ligações que estão na direcção do observador (para a
frente do plano do papel) e as linhas verticais as ligações que se afastam do utilizador (para trás do plano do papel).
A figura 3 representa a projecção de Fisher para a D-glucose.Figura 4 - Projecção de Haworth da β-D-glucose. As
projecções de Haworth, assim denominadas em homenagem ao químico inglês Sir Walter Norman Haworth que as
introduziu, utilizam-se para representar a estrutura tridimensional das formas cíclicas dos monossacarídeos. Nestas
projecções, o anel é representado por um hexágono tendo como vérices um átomo de oxigénio e cinco átomos de
carbono; os substituintes colocam-se nos extremos de segmentos de recta verticais, que partem dos vértices, para
cima ou para baixo, conforme se trate de substituintes acima ou abaixo do plano do hexágono (figura 4).Figura 5 Projecção de Newman das conformações gauche e anti do butano.As projecções de Newman, assim denominadas em
homenagem ao químico norte-americano Melvin Spencer Newman, utilizam-se para representar a disposição
espacial de 6 substituintes em torno de dois carbonos adjacentes. Consistem na projecção das 6 ligações e
substituintes num plano perpendicular à ligação C-C (plano do papel). Utilizam-se para representar confórmeros. Os
substituintes ligados ao átomo de carbono mais próximo do observador são ligados ao centro de um pequeno círculo,
enquanto os substituintes ligados ao carbono mais afastado ligam-se à parte exterior do círculo (figura 5).
Representam aquilo que um observador veria se olhasse a molécula na direcção da ligação C-C.Referências IUPAC
Gold Book: stereochemical formula, consultado em 30/06/2010. IUPAC Gold Book: projection formula, consultado
em 30/06/2010. IUPAC Gold Book: Fischer projection, consultado em 30/06/2010. IUPAC Gold Book: Newman
projection, consultado em 30/06/2010. Chemical Heritage Foundation: Jacobus Henricus van’t Hoff, consultado em
30/06/2010.Criada em 14 de Dezembro de 2010 Revista em 15 de Dezembro de 2010 Aceite pelo editor em 16 de
Dezembro de 2010
Período de semi-desintegração
Referência : Spencer Lima, L. (2010), WikiCiências, 1(12):0212
Autor: Luis Spencer Lima
[1]
Editor: Jorge Gonçalves
O período de semi-desintegração (t1/2) de uma espécie radioactiva (também designado por tempo de semi-vida,
tempo de meia-vida ou período de semi-transformação) representa o intervalo de tempo que é necessário decorrer
para que a sua actividade diminua para metade. Como a actividade (número de desintegrações radioactivas por
unidade de tempo) é directamente proporcional ao número de núcleos atómicos radioactivos, o período de
semi-desintegração é o tempo necessário para diminuir para metade o número de partículas radioactivas.
A velocidade de desintegração radioactiva é directamente proporcional ao número de núcleos presentes não
desintegrados
em que N representa o número de núcleos (não desintegradas) existente no instante t e λ a constante de desintegração
radioactiva (ou de decaimento), o que corresponde a um processo de decaimento primeira ordem. Por integração da
equação diferencial obtém-se a equação que relaciona o número de partículas não desintegradas com o tempo:
Nesta equação, N0 representa o número inicial de núcleos. A constante de desintegração (ou de decaimento) é
característica de cada isótopo radioactivo (radioisótopo) e é independente da temperatura, da pressão e da substância
a que o radioisótopo pertence. A partir da equação (1) pode deduzir-se a equação que permite o cálculo de t1/2. Para
26
Período de semi-desintegração
tal, e atendendo à definição de período de semi-desintegração, substitui-se na equação (1) N por N0/2 e t por t1/2,
após rearranjo e simplificação, obtém-se
o que mostra que t1/2 é constante. Isto significa que, por cada período de semi-desintegração decorrido, o número de
partículas radioactivas reduz-se para metade da anterior:
Isto significa que, ao fim de 5 períodos de semi-desintegração, restam apenas 3,125 % do número inicial de
partículas. É prática corrente considerar que ao fim de 10 períodos de semi-desintegração o produto radioactivo se
esgotou (a quantidade presente é cerca de mil vezes menor do que a inicial).
O período de semi-desintegração é característico de cada isótopo e pode assumir valores tão distintos como alguns
milissegundos (3,4 ms é o t1/2 do meitnério-266, 266Mt) ou milhares de milhões de anos (4,468 109 anos é o t1/2 do
urânio-238, 238U).
Uma das aplicações mais importantes do período de semi-desintegração de um radioisótopo é na datação de objectos
e é com base em t1/2 que é feita a escolha do radioisótopo mais adequado à datação do objecto em questão.
