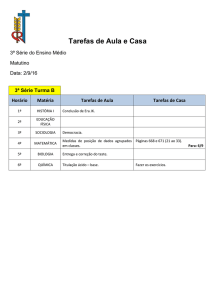
0
10
20
30
40
50
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
ÍNDICE
Índice de Figuras .................................................................................................................... 5
Índice de Tabelas .................................................................................................................... 6
1- ALGARISMOS SIGNIFICATIVOS, ERROS E TRATAMENTO DE DADOS ............. 7
1.1- Algarismos significativos............................................................................................ 7
1.1.1- Algarismos signifivativos na soma e na adição1 .................................................. 8
1.1.2- Algarismos significativos na multiplicação e na divisão1 .................................... 8
1.2- ERROS1....................................................................................................................... 8
1.2.1- ERRO ABSOLUTO............................................................................................. 9
1.2.2- ERRO RELATIVO .............................................................................................. 9
1.3- DESVIO1 ..................................................................................................................... 9
1.4- EXATIDÃO E PRECISÃO1 ..................................................................................... 10
1.5- TIPOS DE ERROS1 .................................................................................................. 10
1.6- REJEIÇÃO DE RESULTADOS1 ............................................................................. 11
1.6.1- Método Q90%. ................................................................................................... 11
1.6.2- Método da Mediana............................................................................................ 12
2- VIDRARIA DE LABORATÓRIO1. ................................................................................ 13
3- LIMPEZA DE VIDRARIA.............................................................................................. 16
4- GRAVIMETRIA1, 2, , . ...................................................................................................... 17
4.1- Solubilidade dos precipitados2, 3. .............................................................................. 20
4.1.1-Efeito do íon comum ........................................................................................... 20
4.1.2- Efeito salino ....................................................................................................... 21
4.1.3- Efeito da formação de complexos ...................................................................... 23
4.1.4- Efeito da concentração do íon hidrogênio (pH) ................................................. 24
4.1.5- Efeito da temperatura ......................................................................................... 27
4.1.6- Efeito da composição do solvente...................................................................... 28
4.1.7- Efeito do tempo .................................................................................................. 29
4.2- Operações da análise gravimétrica1, 3........................................................................ 30
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
4.2.1- Precipitação ........................................................................................................ 30
4.2.2- Filtração.............................................................................................................. 32
4.2.3- Lavagem ............................................................................................................. 32
4.2.4- Dessecação e calcinação de precipitados ........................................................... 34
4.2.5- Esfriamento e pesagem....................................................................................... 36
5- VOLUMETRIA1, 2, 3......................................................................................................... 38
5.1- Classificação dos métodos volumétricos1, 4. ............................................................. 40
5.2- Determinação do ponto final3.................................................................................... 42
5.3- Cálculo dos resultados2 ............................................................................................. 44
5.4- Volumetria de Neutralização2, 3................................................................................. 45
5.4.1- Indicadores de pH1 ............................................................................................. 46
5.4.2- Curvas de neutralização1 .................................................................................... 47
5.5- Volumetria de Precipitação2, 3. .................................................................................. 51
5.5.1- Método de Mohr3 ............................................................................................... 53
5.5.2- Método de Volhard3 ........................................................................................... 54
5.5.3- Método de Fajans2 .............................................................................................. 54
5.6- Volumetria Complexação2, 3...................................................................................... 55
5.6.1- Indicadores Metalocrômicos .............................................................................. 59
5.7- Volumetria de Oxidação-redução1, 2, 3....................................................................... 60
5.7.1- Determinação do Ponto Final............................................................................. 61
5.7.2- Permanganometria.............................................................................................. 64
5.7.3- Química do Iodo: Iodimetria e Iodometria......................................................... 65
6- PARTE EXPERIMENTAL1 ............................................................................................ 70
6.1- Aula Sobre Preparo de Soluções ............................................................................... 70
6.2- Aula Sobre Calibração de Material Volumétrico...................................................... 71
6.3- Aula Sobre Gravimetria (determinação de íons cloretos) ......................................... 72
6.4- Aula Sobre Volumetria de Neutralização (Padronização de uma solução de HCl) .. 73
6.5- Aula Sobre Determinação da Concentração de Ácido Acético no Vinagre.............. 74
6.6- Aula Sobre a Determinação da Concentração de Mg(OH)2 no Leite de Magnésia . 75
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.7- Aula Sobre Determinação do íon Carbonato por Um Ácido Forte (deslocamento) . 76
6.8- Aula Sobre Determinação de íons Cloreto pelo Método de Mohr............................ 77
6.9- Aula Sobre Determinação de íons prata pelo método de Volhard (mét. direto) ....... 78
6.10- Aula Sobre Determinação de Íons Cloreto pelo Método de Volhard (mét. Indireto)
.......................................................................................................................................... 79
6.11- Aula Sobre Determinação de íons Ca2+ no leite ...................................................... 80
6.12- Aula sobre a determinação de peróxido de hidrogênio na Água Oxigernada
(iodometria) ...................................................................................................................... 81
6.13- Aula Sobre Determinação de Ácido Ascórbico por Oxidação-redução (iodimetria)
.......................................................................................................................................... 82
6.14- Aula sobre a Determinação de Fe3+ em medicamentos (oxi-redução).................... 83
7- BIBLIOGRAFIA.............................................................................................................. 84
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Índice de Figuras
Figura 1-Curvas de neutralização de 100 ml de HCl com NaOH de igual molaridade1. ..... 50
Figura 2- Curvas de neutralização de 100 ml de HCl com NaOH de igual normalidade nas
imediações do ponto de equivalência1.......................................................................... 51
Figura 3- Curvas de titulação de um metal M com um ligante L correspondentes à formação
de complexos 1:1 de diferentes estabilidades1. ............................................................ 57
Figura 4- Salto potenciométrico em uma análise volumétrica de oxidação-redução. .......... 63
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Índice de Tabelas
Tabela 1- Valores para Q90%1. ............................................................................................ 12
Tabela 2- Vidraria e material utilizados em laboratório de química analítica...................... 13
Tabela 3- Solubilidade do cloreto de prata e do sulfato de bário em soluções de nitrato de
potássio a 25ºC2. ........................................................................................................... 22
Tabela 4- Solubilidades de cloreto de prata em solução de cloreto de sódio a 25ºC2. ......... 24
Tabela 5- Solubilidade do Sulfato de Bário (BaSO4) em diferentes concentrações de ácido
Clorídrico2. ................................................................................................................... 26
Tabela 6- Variação da solubilidade do sulfato de chumbo na presença do solvente orgânico
etanol. ........................................................................................................................... 28
Tabela 7- Indicadores de pH, sua coloração e zona de transição1, 2. ................................... 47
Tabela 8- Titulação de 100 ml de HCl com NaOH de igual normalidade ........................... 49
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
1- ALGARISMOS SIGNIFICATIVOS, ERROS E TRATAMENTO DE
DADOS1
Todas as medidas físicas possuem um certo grau de incerteza. Quando faz-se uma
medida procura-se manter este grau de incerteza o menor possível, de forma que possua
uma confiabilidade aceitável. Para isto os resultados obtidos devem ser tratados de forma
estatística.
1.1- Algarismos significativos
O valor de uma determinada grandeza pode ser obtido de forma direta
(determinação de massa por pesagem) ou indiretamente (determinação da concentração de
uma solução a partir da massa de soluto e do volume da solução).
Quando fala-se em algarismos significativos, refere-se aos algarismos de um valor
de forma que apenas o último algarismo seja duvidoso.
Imagine um corpo de massa 22,1234g. Quando este é pesado em uma balança cuja
incerteza é de 0,1g a forma correta de expressá-lo é 22,1g. Porém, quando pesado em uma
balança cuja incertez é de 0,0001g, a forma correta de expressá-lo é 22,1234g.
O nº de algarismos significativos é independente do número de casas decimais. Por
exemplo: 1245,4ml possui 5 algarismos significativos, igual a 1,245 litros. Os zeros
também são algarismos significativos quando pertencem ao número, e não quando são
utilizados apenas para expressar casas decimais. Por exemplo: o algarismo 0,0124 possui 3
algarismos significativos; 0,2003 possui 4 algarismos significativos; 1,000 possui 4
algarismos significativos e assim por diante.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
1.1.1- Algarismos signifivativos na soma e na adição1
O nº de algarismos significativos de uma soma ou de uma subtração deve ser igual
ao nº de algarismos significativos que existirem no componente com o menor número de
casas decimais. Na soma: 3,41g + 1,2245g a resposta deve conter apenas 2 algarismos
significativos após a vírgula, ou seja: 4,63g. Na subtração o procedimento é igual. Por
exemplo na subtração: 16,8ml – 2,636ml a resposta deve possuir apenas 1 algarismo
significativo após a vírgula, ou seja 14,2ml.
1.1.2- Algarismos significativos na multiplicação e na divisão1
Nestes casos, o resultado deverá conter tantos algarimos significativos quantos
existirem no componente com o menor nº de algarismos significativo. Por exemplo na
multiplicação: 25,00 x 0,1000. 10-3= 2,500. 10-3; ou ainda 25 x 0,1000. 10-3= 2,5. 10-3.
No caso da realização de operações sucessivas, convén manter um algarimo além do
último algarismo significativo, deixando-se o arredondamento para o resultado final.
A regra para arredondamentos adotada deve ser a seguinte: acima de 5 o nº deve ser
arredondato para cima e até 5 para baixo. Por exemplo: 12,346 ~ 12,35, para cima; 2,323 ~
2,32, para baixo.
1.2- ERROS1
O erro de uma medida pode ser absoluto ou pode ser relativo. Geralmente o erro de
uma análise é expresso em termos relativo.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
1.2.1- ERRO ABSOLUTO
É a diferença entre o valor real e o valor encontrado. Suponha que o valor
verdadeiro de cubre em um dado material seja de 33,30g, porém o resultado de uma análise
acusou 32,90g.
Assim, o erro absoluto é de –0,40g.
1.2.2- ERRO RELATIVO
Considerando os resultados expressados acima, o erro relativo se obtém da seguinte
forma:
33,30
100%
-0,40
X
X = -1,2%
O erro relativo é adimensional, ou seja, não possui unidade.
1.3- DESVIO1
O desvio de uma uma medida, também chamado de erro aparente, é definido pela
diferença entre o valor medido e média.
d i= Xi _ X
Média
Valor
medido
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Lembre-se que a média é a soma dos resultados divididos pelo nº de resultados.
1.4- EXATIDÃO E PRECISÃO1
A exatidão de uma resposta está relacionada com o seu erro absoluto. Ou seja,
quanto mais próximo do valor real, mais exata é a medida.
A precisão, por sua vez, está relacionada com a concordância dos resultados obtidos
entre si. Desta forma, o desvio de uma análise nos fornece sua precisão. Logo, quanto
menor for o desvio padrão de uma análise, maior será sua precisão, mesmo que não
concorde com o valor real.
1.5- TIPOS DE ERROS1
Os erros de uma medida podem ser classificados em dois grupos. Os erros
indeterminados e os erros determinados.
Os erros indeterminados não possuem valor definido, logo não podem ser
eliminados.
Os erros determinados podem ser detectados, medidos e eliminados. Podem ser:
Erro de Método- erro inerente ao método, como a solubilidade dos precipitados, na
gravimetria, ou mesmo a utilização de um indicador inadequado, na volumetria.
Erro Operacional- está ligada a incapacitação do operador, como não remover bem
o precipitado em uma análise gravimétrica.
Erro Pessoal- provém da inaptidão de certas pessoas na diferenciação de cores, por
exemplo, ou ainda, no forjamento de resultados.
Erro Instrumental- proveniente de aparelhos mau calibrados ou reagentes.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
1.6- REJEIÇÃO DE RESULTADOS1
Quando uma análise química é realizada, resultados são obtidos. O analista deve
tratar este resultados para saber se este resultado deve ser rejeitados ou não. Assim, os
resultados obtdos devem ser tratados estatisticamente. Usaremos dois métodos: o método
Q90% e o método da Mediatriz.
1.6.1- Método Q90%.
Este teste rejeita valores críticos com um nível de 90% de confiança. Sua aplicação
é realizada da seguinte maneira.
a. colocar os valores em ordem crescente
b. determinar a diferença existente entre o maior e o menor valor da faixa
c. determinar a diferença existente entre o menor valor e o mais próximo (em módulo)
d. dividir este valor pela diferença da faixa, obtendo-se um valor Q
e. se Q < Q90% (tabela), o valor é aceito, se Q > Q90% o valor é rejeitados
f. se o menor valor for rejeitado, determine a diferenç entre os valore restantes, e em
seguida, testar o maior valor.
g. repetir o processo até o menor e o maior valor serem aceitos.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Tabela 1- Valores para Q90%1.
Número de Resultados (n)
Q90%
2
-
3
0,94
4
0,76
5
0,64
6
0,56
7
0,51
8
0,47
9
0,44
10
0,41
1.6.2- Método da Mediana
Quando se possui de 3 a 5 valores e apenas um valor duvidoso, é aconselhável a
utilizar-se o método da Mediana.
Para determinar o valor por este método, deve primeiramente ordenar os resultados
em ordem crescente. Se o número de resultados for um número ímpar, tomar o valor central
como a Mediana. Caso o número de resultados seja par, deve-se tomar os dois valores
centrais, somá-los e dividi-los por dois (média).
Veja os exemplos abaixo:
a. 4,78; 4,81; 4,89; 4,95; 4,99; a Mediana é= 4,89
b. 35,,44; 35,78; 3581; 36,04; 36,10; 36,19; 36,38; 36,68;
a Mediana é= (36,04 + 36,10)/2= 35,07
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
2- VIDRARIA DE LABORATÓRIO1.
Existe uma relação muita estreita entre o vidro e a química. Na Química Analítica
Quantitativa há uma série de frascos de vidro que possuem grande importância. Este
material pode ser volumétrico ou graduado, sendo que cada um possui uma finalidade
específica. Entre os frascos mais utilizados estão os balões volumétricos, as buretas e as
pipetas.
Abaixo está apresentado o principal material vítrio que é utilizado em Química
Analítica Quantitativa. É importante que não se saiba apenas o nome, mas sua utilização e
modo de operação.
Lembre-se que 1 Litro(l)=1000 mililitros(ml) ou 1,000028 decímetros cúbico (dm3).
Tabela 2- Vidraria e material utilizados em laboratório de química analítica.
BÉQUER
ERLENMEYER
KITASSATO
FUNIL
FUNIL DE BÜCHNER
FUNIL DE FUNDO
ESMERILHADO
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
ALMOFARIZ OU GRAAL
PISTILO
BALÃO VOLUMÉTRICO
0
10
20
BICO DE BUNSEN
30
40
50
TERMÔMETRO
BURETA
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
BASTÃO DE VIDRO
PIPETA GRADUADA
PIPETA VOLUMÉTRICA
45
3
21
6
7
8
9
11
45
3
2
1
6
7
8
9
1
0
CHAPA DE
PERA
Química Analítica Quantitativa
AQUECIMENTO
TRIPÉ
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
3- LIMPEZA DE VIDRARIA2
Toda vidraria utilizada deve estar em perfeitas condições de uso para que os
resultados obtidos nas análises sejam confiáveis. A limpeza deste material é fundamental.
Para saber se o material está limpo, pode ser realizado um teste rápido e fácil. Encha o
frasco com água destilada e esvazie-o logo em seguida. Caso não se forme uma lâmina de
água uniforme, formando gostas nas paredes do frasco, a vidraria não está limpa.
Existem várias formas e métodos de limpeza para vidrarias. Muitos detergentes
comerciais são destinados à limpeza de vidraria de laboratório, inclusive para na remoção
de material radioativo.
Um detergente relativamente brando, porém eficaz, é o Teepol. Uma solução
estoque com a concentração de 10% é comumente preparada em laboratório. Para limpeza,
dilui-se 2 ml desta solução em 50 ml de água destilada.
Outro método muito utilizado é enchendo-se o frasco com uma solução
sulfocrômica (uma solução quase saturada de dicromato de sódio ou de potássio em ácido
sulfúrico). Deixa-se o frasco pernoitar com a solução, após escorre-se bem a solução antes
de lavá-la. Porém evita-se sua utilização devido ao potencial tóxico do cromo, e por este
motivo, não deve ser despejado na pia.
Para obter-se um agente desengordurante de grande eficiência e de ação rápida,
dissolve-se 100g de NaOH em 50ml de água, após esfriar, levar o volumet a um litro com
álcool etílico. Muito cuidado no manuseio desta solução. Esta solução não deve ficar em
contato com a vidraria por longo tempo, pois ataca o vidro.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
4- GRAVIMETRIA1, 2, 3, 4.
Na análise gravimétrica, o constituinte a determinar é separado dos demais
presentes no material em estudo na forma de uma fase pura, que é então pesada. A partir do
peso desta última acha-se o peso do constituinte desejado. A separação do constituinte pode
ser efetuada por meios diversos: precipitação química, eletrodeposição, volatilização ou
extração.
Na gravimetria por precipitação química o constituinte é isolado mediante adição de
um reagente capaz de formar com aquele constituinte uma substância muito pouco solúvel.
O precipitado formado é recolhido em um cadinho filtrante, lavado primeiramente com
água, e depois com etanol, quando possível, secado e pesado. Por exemplo, na
determinação gravimétrica de cloreto, a solução é tratada com nitrato de prata em excesso,
de maneira a assegurar a completa precipitação do constituinte desejado como cloreto de
prata, que é, então, lavado com solução diluída de ácido nítrico, dessecado a 110ºC – 120ºC
e pesado. A a quantidade de cloreto é calculada em função do peso achado de cloreto de
prata.
Nem sempre, o constituinte é pesado sob a mesma forma como foi precipitado. Isto
se deve por, muitas vezes, uma forma de precipitação não representar uma adequada forma
de pesagem, seja por não possuir uma composição definida, seja por não suportar
dessecação por aquecimento. Assim, o ferro (III) pode ser precipitado por hidróxido de
amônio como óxido hidratado, Fe2O3.xH2O, que dessecado, por exemplo, a 100ºC, dá um
resíduo com teor indefinido de água; porém, quando calcinado, é convertido em óxido
anidro, Fe2O3, que se presta para ser pesado. Outro exemplo, é a precipitação de magnésio
como fosfato de amônio e magnésio hexaidratado, NH4MgPO4.6H20. Este precipitado não
é, todavia, uma forma de pesagem muito adequada, posto que ele não pode ser dessecado
por aquecimento, que acarretaria perda de amônia e água de cristalização. A maneira usual
de proceder, no caso, consiste em converter o precipitado, por calcinação, em difosfato de
magnésio, Mg2P2O7, que se presta perfeitamente como forma de pesagem.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
As reações de precipitação, usadas para fins gravimétricos, devem preencher certos
requisitos. Em primeiro lugar, a substância resultante deve ser tão pouco solúvel, nas
condições em que se efetua a precipitação, que esta pode ser considerada como
praticamente completa. A reação de precipitação é tanto mais satisfatória quanto maior sua
seletividade. Uma maior seletividade significa que a precipitação estará menos sujeita a
interferências. O precipitado deve ser facilmente filtrável e lavável. Um precipitado
constituído de grandes partículas pode ser recolhido sobre um filtro bastante poroso e a
filtração é rápida; porém, um sólido finamente dividido requer um filtro denso e,
conseqüentemente, a filtração será lenta. Finalmente, o precipitado deve ser ele próprio
uma forma de pesagem ou, então, fácil e completamente conversível em uma forma de
pesagem adequada.
A forma de pesagem, por sua vez, também tem de atender certos requisitos.
Obviamente, ela deve possuir composição química perfeitamente definida. É desejável que
a substância a ser pesada possa ser precipitada a uma temperatura relativamente baixa e que
a forma de pesagem seja estável a temperaturas elevadas. Desta forma a conversão do
precipitado na forma de pesagem poderá ser realizada mediante aquecimento sem controle
crítico da temperatura. O precipitado na sua forma de pesagem não deve ser
apreciavelmente higroscópica (reter água). Por último, é conveniente que uma pequena
quantidade do constituinte a determinar origine uma quantidade relativamente grande da
forma de pesagem; quanto menor a razão do peso do constituinte para o da forma de
pesagem tanto mais sensível o método.