Por exemplo, se se pretende determinar a idade de uma rocha do período jurássico, ocorrido há mais de 145 milhões
de anos, não se pode utilizar o método de datação com carbono-14 (14C), porque como o seu período de
semi-desintegração é de 5730 anos, significa que se passaram mais de 25300 períodos de t1/2, o que implica que a
quantidade de 14C é praticamente nula. Os radioisótopos mais adequados para este caso seriam, por exemplo, o 238U
(t1/2 = 4,468 109 anos), o 235U (t1/2 = 7,04 108 anos) ou o 40K (t1/2 = 1,248 109 anos). Para determinar a idade
de um vinho, o radioisótopo mais adequado é o trítio, 3H, pois t1/2 = 12,3 anos. Quando o vinho é submetido à
determinação do nível de trítio, juntamente com a água (que fornece o valor inicial de trítio), e se verifica que este
apresenta apenas 30,6 % (por exemplo) da quantidade de inicial de 3H (o que significa que 69,4 % do trítio sofreu
desintegração), tal significa que foi engarrafado há 21 anos.
Criada em 16 de Dezembro de 2010
Revista em 16 de Dezembro de 2010
Aceite pelo editor em 17 de Dezembro de 2010
27
Ligação iónica
Ligação iónica
Referência : Manuel Ferreira Fernandes, R. (2010), WikiCiências, 1(12):0213
Autor: Ricardo Ferreira Fernandes
[1]
Editor: Jorge Gonçalves
A ligação iónica é um tipo de ligação química que ocorre através da atracção electrostática entre iões de carga
oposta.
A ligação iónica foi proposta no início do século XX pelo físico alemão Walther Ludwig Julius Kossel, e foi
interpretada com base na máxima estabilidade alcançada quando os átomos adquirem a configuração electrónica de
um gás nobre, que neste caso é atingida pela transferência de electrões entre os átomos que participam na ligação.
A ligação iónica ocorre entre átomos que apresentam diferenças acentuadas de electronegatividade, isto é, diferenças
apreciáveis na capacidade de atrair electrões dos átomos a que se encontram ligados, como sucede entre os metais e
os não-metais. O átomo não-metálico, que é mais electronegativo, capta os electrões do metal, adquirindo carga
negativa (-) adquirindo a configuração electrónica do gás nobre mais próximo. Por seu lado, o átomo menos
electronegativo - o metal - que perde menos dificilmente os electrões de valência, alcançando a configuração
electrónica do gás nobre mais próximo na tabela periódica, adquire simultaneamente carga positiva (+).
Após se terem formado os respectivos catiões e aniões, surgem forças electrostáticas atractivas entre iões de carga
oposta (mais próximos) e repulsivas entre os iões de carga igual (mais afastados). Como consequência destas
interacções, a força resultante é atractiva e estabelece-se uma ligação, denominada ligação iónica, que mantém os
aniões e os catiões unidos no cristal.
Na prática, todas as ligações iónicas têm algum carácter covalente, sendo possível avaliar a quantidade de carácter
covalente (e iónico) de uma ligação, em vez de se considerar uma ligação como puramente iónica ou puramente
covalente. Assim, o químico norte-americano Linus Pauling propôs uma relação que permite estimar o carácter
iónico de uma ligação que ocorre entre os átomos A e B:
Fracção de carácter iónico
em que χA e χB representam a electronegatividade dos átomos A e B.
28
Ligação iónica
Deste modo, as ligações com elevado carácter iónico
originam compostos, geralmente sólidos, designados
por compostos iónicos, os quais não existem como
moléculas discretas, mas sim como estruturas gigantes
tridimensionais organizadas (estruturas cristalinas). A
figura 1 representa a estrutura cristalina do cloreto de
sódio (NaCl), em que o balanço das forças repulsivas e
atractivas entre os iões Na+ e Cl- conduz a um
empacotamento organizado numa estrutura cristalina.
Os compostos iónicos formam-se igualmente entre iões
com cargas não unitárias como, por exemplo, o cloreto
de magnésio (MgCl2), em que o catião magnésio
(Mg2+), que é um catião bivalente (carga 2+), interage
com dois aniões cloreto (Cl-). Os iões poliatómicos
como, por exemplo, o catião NH4+ e o anião NO3-,
formam também compostos iónicos dando origem,
neste caso, ao nitrato de amónio (NH4NO3).
29
Figura 1 - Estrutura cristalina do composto iónico cloreto de sódio
(NaCl). As esferas violeta representam os catiões sódio (Na+); as
esferas verdes representam os aniões cloreto (Cl-).
As elevadas temperaturas de fusão dos sólidos iónicos
como, por exemplo, o cloreto de potássio (KCl) 1043 K (770 ºC), indica que as ligações iónicas são fortes, sendo
necessário elevar consideravelmente a temperatura para aumentar a agitação de modo a vencer as elevadas forças
electrostáticas entre os iões.
Referências
IUPAC Gold Book: Ionic bond [1], consultado em 11/07/2010.