Os métodos gravimétricos, em virtude da natureza das operações que eles
envolvem, são, em geral, de execução laboriosa e demorada. Além disso, a carência de
reagentes precipitantes específicos, ou mesmo bastante seletivos, faz com que,
freqüentemente, a precipitação do constituinte desejado tenha de ser precedida da separação
prévia de substâncias interferentes. De qualquer maneira, os métodos gravimétricos
conservam um grande valor como métodos finais de referência, suposto que permitam
separar quantitativamente o constituinte sob a forma de uma fase sólida pura. Os métodos
gravimétricos são, em muitos casos, insubstituíveis quando se requer uma elevada exatidão.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Não é raro um nível de exatidão correspondente a um erro relativo de 0,1%. A faixa de
aplicabilidade da análise gravimétrica é limitada pela sensibilidade da balança analítica. A
análise gravimétrica se aplica à determinação dos constituintes maiores ou moderados (0,1
a 100%).
Cálculo dos resultados na análise gravimétrica
Os cálculos na análise gravimétrica são muito simples. A percentagem em peso de
um constituinte em uma amostra é dada por
P "
q
! 100
Q
em que P é a percentagem do constituinte, q o peso do constituinte achado na determinação
e Q o peso da amostra tomada para a análise.
Às vezes, o constituinte é pesado exatamente na forma química em que o resultado
deve ser expresso. Então, bastará entrar com o peso do constituinte, diretamente achado, na
equação anterior. Por exemplo, o cálcio pode ser gravimetricamente determinado mediante
precipitação como oxalato de cálcio, seguida de conversão do precipitado, por calcinação,
em óxido de cálcio e pesagem do resíduo. Se o resultado tiver de ser expresso em CaO, o
peso do resíduo da calcinação dá, imediatamente, o valor de q.
Muitas vezes, porém, a forma de pesagem não coincide com a forma química usada
para expressar o resultado. O constituinte é pesado na forma de um composto que o contém
em uma razão conhecida constante. Em certas determinações gravimétricas, o constituinte
procurado não mais aparece na forma de pesagem. Assim, é possível determinar oxalato
mediante precipitação como oxalato de cálcio e pesagem como óxido de cálcio. Como um
íon-grama de oxalato dá uma molécula-grama de oxalato de cálcio, que, por sua vez, se
transforma em uma molécula-grama de óxido de cálcio, conhecendo-se a massa de óxido de
cálcio sabe-se a massa de oxalato.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
4.1- Solubilidade dos precipitados2, 3.
4.1.1-Efeito do íon comum
Em geral, a solubilidade de um sal pouco solúvel é menor em uma solução contendo
um íon comum do que em água pura. O efeito de diminuição da solubilidade de um sal
pouco solúvel em presença de um íon comum na solução é denominado efeito do íon
comum. O efeito do íon comum é facilmente explicado à luz do princípio do produto de
solubilidade.
Consideremos, por exemplo, uma solução de cloreto de prata em equilíbrio com a
fase sólida. As concentrações dos íons cloreto e prata na solução saturada são reguladas
pelo produto de solubilidade do cloreto de prata. O produto de solubilidade, em termos de
concentração, do cloreto de prata é dado por
K 's " #Ag & $#Cl % $ " 1,7 !10 %10
No caso de uma simples solução saturada de cloreto de prata, tem-se:
[Ag+] = [Cl -] = 1,7. 10-10 = 1,3. 10-5
portanto, a solubilidade do cloreto de prata em água pura é igual a 1,3. 10-5 moles por litro
ou 1,3. 10-5 X 143,3 = 0,001862 g por litro. Se for adicionado nitrato de prata à solução
saturada de cloreto de prata, aumentará a concentração do íon Ag+ e, conseqüentemente, o
produto das concentrações dos íons sobrepassará momentaneamente o produto de
solubilidade do cloreto de prata. O equilíbrio do sistema será reestabelecido com a
precipitação de tanto cloreto de prata quanto for necessária para que o produto das
concentrações dos íons Cl- e Ag+ volte a 1,7. 10-10. Efeito semelhante seria exercido, por
exemplo, pela adição de cloreto de sódio. A diminuição da solubilidade do cloreto de prata
pelo efeito do íon comum pode ser facilmente calculada. Calculemos, por exemplo, a
solubilidade do cloreto de prata em AgNO3 10-4 M.
#Cl $
%
"
1,7 ! 10 %10
" 1,7 ! 10 % 6
%4
10
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
A solubilidade do cloreto de prata baixa, pois, para 1,7. 10-6 X 143,3 = 0,0002436g
por litro, isto é, cerca de seis vezes menor do que em água pura. A solubilidade diminui
cerca de 60 e 600 vezes em AgNO3 10-3 e 10-2 M, respectivamente.
Nos cálculos acima desenvolvidos, não foi levada em conta a concentração de íon
Ag+ fornecida pelo próprio cloreto de prata. A rigor, porém, é preciso considerá-la.
A repressão da solubilidade dos sais poucos solúveis pelo efeito do íon comum tem
uma grande importância na análise gravimétrica por precipitação química. Os processos
gravimétricos fazem sempre uso de um excesso de agente precipitante. Com isso reduz-se,
em geral, a solubilidade do precipitado a um nível negligenciável. Considerado
isoladamente o efeito do íon comum, poderia parecer conveniente usar o maior excesso
possível do agente precipitante para tornar tanto mais quantitativa a precipitação, todavia,
podem ocorrer efeitos negativos paralelos (efeito salino, efeito da formação de complexos),
que recomendem uma certa limitação do excesso. Em regra, o excesso de reagente é limitado a concentrações de 0,01 a 0,05 M. Excepcionalmente, são usados excessos maciços,
como na precipitação de dodecamolibdofosfato de triamônio.
O efeito do íon comum encontra, ainda, aplicação na lavagem de precipitados.
Muitos precipitados não podem ser lavados com água simplesmente, em virtude das
consideráveis perdas por solubilidade que sofreriam. A dificuldade é contornada, quando se
trata da determinação de um cátion, mediante lavagem do precipitado com uma solução
diluída de um sal de amônio possuindo um íon comum com o precipitado.
4.1.2- Efeito salino
A solubilidade de um sal pouco solúvel em presença de eletrólitos não contendo
íons comuns é maior do que em água pura. A tabela a seguir dá as solubilidades do cloreto
de prata e do sulfato de bário em água e soluções de nitrato de potássio a 25ºC. O aumento
da solubilidade do sal pouco solúvel com a elevação da concentração do eletrólito constitui
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
o efeito salino. Ele é devido à variação dos coeficientes de atividade com a força iônica da
solução.
Tabela 3- Solubilidade do cloreto de prata e do sulfato de bário em soluções de nitrato de potássio a
25ºC2.
Molaridade da
AgCl
BaSO4
soIução de KNO3
S (moles/l X 105)
S/S0
S (moles/l X 105)
S/S0
(0)
1,278 (So)
1,00
0,96(S0)
1,00
0,001
1,325
1,04
1,16
1,21
0,005
1,385
1,08
1,42
1,48
0,01
1,427
1,12
1,63
1,70
0,036
—
—
2,35
2,45
A solubilidade de um sal pouco solúvel aumenta com a presença de outros
eletrólitos sem íons comuns presentes na solução. À medida que aumenta a força iônica do
meio diminui o coeficiente de atividade, de modo que a solubilidade deve aumentar a fim
de que o produto de solubilidade Kps possa permanecer constante. Contudo, se a força
iônica se tornar suficientemente grande, o coeficiente de atividade atravessará por um
mínimo e, em seguida, aumentará, paralelamente, a solubilidade de um sal pouco solúvel
até atingir um máximo.
Embora tenhamos considerado o efeito salino exercido por eletrólitos não possuindo
íon comum com o sal pouco solúvel, deve-se observar que ele também é exercido por
eletrólitos tendo íon comum. Porém, então, o efeito salino é, geralmente, superado pelo
efeito contrário do íon comum.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
4.1.3- Efeito da formação de complexos
Conforme foi assinalado no estudo do efeito do íon comum, o uso de um excesso de
precipitante, além de certo limite, pode tornar-se prejudicial em relação à solubilidade do
precipitado. Afora o efeito salino sobre a solubilidade dos precipitados, há ainda a
considerar a possibilidade de os precipitados formarem íons complexos com um excesso de
seus próprios íons.
Um caso extremo de precipitado, formando com excesso de reagente um íon
complexo solúvel, é dado pelo cianeto de prata. Quando se adiciona cianeto a uma solução
de nitrato de prata precipita cianeto de prata: Ag+ + CN-
AgCN. O precipitado dissolve-
se em excesso de cianeto originando íon dicianoargentato: AgCN + CN- ! [Ag(CN)2]-. O
íon dicianoargentato é um complexo muito estável e, praticamente, todo excesso de cianeto
converte-se em dicianoargentato.
Em geral, quando o precipitado está sujeito a um efeito de formação de complexo, o
excesso de precipitante, primeiramente, reprime a solubilidade pelo efeito do íon comum.
Porém, depois de um certo limite, o excesso de precipitante pode fazer com que o efeito da
formação de complexo supere a repressão da solubilidade. Então, aparece uma solubilidade
mínima para um certo excesso de íon comum e, depois disso, a solubilidade passa a aumentar com excesso maior do precipitante.
A tabela a seguir dá as solubilidades de cloreto de prata em soluções de cloreto de
sódio, com diferentes concentrações, a 25ºC. A solubilidade mínima do cloreto de prata é a
que corresponde a uma solução de cloreto de sódio aproximadamente 0,004 M. O aumento
da solubilidade do cloreto de prata, em soluções de cloreto de sódio com concentrações
mais elevadas, deve-se à formação de íon dicloroargentato: AgCl + Cl - ! AgCl2-.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Tabela 4- Solubilidades de cloreto de prata em solução de
cloreto de sódio a 25ºC2.
NaCl (M)
AgCI (10-5 M)
(0)
1,33
0,0039
0,072
0,0092
0,091
0,0176
0,131
0,0365
0,189
0,0884
0,361
0,3556
1,74
0,5112
2,80
0,9747
8,06
4.1.4- Efeito da concentração do íon hidrogênio (pH)
O efeito da concentração do íon hidrogênio sobre a solubilidade dos sais pouco
solúveis depende da constante de ionização do ácido de que deriva o sal e do produto de
solubilidade do próprio sal.