Criada em 17 de Dezembro de 2010
Revista em 17 de Dezembro de 2010
Aceite pelo editor em 17 de Dezembro de 2010
Auto-ionização da água
30
Auto-ionização da água
Referência : Spencer Lima, L. (2010), WikiCiências, 1(12):0214
Autor: Luis Spencer Lima
[1]
Editor: Jorge Gonçalves
A auto-ionização da água consiste na transferência de um protão entre duas moléculas de água com formação dos
iões hidróxido (OH-) e oxónio (H3O+). A transferência é possível pelo facto de a água ser uma substância anfotérica,
isto é, poder actuar como ácido e como base. A equação que ilustra a reacção de auto-ionização da água é a seguinte:
2 H2O(l)
H3O+(aq) + HO-(aq)
A água desionizada, apesar do nome, contém os iões resultantes da sua auto-ionização. O número de aniões
hidróxido formados é igual ao número de catiões oxónio, pelo que, em água pura, a sua concentração é igual: [H3O+]
= [OH-] = 1,0 10-7 mol dm-3, a 298 K (25 ºC) e a 105 Pa (1 bar). Quando [H3O+] = [OH-], a água diz-se neutra
(em termos ácido-base), sendo esta a única condição que define neutralidade. Daqui resulta que o valor do pH da
água neutra é 7,0 à temperatura de 298 K e à pressão de 105 Pa.
A constante de equilíbrio desta reacção é definida da seguinte forma:
em que Kw representa a constante de auto-ionização da água, também denominada produto iónico da água. A 298
K (25 ºC) e a 105 Pa (1 bar), Kw = 1,0 10-14. A temperatura tem uma influência considerável na extensão da
reacção de auto-ionização da água, ao contrário da influência da pressão, que é praticamente nula. Quanto maior for
a temperatura maior é a extensão da reacção, o que significa que a concentração dos iões H3O+ e HO- é maior e, por
conseguinte, menor é o valor de pH da água neutra e maior é o valor de Kw. Por exemplo, a 323 K (50 ºC) e a 105 Pa,
Kw = 5,5 10-14 [1], logo [H3O+] = [HO-] = 2,3 10-7 mol dm-3, ou seja, para água pura neutra a esta temperatura
e pressão, pH = 6,6. Para soluções de concentrações moderadas, o valor de Kw indica os valores mínimo e máximo
de [H3O+] para uma determinada temperatura, isto é, impõe os limites na escala de pH. A 298 K, o valor máximo de
pH é 14, enquanto a 323 K o valor máximo de pH é 13.2.
Referências
1. A. V. Bandura, S. N. Lvov, J. Phys. Chem. Ref. Data 35 (2006) 15-30.
Criada em 17 de Dezembro de 2010
Revista em 17 de Dezembro de 2010
Aceite pelo editor em 17 de Dezembro de 2010
Modelo atómico de Thomson
Modelo atómico de Thomson
AVISO: Não foi possível gerar a página – será produzido texto simples.
As causas potenciais do problema são: (a) um erro do programa responsável pelo PDF (b) sintaxe problemática do
MediaWiki (c) uma tabela demasiado larga
Referência : Corrêa, C. (2010), WikiCiências, 1(12):0215Autor: Carlos CorrêaEditor: Jorge Gonçalves Neste modelo
o átomo é constituído por electrões encastrados numa esfera maciça com carga eléctrica positiva uniformemente
distribuída. O número de electrões é tal que torna o átomo electricamente neutro. O modelo é conhecido como "o do
bolo de passas".MODELO ATÓMICO DE THOMSONDescrição histórica do modeloO modelo atómico de
Thomson (J. J. Thomson, F.R.S., Cavendish Professor de Física Experimental na Universidade de Cambridge) foi
apresentado numa comunicação publicada na revista Philosophical Magazine and Journal of Science, em Março de
1904, com o título “Sobre a Estrutura do Átomo: uma Investigação da Estabilidade e Períodos de Oscilação de um
número de Corpúsculos dispostos com iguais Intervalos numa Circunferência, com aplicação dos resultados à Teoria
da Estrutura Atómica”[1]. Segundo Thomson, “A ideia de que os átomos dos elementos consistem num certo
número de corpúsculos com carga eléctrica negativa embebidos numa esfera com carga eléctrica positiva
uniforme(...), sugere o estudo do movimento de um anel de partículas negativamente electrificadas embebidas numa
esfera uniformemente electrificada”. É este o modelo de Thomson: anéis de electrões igualmente intervalados
movendo-se em movimento circular, embebidos numa esfera maciça com carga positiva uniformemente distribuída
(bolo de passas). Thomson tratou matematicamente este modelo considerando a força atractiva (de um electrão ao
centro das esfera) e as forças repulsivas (dos restantes electrões sobre um electrão), que deveriam ser iguais para que
o conjunto fosse estável. Considerou que os electrões se moveriam, rodando periodicamente no plano da
circunferência e ou vibrando perpendicularmente a este plano, e relacionou as frequências de vibração mecânicas
com as frequências dos espectros atómicos, sem, no entanto ser capaz de prever os valores experimentais das
frequências espectrais (como haveria de suceder com a teoria de Bohr). Thomson estudou sistemas com números
variáveis de electrões e verificou que a estabilidade dos sistemas dependia do número de electrões e da velocidade
angular dos anéis electrónicos. Para sistemas com mais de 6 electrões, poderia conseguir sistemas estáveis se
colocasse um ou mais electrões em circunferências interiores. Calculou mesmo o número mínimo, p, de electrões
internos que tornariam estável um anel de n electrões: n .....5 6 7 8 9 10 15 20 30 40 p .....0 1 1 1 2 3 15 39 101
232~ o que implicava que, para um número elevado de electrões, se formariam vários anéis. Os que se situavam
perto da superfície da esfera teriam maior número de electrões. Para diferentes sistemas (átomos) viria: Número de
Electrões51015202530354045505560Electrões
em
cadaanel58101213151616171819202579101213141516161356810111213134578113A partir da semelhança entre
alguns destes sistemas de corpúsculos, Thomson interpretou a semelhança de propriedades de famílias de elementos,
a sua variação ao longo da tabela de Mendeleieve, a diferente electronegatividade dos elementos bem como a
formação de ligações iónicas entre certos átomos. A existência de elementos radioactivos foi interpretada como
resultado da diminuição da velocidade angular dos corpúsculos abaixo de um certo valor, que tornaria o sistema de
corpúsculos instável com emissão de uma parte do átomo. 1. Phil. Mag., S. 6, Vol. 7, No.39, March. 1904 Criada em
25 de Outubro de 2009 Revista em 17 de Dezembro de 2010 Aceite pelo editor em 17 de Dezembro de 2010
31
Modelo atómico de Thomson
32
pH
Referência : Corrêa, C. (2010), WikiCiências, 1(12):0216
[1]
Autor: Carlos Corrêa
[1]
Editor: Jorge Gonçalves
Tanto em Química como na vida corrente é muitas vezes necessário exprimir quantitativamente a acidez e a
basicidade de soluções de um modo fácil, de preferência por um simples número. Assim, em 1909 o bioquímico
dinamarquês Sorensen, para medir a acidez de soluções aquosas diluídas, introduziu uma grandeza denominada pH,
que quantifica a maior ou menor quantidade de H+(aq) existente por litro de solução, através da relação
.
A escala de pH foi introduzida para simplificar a escrita de concentrações expressas por números muito pequenos.
Assim, em vez de [H+(aq)] = 0,000025 mol dm-3 ou 2,5 x 10-5 mol dm-3, é mais prático escrever pH = 4,60.
A 25 ºC, soluções com pH < 7 dizem-se ácidas e soluções com pH > 7 dizem-se alcalinas ou básicas; as soluções
neutras, a 25 ºC, têm pH = 7,0.
Solução
pH
Solução
pH
~ 1,3 - 2,5
Leite
~ 6,6
~2-3
Saliva
~ 6,8 - 7,3
~4
Bílis
~ 7,6 - 8,5
Urina
~ 4,7 - 7,4
Água do mar
~8
Café
~ 5,0
Suco pancreático
~9
Chuva normal
~ 5,6
Amónia
~ 11
Suco gástrico
Vinagre
Sumo de tomate
Tabela 1 - Exemplo de soluções aquosas ácidas e alcalinas (25 ºC).
Em soluções aquosas diluídas (até cerca de 0,1 mol dm-3), a acidez é tanto maior quanto maior for [H+(aq)].
Para soluções mais concentradas, em que a abundância de moléculas de água para solvatar os iões H+ é mais escassa,
os iões H+ encontram-se menos ligados a moléculas de água e a acidez do meio (capacidade para doar protões) é
superior e não pode ser medida pelo pH. Por esta razão é vulgar apresentar-se a escala de pH compreendida entre 0 e
14, pois dada a relação
pH
33
se [H+(aq)] = 1 mol dm-3 = 100 mol dm-3, vem pH = 0. Se [HO–(aq)] = 1 vem [H+(aq)] = 10-14 mol dm-3 e pH =
14.
O pH é medido utilizando eléctrodos de vidro, que deixam de dar resultados aceitáveis quando [H+(aq)] e [HO–(aq)]
se tornam superiores a 0,1 mol dm-3 (pH fora do intervalo 1 - 13)a.
Na definição rigorosa de pH, em vez de concentração, utiliza-se a actividade,
A actividade é uma grandeza termodinâmica que se torna igual à concentração em soluções bastante diluídas. A
acidez e a alcalinidade de soluções de ácidos e de bases muito concentrados é medida por outras funções de acidez,
com as Funções de Acidez de Hammett.