No caso de sais pouco solúveis derivados de ácidos fortes, o efeito da presença de
um ácido sobre a solubilidade é comparável ao de um qualquer outro eletrólito indiferente e
se resume ao efeito salino. Assim, o efeito do ácido nítrico sobre a solubilidade do cloreto
de prata é mais ou menos o mesmo que o exercido pelo nitrato de potássio.
Quando o sal pouco solúvel deriva de um ácido fraco, então o efeito de um ácido
forte sobre a solubilidade é muito grande, embora se deva assinalar o papel importante
exercido pelo valor do produto de solubilidade do sal. A ação dissolvente do ácido é tanto
maior quanto menor a constante de ionização do ácido fraco e quanto maior o produto de
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
solubilidade. São solúveis em ácidos minerais todos os carbonatos e a maior parte dos
fosfatos, arsenatos, cromatos, fluoretos, sulfetos e oxalatos.
É fácil relacionar a solubilidade de um sal pouco solúvel, para uma certa
concentração de íon hidrogênio, com o produto de solubilidade do sal e a constante de
ionização do ácido. Examinemos o caso do oxalato de cálcio. A solução saturada de oxalato
de cálcio envolve o sistema:
Ca2+ + C2O42-
CaC2O4(s)
O produto de solubilidade é K, = 2 X 10-9 (25ºC). A solubilidade do oxalato de
cálcio aumenta com a elevação da concentração do íon hidrogênio devido a remoção de
íons C2O42- do sistema; os íons C2O42- são convertidos em íons HC2O4- e, em soluções de
acidez mais alta, em moléculas H2C2O4. Suponhamos que se queira achar a solubilidade do
oxalato de cálcio em pH igual a 4. Para uma solução saturada tem-se a seguinte relação:
[Ca2+] = [C2O42-] + [HC2O4-].
Além disso, podemos escrever
#H $#C O $
"
#HC O $
%2
4
"
2
K2
2
%
4
" 6 ! 10 %5
Para pH= 4, tem-se, então:
[H+] = 10-4
e
[HC2O4-] = 1,7 [C2O4-].
Agora, combinando as equações anteriores, acha-se
[Ca2+] = 2,7 [C2O42-]
Por outro lado,
Ks = [Ca2+] [C2O42-] = 2,0. 10-9
Finalmente, combinando as duas últimas equações, teremos:
[Ca2+]2 = 2,7 Ks
donde,
[Ca2+] =
Química Analítica Quantitativa
K s = 7,0. 105
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Posto que
K s representa a solubilidade do oxalato de cálcio em água pura (pH=
7), conclui-se que a elevação da acidez da solução para pH= 4 aumenta a solubilidade do
precipitado 1,6 vezes.
Na determinação gravimétrica de cálcio, o oxalato de cálcio é precipitado com um
excesso de oxalato, havendo então uma repressão muito grande da solubilidade. A
precipitação é quantitativa ainda ao pH= 4, por exemplo, em presença de ácido acético
diluído, mas a precipitação deixa de ser completa quando a solução contém mesmo que
quantidades relativamente pequenas de ácido mineral. Contrastando com o comportamento
do oxalato de cálcio (Ks= 2,0. 10-9), tem-se, por exemplo, o oxalato de Cério (III) (Ks = 2,6
X 10-29), que pode ser precipitado em presença de ácido clorídrico em virtude do valor
extremamente baixo de seu produto de solubilidade.
A precipitação de fosfato de amônio e magnésio e da maior parte dos fosfatos já não
é quantitativa nem mesmo em solução apenas fracamente ácida; a precipitação deve ser
efetuada em meio fracamente alcalino (amoniacal). São, entretanto, precipitáveis em meio
nitidamente ácido os fosfatos de zircônio, háfnio e titânio.
A ação dissolvente dos ácidos é considerável mesmo no caso dos sulfatos pouco
solúveis. Os sulfatos de bário, estrôncio e chumbo são mais solúveis em soluções de ácidos
fortes do que em água. Eis, por exemplo, alguns dados sobre a solubilidade do sulfato de
bário em ácido clorídrico:
Tabela 5- Solubilidade do Sulfato de Bário (BaSO4) em diferentes
concentrações de ácido Clorídrico2.
HCl M
BaSO4 (mg/100mL)
0,00
0,40
0,10
1,00
0,30
2,90
0,50
4,70
1,00
8,70
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
O aumento da solubilidade pode ser atribuído apenas parcialmente ao efeito salino.
O ácido sulfúrico é um ácido forte com referência ao primeiro hidrogênio, mas não mais
do que moderadamente com referência ao segundo (K2 = 1,2. 10-2). Por conseguinte,
em uma solução ácida saturada de sulfato de bário, coexistem os seguintes equilíbrios:
BaSO4(s)
SO4-2 + H+
Ba2+ + SO4-2
HSO4-
O aumento da acidez do meio favorece a formação de íons HSO4- e, portanto, torna
maior a solubilidade do sulfato de bário. Não obstante, o sulfato de bário pode ser
convenientemente precipitado em meio contendo ácido clorídrico em concentração não
superior a certo limite (0,05 M), pois o efeito dissolvente do ácido é compensado pelo
efeito do íon comum do excesso de precipitante.
É muito importante, na análise gravimétrica, conhecer o efeito da concentração do
íon hidrogênio sobre a solubilidade dos precipitados. Em geral, a precipitação de um sal
pouco solúvel é efetuada ao mais baixo pH em que a mesma possa ainda ocorrer
quantitativamente. Isso porque nos processos gravimétricos as interferências são quase
sempre mais numerosas à medida que se tende para a região alcalina.
4.1.5- Efeito da temperatura
Para a maioria das substâncias sólidas, o calor requerido para afastar as moléculas
do soluto é maior do que o calor desprendido pela solvatação do soluto. Desta forma, o
processo de dissolução é acompanhado de absorção de calor. De acordo com o principio de
Le Chatelier, a solubilidade de tais substâncias aumenta com a temperatura. Há algumas
substâncias, a exemplo do cloreto de sódio, para as quais os dois mencionados efeitos quase
que se equilibram; neste caso, a solubilidade varia pouco com a temperatura. Finalmente,
umas poucas substâncias, como sulfato de cálcio, têm sua solubilidade reprimida com a
elevação da temperatura.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Quando possível a filtração e a lavagem dos precipitados, na análise gravimétrica,
são convenientemente conduzidas a quente. A filtração e a lavagem se processam mais
rapidamente a quente, em virtude da diminuição da viscosidade das soluções. Por outro
lado, a lavagem a quente é mais eficiente. A possibilidade de efetuar a filtração e a lavagem
a quente depende da solubilidade do precipitado e do efeito da temperatura sobre a
solubilidade. O sulfato de bário, por exemplo, é pouco solúvel e a solubilidade aumenta
apenas moderadamente com a temperatura, de sorte que a filtração e a lavagem podem ser
realizadas a quente. No caso do cloreto de prata, também pouco solúvel, mas cuja
solubilidade aumenta grandemente com a elevação da temperatura, a filtração e a lavagem
são feitas à temperatura ambiente. Há precipitados tão pouco solúveis, como são os
hidróxidos de ferro (III), alumínio e cromo, que a filtração e a lavagem podem ser efetuadas
a quente sem consideração do efeito relativo da temperatura. Às vezes, o decréscimo da
solubilidade com a diminuição da temperatura é usado para reduzir as perdas na lavagem de
precipitados relativamente solúveis. Serve de exemplo o fosfato de amônio e magnésio, que
pode ser lavado com solução apropriada devidamente refrigerada.
4.1.6- Efeito da composição do solvente
Geralmente os sais inorgânicos são marcadamente menos solúveis em solventes
orgânicos do que em água pura. A solubilidade de um sal em uma mistura de dois solventes
se situa, freqüentemente (mas, nem sempre), entre as solubilidades nos solventes isolados.
Os dados abaixo mostram a diminuição da solubilidade do sulfato de chumbo em misturas
de água-etanol com a crescente concentração de etanol.
Tabela 6- Variação da solubilidade do sulfato de chumbo na presença do solvente orgânico etanol.
Etanol (%)
0
10
20
30
40
50
60
70
PbSO4 (mg/l)
45
17
6,3
2,3
0,77
0,48
0,30
0,09
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
A adição de solventes orgânicos miscíveis com água é, às vezes, usada na análise
gravimétrica, para tornar praticamente completa a precipitação de substâncias relativamente
solúveis em meio aquoso. Assim, o sulfato de cálcio precipita quantitativamente em mistura
de água-etanol com 50% do solvente orgânico. Quando se faz uso da adição de solvente
orgânico para diminuir a solubilidade de um precipitado, é preciso ter em conta a possibilidade de que também precipitem outros sais além do desejado.
Outras vezes, recorre-se a um solvente orgânico para separar certos componentes
em uma mistura. A determinação gravimétrica de potássio como KCIO4 faz uso de
solventes orgânicos. Os percloratos de potássio, rubídio e césio são muito pouco solúveis
em etanol, n-butanol e acetato de etila, ao passo que os de tálio e amônio são
moderadamente solúveis e os dos demais metais, solúveis. A determinação de potássio se
baseia na fraca solubilidade do perclorato de potássio e na fraca solubilidade do perclorato
de sódio nos mencionados solventes orgânicos. A técnica mais empregada consiste em
extrair os percloratos anidros com uma mistura de n-butanol e acetato de etila em partes
iguais. O método requer a ausência de sulfato, pois o sulfato de sódio é insolúvel nos
solventes orgânicos.
4.1.7- Efeito do tempo
O tempo não tem efeito sobre um verdadeiro equilíbrio. Entretanto, a velocidade
com que a reação alcança o estado de equilíbrio é uma importante consideração a ser feita
quanto à formação de certos precipitados. Nenhuma conclusão se pode tirar a respeito da
velocidade de uma reação partindo da constante de equilíbrio. Às vezes, uma reação com
constante de equilíbrio favorável somente alcança a condição de equilíbrio a uma
velocidade quase imperceptível.
As reações de precipitação, usadas na análise gravimétrica, são muitas vezes lentas,
requerendo muitos minutos e mesmo horas para se completarem. Em regra, a filtração é
realizada uma a dez horas após a adição dos reagentes precipitantes. Alguns precipitados
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
gravimétricos apresentam acentuada tendência para a formação de soluções supersaturadas.