Criada em 20 de Maio de 2010
Revista em 17 de Dezembro de 2010
Aceite pelo editor em 17 de Dezembro de 2010
Campo Eléctrico
Referência : F., M. (2010), WikiCiências, 1(12):0217
Autor: Miguel F.
Editor: Joaquim Agostinho Moreira
[1]
O campo eléctrico é uma realidade física com origem em cargas eléctricas ou em variações temporais de um campo
magnético. No que se segue, apenas será discutido o campo eléctrico com origem em cargas estacionárias.
Quando um corpo electricamente carregado é colocado na região do espaço onde existe um campo eléctrico criado
por um conjunto de cargas estacionárias, esse corpo fica sujeito à acção de uma força eléctrica. Considere-se que
num ponto do espaço onde existe um campo eléctrico se coloca uma partícula carregada positivamente, mas cujo
valor é muito pequeno, designada por carga de teste. Esta condição garante que a carga de prova ou de teste não
influência significativamente a distribuição de cargas que cria o campo eléctrico. Por definição, o campo eléctrico
num ponto do espaço é igual à força eléctrica que actua por unidade de carga positiva colocada nesse ponto, no
limite em que o valor da carga tende para zero:
.
Por simplicidade, iniciaremos a discussão do campo eléctrico criado por uma única carga pontual, Q, colocada na
origem de um referencial. Para caracterizar o campo produzido pela carga Q, utiliza-se uma carga de prova q. A
força eléctrica que actua na carga de prova, quando esta se encontra na posição definida pelo vector de posição , é
dada pela lei de Coulomb, que pode ser formulada matematicamente do seguinte modo:
.
Utilizando a definição operacional apresentada anteriormente, o campo eléctrico criado pela carga pontual Q, num
ponto na posição , é:
,
sendo o versor da direcção definida pelo vector
.
Da expressão anterior podemos concluir que o campo eléctrico criado por uma carga pontual é radial, decai com o
quadrado da distância entre o ponto considerado e a carga criadora de campo, pelo que a sua intensidade é igual em
todos os pontos à mesma distância da carga criadora de campo. O campo eléctrico é centrípeto se a carga criadora for
Campo Eléctrico
34
negativa, e centrífugo se a carga for positiva.
A força de Coulomb obedece ao princípio da sobreposição; isto é, a força que várias cargas exercem sobre uma carga
q é igual à soma vectorial das forças individuais que cada carga do conjunto exerce sobre q. De acordo com a
definição operacional, o campo eléctrico também obedece ao princípio da sobreposição. Matematicamente, o campo
criado por um conjunto de n cargas pontuais é dado por:
.
A expressão matemática que descreve o campo eléctrico criado por uma distribuição contínua de carga é mais
complexa do que esta. Contudo, as ideias de base envolvem o princípio da sobreposição de campo criados por
elementos de carga da distribuição.
No sistema internacional de unidades, o campo eléctrico pode ser expresso newton por coulomb (N/C) ou volt por
metro (V/m). A unidade recomendada é o volt por metro. 1 V/m é a intensidade de um campo eléctrico uniforme tal
que a diferença de potencial entre duas superfícies equipotenciais separadas de 1 m, é 1 V.
Linhas de Campo
Uma maneira útil de representar graficamente o campo eléctrico é através de
linhas imaginárias, paralelas ao vector campo eléctrico em todos os pontos.
Estas linhas têm o nome de linhas de campo. A representação de um campo
eléctrico por linhas de campo permite visualizar a direcção e sentido do
campo eléctrico em cada ponto do espaço, e permite comparar a intensidade
do campo eléctrico em duas regiões do espaço distintas. Ao representar-se um
campo eléctrico através das linhas de campo, a sua densidade espacial deve
ser proporcional à intensidade do campo eléctrico: em zonas onde o campo
eléctrico é mais intenso, as linhas devem estar mais próximas umas das
outras. Para além disso, as linhas nunca se podem cruzar porque nesse caso
haveria uma ambiguidade na determinação do vector campo eléctrico nesse
ponto [ver figura].
Dependência do módulo do campo
eléctrico criado por uma carga pontual na
distância à fonte.
Campo eléctrico criado por uma carga
positiva.
Campo eléctrico criado por uma carga
negativa.
Campo Eléctrico
Carga pontual positiva. As linhas
estendem-se até ao infinito e têm a
mesma direcção e sentido do vector
campo eléctrico em todos os pontos
do espaço.
35
Duas carga pontuais positivas. As
linhas de campo não existem onde o
campo é nulo.
Cargas pontuais de sinais opostos. As
linhas de campo começam na carga
positiva e terminam na negativa.
está bem definido: é o único vector
tangente à linha de campo naquele ponto.
Não é possível definir o vector
porque no ponto em questão as linhas de
campo se cruzam.
Campo Eléctrico em Condutores
Um material condutor caracteriza-se por ter cargas eléctricas que se podem mover sob a acção de um campo
eléctrico aplicado, dando origem a uma corrente eléctrica. Diz-se que um condutor está em equilíbrio electrostático
quando não há movimento organizado de carga, mesmo na presença de um campo eléctrico externo.
No interior de materiais condutores em equilíbrio electrostático, o campo eléctrico é nulo. De facto, enquanto o
campo eléctrico não for nulo no interior do condutor, haverá movimento organizado de carga no sentido de o anular.
Por exemplo, quando um condutor é colocado num campo eléctrico externo, as cargas livres tendem a reorganizar-se
de maneira a anular o campo eléctrico no interior do condutor criando um outro campo eléctrico de intensidade igual
e sentido oposto ao campo eléctrico externo. De acordo com o princípio da sobreposição, na região interna do
condutor os campos somam-se vectorialmente e o resultado é um campo nulo.
Uma vez que o campo eléctrico no interior do condutor é nulo, o seu volume e superfície encontram-se ao mesmo
potencial eléctrico.
O campo eléctrico na superfície de um material condutor em equilíbrio electrostático, é perpendicular a essa
superfície. A condição de se considerar uma situação electrostática é fundamental para se compreender a razão pela
qual o campo só pode ser perpendicular. Se o campo não fosse perpendicular, isto é, se fosse possível decompor o
campo numa componente paralela à superfície do condutor, haveria um movimento de cargas na superfície, o que
contradiz a condição de equilíbrio electrostático.
É possível provar [ver Leitura Recomendada],que o excesso de carga eléctrica num condutor em equilíbrio
electrostático se encontra distribuída na sua superfície externa. Quer isto dizer que não há carga livre no interior do
condutor.
Campo Eléctrico
36
1. Condutor em equilíbrio electroestático; 2. Liga-se um campo eléctrico externo
reorganizam-se e criam um campo
e passa a haver campo no interior do condutor; 3. As cargas
; 4. O movimento de cargas pára quando o campo eléctrico que criam tem o mesmo valor que o campo
eléctrico exterior e o anula no interior do condutor.
Blindagem Electroestática
Um material condutor, que envolve por completo uma dada região do espaço (cavidade) é capaz de a isolar da
influência de campos eléctricos exteriores
. Prova-se que o campo eléctrico no interior de uma cavidade vazia
de um condutor é nulo. Imagine-se que assim não é. Isso significa
que no interior da cavidade podemos traçar linhas de campo com
origem num ponto da superfície da cavidade para outro.
Consideremos um percurso fechado que é contituído por uma das
linhas de campo na cavidade e por um percurso totalmente
inserido np condutor. Uma vez que o campo eléctrico é
conservativo e o campo eléctrico no interior do condutor é nulo, a
existência de linhas de campo na cavidade permite concluir que o
O campo eléctrico numa cavidade de um condutor é
independente do campo eléctrico no seu exterior.
trabalho realizado para transportar uma carga no percuro fechado
definido atrás não é nulo. Assim sendo, o campo eléctrico deve ser
nulo na cavidade vazia do condutor. Para que isso aconteça, as cargas no condutor reorganizam-se de modo a anular
o campo eléctrico externo.
Campo Eléctrico
37
Descontinuidade do Campo Eléctrico numa superfície electricamente
carregada
Considere-se uma superfície electricamente carregada. O vector campo eléctrico num ponto dessa superfície pode ser
decomposto numa componente perpendicular e numa componente paralela à superfície condutora. É possível
mostrar [ver Leitura Recomendada] que a componente perpendicular à
superfície condutora é descontínua, enquanto que a componente paralela é contínua. Isto
quer dizer que se se medir o campo eléctrico nos dois lados da uma superfície carregada,
o valor da componente normal é diferente, e a sua diferença é uma constante que se
relaciona com a densidade superficial de carga da superfície. De facto, é possível mostrar
que
, em que é a densidade superficial de carga.
Exemplos de Campos Eléctricos
1. Campo eléctrico criado por uma esfera maciça de raio R, carregada uniformemente
com carga :
, para
Decomposição do vector
campo eléctrico em duas
componentes:
é
perpendicular à superfície e
,
é paralela.
2. Campo eléctrico criado por uma superfície esférica de raio R
uniformemente carregada, com carga , fora da esfera:
,
3. Campo eléctrico produzido por um filamento rectilíneo de comprimento
ilimitado, com densidade linear de carga constante :
Campo eléctrico criado por uma esfera
maciça carregada com carga Q.
4. Campo eléctrico criado por um plano infinito com densidade superficial de
carga :
, em que
perpendicular ao plano
é o versor que aponta na direcção
Campo eléctrico criado por um filamento
muito comprido.