São exemplos, o fosfato de amônio e magnésio, o dodecamolibdofosfato de triamônio e o
acetato de sódio magnésio (ou zinco) e uranilo. Então, o tempo necessário para assegurar
uma completa precipitação pode ser maior do que o usual. Em certos casos, a precipitação é
acelerada mediante vigorosa agitação da solução por algum tempo.
Ocasionalmente, uma lenta velocidade de reação serve de base para a separação de
substâncias com solubilidades comparáveis. Por exemplo, o cálcio e o magnésio podem ser
separados mediante precipitação do primeiro como oxalato. Na verdade, tanto o oxalato de
cálcio como o oxalato de magnésio são pouco solúveis. Entretanto, o oxalato de cálcio
precipita rapidamente, ao passo que o equilíbrio da formação do oxalato de magnésio é
alcançado muito lentamente. Assim sendo, o oxalato de cálcio, se filtrado imediatamente
após sua precipitação, é essencialmente livre de contaminação com magnésio.
4.2- Operações da análise gravimétrica1, 3.
4.2.1- Precipitação
As reações de precipitação para fins gravimétricos costumam ser realizadas em
copos que são recipientes com forma apropriada para a condução subseqüente da filtração.
A precipitação é, comumente, efetuada mediante adição de uma solução contendo o
reagente precipitante. Na técnica da precipitação em meio homogêneo, o precipitante é
gerado gradualmente no seio da própria solução.
Em certos casos é conveniente que a solução do precipitante seja adicionada
lentamente e com constante agitação da solução em análise. Com isso, tem-se em vista
diminuir tanto quanto possível uma excessiva concentração local do reagente, condição que
favoreceria uma maior contaminação do precipitado. Favorece, ainda, a formação de um
número menor de núcleos, que pelo crescimento de Ostwald, resultará em precipitados
maiores. A solução do reagente pode ser adicionada, eventualmente, de um copo ou
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
proveta, mas se a adição deve ser feita gradualmente, gota a gota, convém fazê-lo com o
auxílio de pipeta ou bureta. Em qualquer caso, para evitar respingos, a solução do
precipitante é deixada escoar cuidadosamente pelas paredes do copo. Comumente, as
precipitações são efetuadas a quente; trata-se de uma condição favorável à formação de um
precipitado facilmente filtrável, composto de grandes cristais e a uma menor contaminação
do precipitado. Não há necessidade, entretanto, de levar o aquecimento até à ebulição; o
mais indicado é adicionar o precipitante à solução aquecida levemente abaixo do ponto de
ebulição, pois, assim, não se corre o risco de perda provocada por uma brusca evolução de
vapor em conseqüência de ruptura de uma condição de superaquecimento. Enquanto a
solução é agitada evita-se que o bastão de vidro atrite contra as paredes ou o fundo do copo
para não riscar o vidro.
Um considerável excesso de precipitante é invariavelmente usado nas precipitações.
Todavia, em geral, evita-se um excesso demasiadamente grande, que poderia aumentar a
solubilidade ou a contaminação do precipitado. Às vezes pode-se formar uma idéia bastante
aproximada da quantidade requerida do precipitante com base no peso da amostra tomada
para a análise. Depois que o precipitado tenha sedimentado adicionam-se mais algumas
gotas da solução do reagente para verificar se ainda ocorre alguma precipitação.
A maioria dos precipitados não é recolhida por filtração imediatamente após a
formação. É que o equilíbrio de solubilidade não é atingido logo, particularmente quando se
trata de substâncias tendentes a formar soluções supersaturadas. O tempo entre a
precipitação e a filtração varia grandemente com os diferentes casos. Às vezes, a filtração
pode ser efetuada sem demora e, outras vezes, é realizada somente depois de uma a quatro
horas ou mais. Quando é requerida uma demora maior, uma boa prática é a que consiste
realizar a precipitação à tarde e deixar o precipitado em repouso durante a noite. Nos casos
em que se manifesta a tendência para a formação de soluções supersaturadas, pode ser
necessário agitar a solução, mas, então, deve-se cuidar para não atritar o bastão contra as
paredes e o fundo do copo. Certos precipitados são obtidos como partículas maiores e mais
facilmente filtráveis quando deixados em contato com a solução-mãe durante algum tempo.
Além disso, o crescimento, e o aperfeiçoamento dos cristais são, por vezes, acompanhados
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
de uma sensível diminuição da contaminação do precipitado por coprecipitação. Estas são
as razões pelas quais se recomenda, muitas vezes, digerir o precipitado, antes da filtração,
deixando-o em contato com a solução sobre o banho de vapor durante horas. Outras vezes,
um semelhante procedimento não traz nenhuma vantagem e, portanto, não há motivo para
usá-lo. Por outra parte, há mesmo casos em que o contato do precipitado com a solução por
algum tempo seria positivamente prejudicial (posprecipitação).
Quando a solubilidade do precipitado é apreciável, pode ser necessário esfriar a
solução à temperatura ambiente antes de iniciar a filtração.
4.2.2- Filtração
A filtração é a operação, comumente usada na análise gravimétrica, para recolher o
precipitado, separando-o tanto quanto possível da solução em que teve origem. Os meios de
filtração usuais são o papel e os cadinhos filtrantes (de Gooch, de vidro sinterizado e de
porcelana porosa). O uso dos diferentes meios de filtração depende da natureza do
precipitado e do tratamento posterior a que este terá de ser submetido.
Sempre que a solubilidade e a natureza química do precipitado o permitam, a
filtração é efetuada com vantagem a quente. A quente, a viscosidade das soluções é menor
e, conseqüentemente, a filtração mais rápida do que a frio.
4.2.3- Lavagem
A lavagem é uma operação por meio da qual se procura livrar o precipitado, após a
filtração, das substâncias presentes na pequena porção de solução retida mecanicamente
pelas partículas do precipitado. Antes de abordar os aspectos técnicos da operação, é
importante considerar o problema da escolha do líquido de lavagem.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
A espécie de líquido de lavagem a usar depende, fundamentalmente, da natureza do
precipitado e, em menor extensão, das substâncias presentes na solução. Os principais
fatores a considerar são a solubilidade e a peptização (absorção de água) do precipitado.
Quando o precipitado é apreciavelmente solúvel em água, não é possível lavá-lo com água,
simplesmente. Se o caso envolver a determinação de um cátion, pode-se resolver a
dificuldade lavando o precipitado com uma solução diluída de um sal de amônio possuindo
um íon comum com o precipitado. A parte do sal de amônio retida pelo precipitado, após a
lavagem, é volatilizada mediante aquecimento a baixa temperatura. Assim, o precipitado de
oxalato de cálcio, na determinação gravimétrica de cálcio, é lavado com uma solução
diluída do oxalato de amônio. Em certos casos a solubilidade em água é diminuída por
adição de etanol. Outro recurso, às vezes cabível, é o uso de uma solução saturada da
substância pouco solúvel em água como líquido de lavagem. Quando o precipitado é
hidrolisável, faz-se uso de um líquido de lavagem contendo uma substância que reprima a
hidrólise; por exemplo, o precipitado de fosfato de amônio e magnésio hexaidratado, na
determinação de magnésio ou de fosfato, é lavado com solução diluída de hidróxido de
amônio para impedir a hidrólise do precipitado a hidrogenofosfato de magnésio. Às vezes,
é necessário evitar a hidrólise de uma substância presente na solução para que não haja
contaminação do precipitado; por exemplo, para que não ocorra a hidrólise de sais de ferro
(III) a sais básicos, na determinação gravimétrica de sílica, usa-se como líquido de lavagem
uma solução diluída de ácido clorídrico. Certos precipitados, embora muito pouco solúveis,
não podem ser lavados simplesmente com água, em virtude da ação peptizante desta, que
faz com que as partículas do precipitado se dispersem atravessando o filtro em estado
coloidal. A peptização ocorre com precipitados de origem coloidal que tenham sido
floculados pela ação dos eletrólitos presentes na solução. A lavagem com água pura removeria gradativamente os eletrólitos, facilitando a peptização. Esta absorção de água é
impedida realizando a lavagem com uma solução de um eletrólito, que seja facilmente
eliminável no tratamento do precipitado que precede a pesagem. Por exemplo, o precipitado
de hidróxido do ferro (III) é lavado com uma solução diluída de nitrato de amônio. Quando
não há indicação em contrário, o líquido de lavagem é usado quente.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
A lavagem deve ser iniciada tão logo o precipitado tenha sido transferido para o
filtro. Se o precipitado fosse mantido no filtro por algum tempo sem lavar, a sua massa
fenderia e se aglutinaria tornando ineficiente qualquer tentativa de lavagem posterior. A
lavagem do precipitado começa sendo feita por decantação. Um jato do líquido de lavagem
é lançado do frasco lavador contra as paredes do copo obrigando as partículas aderentes a
descerem para o fundo do copo; revolve-se o precipitado e, depois, deixa-se que ele
sedimente. Quando o líquido sobrenadante aparecer claro, verte-se-o no filtro, conservando
o precipitado tanto quanto possível no copo. Em seguida, adiciona-se outra porção do
líquido de lavagem ao copo e repete-se o processo. De cada vez, deixa-se escoar
completamente o líquido do filtro. A operação é repetida 3 a 4 vezes ou quantas forem
julgadas convenientes.
4.2.4- Dessecação e calcinação de precipitados
O problema apresenta-se diferentemente conforme o precipitado tenha sido
recolhido em papel de filtro ou cadinho filtrante.
Calcinação de precipitados recolhidos sobre papel: o papel de filtro e seu conteúdo,
depois de completada a lavagem, são transferidos do funil para um cadinho previamente
pesado. Em virtude da fraca resistência do papel sem cinza quando úmido, a operação
requer bastante cuidado.
O perigo de ruptura é consideravelmente reduzido quando se desseca parcialmente o
papel ainda no funil. Neste caso, cobre-se o funil contendo o filtro com um disco de papel
de filtro comum dobrado em torno da borda do funil e, em seguida, leva-se o conjunto para
uma estufa e desseca-se a 100ºC. A dessecação prévia, sem ser indispensável, pode ser
favorável quando se trata de um precipitado gelatinoso capaz de reter muita água. Quase
sempre um precipitado bem drenado pode ser levado logo para o cadinho a ser usado na
calcinação.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
A calcinação é realizada com cadinho de platina ou porcelana. Os cadinhos de
platina esfriam mais rapidamente do que os de porcelana; além disso, suportam
temperaturas mais altas. Em certos casos é imperioso o uso de cadinho de platina; por
exemplo, na determinação de sílica, em que o resíduo da calcinação tem de ser tratado com
ácido fluorídrico. O uso de cadinho de platina deve ser evitado sempre que haja perigo de
ataque do metal. Os cadinhos de porcelana, de menor custo, encontram largo uso.
Dessecação e calcinação de precipitados em cadinhos filtrantes: os precipitados
recolhidos em cadinhos filtrantes costumam ser livrados completamente da água por eles
retida mediante aquecimento. Em casos isolados, o precipitado é dessecado à temperatura
ambiente mediante eliminação da água com solventes orgânicos voláteis.
Quando a dessecação de um precipitado deve ser efetuada a baixas temperaturas, o
cadinho filtrante é livrado tanto quanto possível da água por sucção e, então, levado para
dentro de um pequeno copo frouxamente coberto com um vidro de relógio para ficar
protegido das poeiras. O copo é colocado em uma estufa elétrica ajustada à temperatura
requerida. Comumente, aplica-se um primeiro período de aquecimento com duração de
uma hora a uma hora e meia. Os subseqüentes aquecimentos até constância de peso
limitam-se a períodos de meia hora.
Quando os precipitados recolhidos em cadinhos filtrantes devem ser aquecidos a
temperaturas mais altas, faz-se uso, preferentemente, de forno de mufla com aquecimento
elétrico. Caso se use uma chama, o cadinho filtrante devidamente coberto é colocado em
um cadinho de porcelana, do tipo comum; os cadinhos filtrantes não são aquecidos
diretamente sobre a chama de um bico de gás. Se o precipitado for úmido, o aquecimento
deverá ser gradual, de início, até que a água seja completamente expulsa.
Há precipitados que, recolhidos em cadinhos filtrantes e tendo sido lavados com
água ou solução aquosa apropriada, podem ser dessecados mediante passagem de
sucessivas porções de etanol e, então, éter ou acetona. Finalmente o cadinho é deixado ao ar
para que evaporem os últimos traços do solvente ou faz-se passar uma corrente de ar pelo
cadinho com a mesma finalidade. O método de dessecação à temperatura ambiente é
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
satisfatório apenas com certos precipitados. Eles não podem ser finamente divididos e que
não adsorvam ou retenham por oclusão quantidades consideráveis de água.
4.2.5- Esfriamento e pesagem
Os precipitados na análise gravimétrica são simplesmente dessecados ou, quando
necessário, convertidos em formas de pesagem adequadas. A pesagem do cadinho com o
seu conteúdo é feita somente após ter sido o conjunto esfriado à temperatura do laboratório
em ambiente seco. A prática usual consiste em esfriar o cadinho com seu conteúdo em
dessecadores hermeticamente fechados e carregados com agentes dessecantes apropriados,
como sílica-gel ou cloreto de cálcio anidro.
O cadinho é levado ainda quente, coberto com a respectiva tampa, para dentro do
dessecador. Se o cadinho tiver sido submetido a forte aquecimento, espera-se que ele esfrie
ao ar abaixo do rubro antes de transferi-lo para o dessecador. Deixa-se a tampa do
dessecador por alguns instantes levemente entreaberta para que o ar no interior do aparelho,
aquecido em contato com o cadinho quente, possa expandir-se para fora. Depois disso,
fecha-se o dessecador e deixa-se o cadinho com seu conteúdo esfriar à temperatura
ambiente e finalmente pesa-se o conjunto.
O tempo de esfriamento do cadinho no dessecador antes da pesagem, não pode ser
especificado de uma maneira geral, pois o tempo requerido depende de material do
cadinho, do tamanho deste e, ainda, da temperatura do cadinho ao dar entrada no
dessecador. Os cadinhos de platina requerem um tempo de esfriamento menor do que os
cadinhos de porcelana. Em geral, os cadinhos de porcelana são mantidos no dessecador por
meia hora antes da pesagem. Após a pesagem, o aquecimento é repetido por 10 a 15
minutos. O aquecimento, o esfriamento e a pesagem são repetidos até alcançar-se
constância de peso. Considera-se atingido o peso constante quando duas sucessivas
pesagens não diferirem em mais do que 0,2 mg. Na transferência do cadinho para a balança,
a operação deve ser sempre realizada com o auxilio de uma tenaz para cadinho.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Ao abrir o dessecador, para dele remover o cadinho, é preciso fazê-lo com o devido
cuidado. A transferência do cadinho ainda quente para o dessecador provoca a expansão do
ar contido no interior deste, uma parte do qual é expulse no intervalo em que a tampa é
mantida ligeiramente entreaberta. O esfriamento posterior causa a contração do ar retido no
dessecador, produzindo-se, assim, uma atmosfera de pressão reduzida. A rápida abertura da
tampa pode dar lugar à entrada de uma brusca corrente de ar no dessecador. A tampa deve
ser aberta cuidadosamente pelo lado oposto àquele em que se acha colocado o cadinho. Não
é recomendável o uso de um dessecador para o esfriamento simultâneo de vários cadinhos.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
5- VOLUMETRIA1, 2, 3.
Na análise volumétrica, o constituinte desejado é determinado medindo sua
capacidade de reação com uma solução de um reagente adequado em concentração
exatamente conhecida. As soluções de concentração exatamente conhecida usadas na
volumetria são chamadas soluções padrões. A solução padrão é livrada progressivamente
para dentro da solução contendo o constituinte até completar-se a adição de uma quantidade
de reagente equivalente à quantidade presente da substância sob determinação. Esta
operação, que é realizada com o auxilio de uma bureta, denomina-se titulação, também
conhecida como titrimetria. A quantidade do constituinte envolvida na determinação é,
finalmente, calculada a partir do volume de solução padrão gasto na titulação.
A etapa crítica de uma titulação é a parte final, em que se procura localizar o ponto
correspondente à adição exatamente suficiente do reagente, conhecido como ponto de
equivalência (PE). Qualquer propriedade que varie bruscamente nas imediações do ponto
de equivalência pode servir, em princípio, para a localização deste. Um meio muito usado é
o que consiste em adicionar ao sistema um reagente auxiliar ou indicador, capaz de
produzir mudança de coloração ou turvação em um ponto em que a reação já se tenha
virtualmente completado. O ponto em que isso ocorre é denominado ponto final da
titulação. O ponto de equivalência e o ponto final não coincidem necessariamente. A
diferença, cuja magnitude depende das propriedades químicas do sistema, constitui o erro
de titulação, que pode ser determinado experimentalmente.
Uma reação química, para que possa ser usada como base de método volumétrico,
deve atender vários requisitos:
a) A reação entre o reagente e o constituinte deve processar-se quantitativamente
segundo a equação estequiométrica. Em outras palavras, não devem ocorrer
reações paralelas.
b) A reação deve ser rápida. Influem na velocidade de reação a natureza e a
concentração dos reagentes, a temperatura, a composição do meio e a possível
presença de catalisadores ou inibidores. As reações de neutralização e de
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
formação de complexos ou compostos pouco ionizados, que envolvem a simples
combinação de íons, são praticamente instantâneas. Alguns precipitados cristalinos se separam bastante lentamente das suas soluções supersaturadas. A precipitação pode ser acelerada, às vezes, mediante a modificação do meio, por
exemplo, com a adição de etanol. Muitas reações de oxidação-redução não têm
lugar
instantaneamente
então,
é
possível,
muitas
vezes,
acelerar
convenientemente a velocidade de reação por meio de aquecimento ou adição de
um catalisador adequado.
c) A reação deve oferecer um meio satisfatório para a localização do ponto final. O
meio usual são os indicadores visuais, porém, às vezes, o ponto final pode ser
achado com métodos baseado em certas propriedades físico-químicas, que
variam bruscamente perto do ponto de equivalência.
As condições acima limitam, evidentemente, o número de reações utilizáveis como
base de métodos volumétricos. Em geral, reações que melhor atendem aquelas condições
são reações reversíveis. São exemplos, as reações de neutralização, que se processam direta
e rapidamente e apresentam a possibilidade de exata determinação do ponto final com
indicadores de pH.
As técnicas volumétricas são mais simples do que as técnicas gravimétricas que
envolvem a formação, a filtração, a lavagem e a dessecação ou a calcinação de precipitados
e, finalmente, a pesagem do resíduo da dessecação ou calcinação (precipitado seco). A
maior simplicidade das técnicas volumétricas torna as análises de execução, geralmente,
mais fácil e rápida. Comumente, a análise volumétrica aplicada na escala de
macroquantidades, porém, em muitos casos, ela também pode ser aplicada com técnica
microanalítica. Na macrovolumetria, a exatidão de muitos métodos alcança o nível de
0,1%.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
5.1- Classificação dos métodos volumétricos1, 4.
Os métodos da análise volumétrica podem ser classificados de acordo com a
natureza das reações em que se baseiam. As reações usadas são dos seguintes tipos gerais:
a) Reações de combinação de íons, que compreendem as reações de neutralização, de
precipitação e de complexação.
b) Reações de oxidação-redução.
Volumetria de neutralização1, 4.
Em principio, a reação de neutralização consiste na combinação de íons H+ e OHcom formação da água. A volumetria de neutralização se aplica à determinação de
substâncias de caráter ácido ou básico. Os ácidos são determinados por titulação com
soluções padrões alcalinas e as bases, mediante titulação com soluções padrões ácidas; no
primeiro caso tem-se a acidimetria e, no segundo, a alcalimetria. A volumetria de
neutralização se estende à titulação de sais de bases muito fracas com solução padrão
alcalina e de sais de ácidos muito fracos com solução padrão ácida (titulações de
deslocamento). Abaixo está apresentada as equações que expressam as reações de
neutralização:
H++ A-
HA
B+ + OH-
BOH.
Volumetria de precipitação1, 4.
Certos cátions e ânions reagem formando compostos pouco solúveis, por exemplo:
Ag+ + ClQuímica Analítica Quantitativa
AgCI.
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Na volumetria de precipitação, o íon a determinar é precipitado por um outro de
carga oposta oriundo da solução padrão. O mais importante método volumétrico de
precipitação é a argentimetria, que utiliza uma solução padrão de nitrato de prata.