Campo Eléctrico
38
Leitura Recomendada
1. Alonso, M. e Finn, E. J., Física, Addison Wesley, 1999
2. Purcell, E. M., Electricity and Magnetism, McGraw Hill, 1985
3. Brito, L., Fiolhais, M. e Providência, C., Campo Electromagnético, McGraw Hill, 1999
Criada em 03 de Outubro de 2010
Revista em 28 de Dezembro de 2010
Aceite pelo editor em 28 de Dezembro de 2010
Massa
Referência : de Araújo, M. (2010), WikiCiências, 1(12):0218
Autor: Mariana de Araújo
Editor: Joaquim Agostinho Moreira
[1]
Em ciência, o termo "massa de um corpo" pode referir-se à sua massa inercial, bem como à sua massa gravitacional,
apesar de estar verificado que as duas são equivalentes. Também é comum, no contexto da relatividade restrita,
designar por "massa" a massa em repouso do corpo.
A massa inercial (mi) de um corpo é a constante de proporcionalidade entre a força resultante que actua nele e a sua
aceleração (excepto se considerarmos um sistema de massa variável). A massa inercial pode ser medida através de
um método dinâmico, fazendo colidir frontalmente o corpo com outro em repouso, cuja massa é tomada como
unidade. Temos então que:
.
Medindo as velocidades finais dos dois corpos e conhecida a massa do corpo de referência, podemos calcular a
massa inercial do outro.
A massa gravitacional (mg)é a propriedade dos corpos responsável pela interacção gravítica entre eles, tal como a
carga é reponsável pela interacção eléctrica e magnética. A massa gravitacional é medida através de um método
estático, utilizando uma balança de dois pratos em equilíbrio. Quando os pratos da balança estão equilibrados, a
força gravítica exercida pela Terra em cada corpo colocado nos pratos da balança é igual.
A equivalência entre massa inercial e gravitacional foi observada pela primeira vez por Galileo, ao verificar que
corpos com massas diferentes em queda livre têm a mesma aceleração. Pela segunda lei de Newton e pela lei da
gravitação universal (próximo da superfície da Terra) temos:
pelo que a aceleração de um corpo em queda livre é sempre igual a g se para todos os corpos
. Esta relação
é chamada princípio da equivalência, de grande importância na teoria da relatividade geral. Actualmente a razão
entre massa inercial e gravítica está confirmada com uma precisão de 10-13 [1].
Massa
39
Referências
1. Adelberger, E.G., “New tests of Einstein’s equivalence principle and Newton’s inverse-square law”
Quantum Grav., 18, 2397–2405, (2001).
Criada em 06 de Setembro de 2010
Revista em 28 de Dezembro de 2010
Aceite pelo editor em 28 de Dezembro de 2010
[1]
, Class.
Fontes e Editores da Página
Fontes e Editores da Página
Leis de Kepler Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=22794 Contribuidores: Admin
Leis da dinâmica de Newton Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=5903 Contribuidores: Jamoreir
Campo Eléctrico Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=5922 Contribuidores: Admin
Constante de Avogadro Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=5940 Contribuidores: Jmgoncalves
Efeito fotoeléctrico Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=7122 Contribuidores: Admin
Electrólise Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=7144 Contribuidores: Admin
Reagente limitante Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=7157 Contribuidores: Admin
Modelo atómico de Bohr Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=24111 Contribuidores: Admin
Modelo atómico de Rutherford Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=17094 Contribuidores: Admin
Fórmula de estrutura Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=7158 Contribuidores: Admin
Aminoácido Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=5994 Contribuidores: Admin, Jmgoncalves
Fórmula estereoquímica Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=7159 Contribuidores: Admin
Período de semi-desintegração Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=7184 Contribuidores: Admin
Ligação iónica Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=7160 Contribuidores: Admin
Auto-ionização da água Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=7187 Contribuidores: Admin
Modelo atómico de Thomson Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=17093 Contribuidores: Admin
pH Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=17095 Contribuidores: Admin
Campo Eléctrico Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=6257 Contribuidores: Admin
Massa Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?oldid=7223 Contribuidores: Admin
40
Fontes, Licenças e Editores da Imagem
41
Fontes, Licenças e Editores da Imagem
Ficheiro:Elipse.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Elipse.png Licença: desconhecido Contribuidores: Marianaraujo
Ficheiro:Binario.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Binario.png Licença: desconhecido Contribuidores: Marianaraujo
Ficheiro:Graf.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Graf.png Licença: desconhecido Contribuidores: Jamoreir
Ficheiro:Cpos.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Cpos.png Licença: desconhecido Contribuidores: Jamoreir