Volumetria de formação de complexos ou compostos levemente ionizados1, 4.
Compreende os métodos baseados em reações em que o íon a determinar e um íon
da solução padrão se combinam para formar um íon complexo ou um composto levemente
ionizado, por exemplo:
2CN- + Ag+
Ag(CN)2-
2Cl- + Hg2+
HgCl2.
As reações acima servem de base, a primeira, para a titulação de cianeto com nitrato
de prata, e, a segunda, para a titulação de cloreto com nitrato de mercúrio (II)
(mercurimetria).
Volumetria de oxidação-redução1, 4.
Importantes métodos da análise volumétrica são métodos de oxidação-redução. Um
agente redutor pode ser titulado com um agente oxidante apropriado e vice-versa. Alguns
dos agentes oxidantes mais usados como reagentes volumétricos são: permanganato de
potássio, dicromato de potássio, iodo, iodeto de potássio e bromato de potássio. Agentes
redutores freqüentemente usados são: tiossulfato de sódio, sulfato de ferro (II), trióxido de
arsênio, cloreto de estanho (II) e sais de cromo (II). Os métodos volumétricos de oxidaçãoredução
costumam
ser
denominados
de
acordo
com
o
reagente
empregado:
permanganimetria, dicromatometria, iodometria, iodatometria, etc.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
5.2- Determinação do ponto final3.
Conforme já foi anteriormente assinalado, qualquer propriedade do sistema que
exiba uma variação brusca perto do ponto de equivalência pode servir para acusar o ponto
final da titulação.
O método mais usado é o dos indicadores, que permitem localizar o ponto final
visualmente. O indicador (um dos reagentes, o produto da reação ou uma substância
adicionada propositadamente) é uma substância que acusa o ponto final modificando a
aparência do meio através de uma mudança de coloração ou aparecimento de turvação.
Na permanganimetria, por exemplo, o próprio reagente da solução padrão atua
como indicador. As soluções padrões de permanganato de potássio possuem coloração
violeta intensa, de sorte que o ponto final é acusado pelo aparecimento de coloração rósea
devida a um leve excesso do reagente. Na titulação de cianeto com nitrato de prata, produzse íon dicianoargentato e o ponto final é acusado pelo aparecimento de turvação devida à
formação de dicianoargentato de prata com excesso de nitrato de prata:
Ag(CN)2- + Ag+
Ag[Ag(CN)2].
Comumente, entretanto, o indicador é uma substância propositadamente adicionada
à solução para atuar como tal. Muitos indicadores volumétricos são mais ou menos
específicos. Por exemplo, na titulação de cloreto com nitrato de prata pode-se usar cromato
de potássio como indicador (método de Mohr); o cloreto é precipitado como cloreto de
prata, branco, e um excesso de solução padrão origina um segundo precipitado, de cromato
de prata, vermelho, cujo aparecimento acusa o ponto final. O amido, que dá um produto de
adsorção azul com iodo, é usado como indicador nas titulações baseadas no emprego de
uma solução padrão de iodo.
Há, por outro lado, grandes classes de indicadores, com mecanismos de
funcionamento comuns para cada tipo de reação. Assim, a volumetria de neutralização faz
uso de indicadores de pH, que são substâncias orgânicas, de caráter ácido ou básico, com a
propriedade de mudar de coloração dentro de uma estreita faixa de pH. Na volumetria de
precipitação, têm-se os indicadores de adsorção. São substâncias orgânicas que,
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
sobrepassado o ponto de equivalência, passam da solução para a superfície do precipitado,
causando o descoramento da solução e, ao mesmo tempo, a mudança de cor do precipitado.
Por exemplo, uma solução contendo cloreto pode ser titulada com nitrato de prata em
presença de fluoresceína: o ponto final é dado pela adsorção do corante pelas partículas de
cloreto de prata, que se coram de vermelho no primeiro excesso de íons prata. Na
volumetria de oxidação-redução, pode-se lançar mão dos indicadores de oxidação-redução,
que são substâncias que mudam de coloração conforme o caráter oxidante ou redutor do
meio.
O erro de titulação é, essencialmente, o erro devido ao indicador acusar o ponto
final um pouco antes ou depois do ponto de equivalência. O erro de titulação depende de
vários fatores como a sensibilidade do indicador, a natureza do sistema em estudo, a
temperatura e a diluição. O erro de titulação é nulo somente quando o indicador acusa o
ponto final exatamente em coincidência com o ponto de equivalência. Quanto mais sensível
o indicador, menor o erro de titulação. Por outro lado, quanto menor a constante de
equilíbrio da reação básica (ou o produto de solubilidade quando se trata de um
precipitado), tanto mais brusca é a variação da concentração iônica perto do ponto de
equivalência. Desta forma, o uso de um indicador adequado é capaz de causar um ponto
final nítido. Porém, quando a variação de concentração perto do ponto de equivalência é
relativamente pequena, o ponto final é gradual e pouco exato. O erro de titulação (relativo)
aumenta com a diluição.
Em certos casos, pode ser conveniente lançar mão de métodos físico-químicos para
a localização do ponto final. Há reações para as quais não se têm indicadores adequados e a
percepção da mudança do indicador é dificultada ou mesmo impossibilitada. Quando a
solução é corada ou turva o erro de titulação pode toma-se muito grande para soluções
diluídas. Ainda há o possibilidade da presença do indicador causar interferências. Em
princípio, qualquer propriedade físico-química que varie bruscamente nas indicações do
ponto de equivalência pode servir de base para a determinação do ponto final. São muito
importantes, neste sentido, os métodos eletrométricos, que se baseiam na medida do
potencial de um elétrodo indicador (potenciometria), da condutância da solução
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
(condutometria) e da corrente de difusão com microeletrodos polarizáveis (amperometria)
ou ainda na medida da absorbância no curso da titulação, como no caso da volumetria
fotométrica.
5.3- Cálculo dos resultados2
O cálculo dos resultados na análise volumétrica quando se usam soluções padrões
com título expresso em termos de molaridade é muito simples. O princípio do cálculo é o
fato de o número de moles de reagente da solução padrão reagir de forma estequiométrica
segundo a equação reacional balanceada.
Suponhamos que, na análise de um material contendo carbonato de sódio, tenham
sido gastos 41,23 ml de ácido clorídrico 0,1015 M para titular uma solução preparada com
uma amostra de material pesando 0,4501g. A estequiometria e os dados do problema
permitem estabelecer as seguintes relações:
1 ml HCl 0,1015M ' 0,005300 g de Na2CO3
41,23 X 0,1015 ' 4,185 mmoles de HCl
que corresponde a 0,2218 g de Na2CO3.
Portanto, a amostra tomada para a análise contém 0,2218g de Na2CO3 e a
percentagem de Na2CO3 na amostra é
0,2218 X 100/0,4501= 49,28%.
O cálculo da percentagem do constituinte é feito facilmente utilizando-se regra de
três a partir da equação balanceada.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
5.4- Volumetria de Neutralização2, 3.
Em principio, a volumetria de neutralização baseia-se na reação de combinação dos
íons hidrogênio e hidróxido com formação de água, segundo a reação abaixo:
H+ + OH-
H2O
ou
H3O+ + OH-
2H2O
Com soluções padrões ácidas podem ser tituladas substâncias de caráter alcalino. Da
mesma forma, com soluções padrões alcalinas são tituladas substâncias de caráter ácido.
Têm-se, assim, duas variantes da volumetria de neutralização, a alcalimetria e a acidimetria,
respectivamente. A volumetria de neutralização também inclui as chamadas titulações de
deslocamento, em que o ânion de um ácido fraco é deslocado de seu sal mediante titulação
com um ácido forte ou, então, o cátion de uma base fraca é deslocada de seu sal mediante
titulação com uma base forte.
Comumente, o ponto final na volumetria de neutralização é identificado com o
auxílio de indicadores de pH. Desta forma é muito importante uma adequada escolha do
indicador e conhecer o ponto da escala do pH em que se situa o ponto de equivalência da
titulação. Também é importante conhecer a maneira como varia o pH nas imediações do
ponto de equivalência. Na volumetria de neutralização, o ponto de equivalência coincide
com o ponto de neutralidade (pH 7) sempre que a tituação envolva uma reação entre um
ácido forte e uma base também forte. Nos demais casos, em virtude da hidrólise dos
produtos da reação, o ponto de equivalência costuma deslocar-se para as regiões ácida ou
alcalina. Mais decisivo para uma apropriada escolha do indicador é, entretanto, o
conhecimento da variação do pH nas imediações do ponto de equivalência. A curva dando
a variação do pH com o volume de reagente adicionado durante a titulação é conhecida
como curva de neutralização. A curva de titulação pode ser traçada com dados
experimentais da análise potenciométrica ou, então, com base em considerações teóricas,
que é o método adotado nas seções subseqüentes.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
5.4.1- Indicadores de pH1
Os indicadores de pH são substâncias orgânicas fracamente ácidas (indicadores
ácidos) ou fracamente básicas (indicadores básicos), que mudam gradualmente de
coloração dentro de uma faixa de pH relativamente estreita, chamada zona de transição.
A tabela a seguir apresenta uma relação selecionada de indicadores de pH, com as
colorações em meios ácido e alcalino, as zonas de transição, os solventes usados na
preparação das soluções (todos com concentração de 0,1g/100ml). Quase todos os
indicadores de pH são de dupla coloração, isto é, têm uma coloração para o meio ácido e
outra para o meio alcalino. Porém, há alguns indicadores com coloração única, por
exemplo, fenolftaleína e timolftaleína, que são incolores em meio ácido e vermelhos em
meio alcalino. As zonas de transição dos indicadores abarcam, em geral, uma faixa de cerca
de duas unidades de pH.
Os indicadores de pH são quase todos usados como soluções a 0,1% em água ou
etanol. São solúveis em água, por exemplo, alaranjado de metila, p-nitrofenol,
alizarinossulfonato de sódio, azul do Nilo, amarelo de alizarina, diazovioleta e tropeolina.