Ficheiro:Cneg.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Cneg.png Licença: desconhecido Contribuidores: Jamoreir
Image:Ce5.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Ce5.png Licença: desconhecido Contribuidores: Miguel.cfer
Image:Ce2.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Ce2.png Licença: desconhecido Contribuidores: Miguel.cfer
Image:Ce1.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Ce1.png Licença: desconhecido Contribuidores: Miguel.cfer
Image:Ce8.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Ce8.png Licença: desconhecido Contribuidores: Miguel.cfer
Ficheiro:Ce9.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Ce9.png Licença: desconhecido Contribuidores: Miguel.cfer
Ficheiro:Ce7.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Ce7.png Licença: desconhecido Contribuidores: Miguel.cfer
Ficheiro:Ce6.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Ce6.png Licença: desconhecido Contribuidores: Miguel.cfer
Ficheiro:EsfgrafRR.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:EsfgrafRR.png Licença: desconhecido Contribuidores: Jamoreir
Ficheiro:Ce3.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Ce3.png Licença: desconhecido Contribuidores: Miguel.cfer
Ficheiro:Avogadro.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Avogadro.png Licença: desconhecido Contribuidores: Rmfernandes
Ficheiro:Electrólise - Figura 1.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Electrólise_-_Figura_1.png Licença: desconhecido Contribuidores:
Rmfernandes
Ficheiro:Fórmulas_de_estrutura_de_isómeros_constitucionais-_(a)_etanol;_(b)_metoximetano.png Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Fórmulas_de_estrutura_de_isómeros_constitucionais-_(a)_etanol;_(b)_metoximetano.png Licença: desconhecido
Contribuidores: Rmfernandes
Ficheiro:Figura_2_-_Representação_do_anel_do_benzeno_retirado_de_Kekulé's_Lehrbuch_der_organischen_Chemie_(1861–1867).jpg Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Figura_2_-_Representação_do_anel_do_benzeno_retirado_de_Kekulé's_Lehrbuch_der_organischen_Chemie_(1861–1867).jpg
Licença: desconhecido Contribuidores: Rmfernandes
Ficheiro:Fórmulas_de_Lewis-_(a)_molécula_de_H2_(ligação_covalente_simples);_(b)_molécula_de_O2_(ligação_covalente_dupla);_(c)_molécula_de_N2_(ligação_covalente_tripla).png
Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Fórmulas_de_Lewis-_(a)_molécula_de_H2_(ligação_covalente_simples);_(b)_molécula_de_O2_(ligação_covalente_dupla);_(c)_molécula_de_N2_(lig
Licença: desconhecido Contribuidores: Rmfernandes
Ficheiro:Isómeros_constitucionais-_(a)_etanol;_(b)_éter_dimetílico.png Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Isómeros_constitucionais-_(a)_etanol;_(b)_éter_dimetílico.png Licença: desconhecido Contribuidores: Rmfernandes
Ficheiro:Aminoacido figura 1.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Aminoacido_figura_1.png Licença: desconhecido Contribuidores:
Rmfernandes
Ficheiro:Aminoacido figura 2.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Aminoacido_figura_2.png Licença: desconhecido Contribuidores:
Rmfernandes
Ficheiro:Figura_1_-_Formulas_estereoquímicas_de_um_par_de_enantiómeros.png Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Figura_1_-_Formulas_estereoquímicas_de_um_par_de_enantiómeros.png Licença: desconhecido Contribuidores:
Rmfernandes
Ficheiro:Fórmulas_estereoquímicas_de_isómeros_cis-trans_do_1,2-dicloroeteno.png Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Fórmulas_estereoquímicas_de_isómeros_cis-trans_do_1,2-dicloroeteno.png Licença: desconhecido Contribuidores:
Rmfernandes
Ficheiro:Figura_3_-_Projecção_de_Fisher_da_D-glucose.png Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Figura_3_-_Projecção_de_Fisher_da_D-glucose.png Licença: desconhecido Contribuidores: Rmfernandes
Ficheiro:Figura_4_Projecção_de_Haworth_da_D-glucose.png Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Figura_4_Projecção_de_Haworth_da_D-glucose.png Licença: desconhecido Contribuidores: Rmfernandes
Ficheiro:Figura_5_Projecção_de_Newman_das_conformações_gauche_e_anti_do_butano.png Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Figura_5_Projecção_de_Newman_das_conformações_gauche_e_anti_do_butano.png Licença: desconhecido
Contribuidores: Rmfernandes
Ficheiro:Tempo_de_meia_vida_diagrama_decaimento.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Tempo_de_meia_vida_diagrama_decaimento.png
Licença: desconhecido Contribuidores: Luisspencerlima
Ficheiro:Figura_1_-_Estrutura_cristalina_do_composto_iónico_cloreto_de_sódio_(NaCl)..png Fonte:
http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Figura_1_-_Estrutura_cristalina_do_composto_iónico_cloreto_de_sódio_(NaCl)..png Licença: desconhecido
Contribuidores: Rmfernandes
FICHEIRO:Mat.png Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:Mat.png Licença: desconhecido Contribuidores: Admin
Ficheiro:PH figura.PNG Fonte: http://wikiciencias.casadasciencias.org/wiki/index.php?title=Ficheiro:PH_figura.PNG Licença: desconhecido Contribuidores: Jrpinto
Licença
Licença
Creative Commons - Atribuição - Uso Não Comercial - Partilha nos Mesmos Termos
http:/ / creativecommons. org/ licenses/ by-nc-sa/ 3. 0/
42