As soluções de alguns indicadores, como amarelo de metila, vermelho neutro,
fenolftaleína e timolftaleína, não são diretamente solúveis em água. Devido a isto, são
primeiramente dissolvidos em etanol e posterior diluição com água conforme as indicações
da tabela. As sulfonoftaleínas dissolvem-se em água quando presente hidróxido de sódio
suficiente para neutralizar o grupo —S03H. As soluções dos referidos indicadores são
preparadas triturando 100 mg do reagente, em um gral de ágata, com o volume especificado
de solução de hidróxido de sódio 0,1 M e posterior diluição com água a 100 ml. A solução
de vermelho de metila pode ser preparada por dissolução de 1 g do corante em 300 ml de
etanol e posterior diluição com 200 ml de água.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Tabela 7- Indicadores de pH, sua coloração e zona de transição1, 2.
Indicador
Azul de timol (A) (faixa ácida)
Alaranjado de metila (E)
Azul de bromofenol (A)
Vermelho de (-naftila (B)
70%
Verde de bromocresol (A)
Vermelho de metila (A)
Vermelho de clorofenol (A)
Azul de bromotimol (A)
p-Nitrofenol (A)
Vermelho de fenol (A)
Vermelho neutro (B)
70%
Vermelho de cresol (A)
(-Naftolftaleína (A)
70%
Tropeolina 000 (E)
Azul de timol (A) (faixa alcalina)
(-Naftolbenzeína (A)
90%
Timolftaleína (A)
90%
Azul do Nilo
Amarelo de alizarina (A)
Amarelo de salicil (A)
90%
Diazovioleta
Tropeolina O (E)
Nitramina (B)
70%
Azul de Poirier
Acido trinitrobenzójco
Coloração
Zona de
Solução ácida
Solução Básica transição (pH)
Vermelho
Amarelo
1,2-2,8
Vermelho
Amarelo
3,1-4,4
Amarelo
Azul-violeta
3,0-4,6
Vermelho
Amarelo
3,7-5,0
Solvente
Água
Água
Água
Etanol
Amarelo
Vermelho
Amarelo
Amarelo
Incolor
Amarelo
Vermelho
Azul
Amarelo
Vermelho
Azul
Amarelo
Vermelho
Amarelo
4,0-5,6
4,4-6,2
5,4-6,8
6,2-7,6
5,0-7,0
6,4-8,0
6,8-8,0
Água
Água
Água
Água
Água
Água
Etanol
Amarelo
Róseo
Vermelho
Verde
7,2-8,8
7,3-8,7
Água
Etanol
Amarelo
Amarelo
Amarelo
Róseo-vermelho 7,6-8,9
Azul
8,0-9,6
Azul
9,0-11,0
Água
Água
Etanol
Incolor
Azul
9,4-10,6
Etanol
Azul
Amarelo
Amarelo
Vermelho
Lilás
Alar.-marrom
10,1-11,1
10,0-12,0
10,0-12,0
Água
Água
Etanol
Amarelo
Amarelo
Incolor
Violeta
Alar.-marrom
Alar.-marrom
10,1-12,0
11,0-13,0
11,0-13,0
Água
Água
Etanol
Azul
Incolor
Viol.-vermelho 11,0-13,0
Alar-vermelho 12,0-13,4
Água
Água
(A) = indicador ácido; (B) = indicador básico
5.4.2- Curvas de neutralização1
A título ilustrativo, apresentamos a seguir uma curva de titulação de ácido forte por
base forte.
Na titulação de ácidos fortes com bases fortes a solução resultante, no ponto de
equivalência, é neutra, pois o ânion do ácido e o cátion da base não sofrem hidrólise:
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
H+ + A- + M+ + OH-
H20 + M+ + A-
O pH no curso da titulação pode ser facilmente calculado tendo em conta a porção
remanescente do ácido ou o excesso da base.
Seja a titulação de 100 ml de ácido clorídrico 0,1 M com hidróxido de sódio 0,1 M.
A concentração do íon hidrogênio na solução original, posto que o ácido se ioniza
completamente, é
[H+] = 0,1 ou 10-1 , logo, pH = 1,0.
Após a adição de 50 ml de hidróxido de sódio, restará metade do ácido
originariamente presente e o volume da solução terá aumentado para 150 ml. Então:
[H+] = 50 x 0,1/150 = 3,3 X 10-2
e pH= 1,5
Semelhantemente, pode-se calcular o pH da solução para qualquer altura da
titulação até o ponto de equivalência.
O ponto de equivalência é alcançado com a adição de 100 ml de NaOH 0,1 M. Onde
tem-se, então, uma solução de cloreto de sódio. Como este sal não sofre hidrólise, o pH da
solução no ponto de equivalência é igual a 7.
A adição de excesso de hidróxido de sódio torna a solução alcalina. O pH da
solução, depois do ponto de equivalência, pode ser calculado tendo em conta o excesso de
NaOH 0,1 M e o volume da solução. Por exemplo, após a adição de 101 ml da solução de
hidróxido de sódio, tem-se
[0H-] =
1
0,1
= 5 X 10-4
201
ou
pOH= 3,3 e
pH= 10,7.
A tabela a seguir dá os valores de pH, na titulação de 100 ml de HCl 0,1 M com
NaOH 0,1 M, para a adição de diferentes volumes da solução de hidróxido de sódio. A
tabela inclui, também, os dados referentes à titulação de 100 ml de HCl 0,01 M com NaOH
0,01 M e 100 ml de HCI 1 M com NaOH 1 M.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
As curvas de neutralização mostram que, na fase inicial da titulação, o pH se eleva
gradualmente. Em uma segunda fase, o pH aumenta mais rapidamente para iguais
incrementos de hidróxido de sódio. Nas imediações do ponto de equivalência, o pH da
solução aumenta bruscamente com a adição de pequenas quantidades de hidróxido de
sódio.
Tabela 8- Titulação de 100 ml de HCl com NaOH de igual normalidade
NaOH adicionado
Solução 1 M
Solução 0,1 M
Solução 0,01 M
ml
pH
pH
pH
0
0,0
1,0
2,0
50
0,5
1,5
2,5
75
0,8
1,8
2,8
90
1,3
2,3
3,3
98
2,0
3,0
4,0
99
2,3
3,3
4,3
99,5
2,6
3,6
4,6
99,8
3,0
4,0
5,0
100,0
7,0
7,0
7,0
100,1
10,7
9,7
8,7
100,2
11,0
10,0
9,0
100,5
11,4
10,4
9,4
101
11,7
10,7
9,7
102
12,0
11,0
10,0
110
12,7
11,7
10,7
125
13,0
12,0
11,0
150
13,3
12,3
11,3
200
13,5
12,5
11,5
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
A figura a seguir dá as correspondentes curvas de neutralização, apresentando os
pontos de equivalências existentes.
Ponto de
Equivalência
Volume de NaOH (ml)
Figura 1-Curvas de neutralização de 100 ml de HCl com NaOH de
igual molaridade1.
A figura a seguir representa exatamente esta parte das curvas de
neutralização em escala maior, bem como as zonas de transição de alguns indicadores de
pH mais comuns. O ponto final na titulação de ácidos fortes pode ser determinado com o
auxílio de indicadores cujas zonas de transição se situam dentro da faixa de pH
correspondente às modificações bruscas.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Figura 2- Curvas de neutralização de 100 ml de HCl com NaOH de igual
normalidade nas imediações do ponto de equivalência1.
5.5- Volumetria de Precipitação2, 3.
A volumetria de precipitação baseia-se em reações de formação de compostos
pouco solúveis. A reação de precipitação deve processar-se quantitativamente, tornar-se
completa em um tempo relativamente curto e oferecer condições para uma conveniente
sinalização do ponto final.
Na prática, as condições acima apontadas limitam seriamente o número de reações
de precipitação utilizáveis como base de métodos volumétricos. Muitas reações de
precipitação não podem ser usadas com esta finalidade por falta de uma maneira adequada
para a localização do ponto de equivalência. Em número muito limitado de casos, o ponto
de equivalência pode ser localizado visualmente conduzindo a titulação até o ponto em que
a formação de precipitado deixa de ocorrer. Mais comumente, apela-se para o uso de
indicadores. Muitos métodos volumétricos de precipitação empregam indicadores mais ou
menos específicos. Existem indicadores de precipitação (formam um precipitado colorido),
indicadores por complexação (formam um complexo solúvel e colorido) e há uma classe
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
especial de indicadores, os indicadores de adsorção (se adsorver sobre o precipitado
formado). As possibilidades de aplicação das reações de precipitação na análise
volumétrica se ampliam consideravelmente com a determinação gráfica do ponto de
equivalência baseada em métodos eletrométricos (potenciometria, condutometria ou
amperometria) ou fotométricos.
Outro aspecto a considerar é que muitas reações de precipitação se processam a uma
velocidade relativamente lenta e deveriam que ser conduzidas muito morosamente para
assegurar o estabelecimento do equilíbrio de solubilidade. Freqüentemente é possível
acelerar convenientemente a velocidade da reação mediante uma judiciosa adição de etanol
ou acetona.
Na análise volumétrica em geral, a nitidez do ponto final da titulação depende
grandemente do grau com que se completa a reação envolvida. Na volumetria de
precipitação, os fatores que decidem a questão são o produto de solubilidade e a
concentração sob a qual se efetua a titulação. Quanto mais concentrados os reagentes
utilizados mais evidenciado o ponto de equivalência.
Numerosos métodos volumétricos de precipitação têm apenas aplicação especifica.
A titulo de ilustração, podem ser citados os seguintes exemplos:
a) Titulação de zinco com hexacianoferrato (II) do potássio em solução contendo
um pouco de hexacianoferrato (III) em presença de difenilamina (indicador de oxidaçãoredução).
b) Titulação de sulfato com solução de cloreto do bário com rodizonato de sódio ou
tetraidroxiquinona como indicador (formação de um segundo precipitado corado).
c) Titulação de sulfato com solução de nitrato de chumbo em presença de critrosinaB (indicador de adsorção).
d) Titulação do chumbo com molibdato de magnésio em presença de vermelho de
solocromo-B (indicador de adsorção) ou com cromato de potássio em presença de
ortocromo-T (indicador de adsorção).
e) Titulação de fluoreto com solução de nitrato de tório em presença de alizarina,
que origina uma laca vermelha com o produto da hidrólise do excesso do tório.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
f) Titulação de cloreto ou brometo com solução do nitrato de mercúrio (I) em
presença de indicadores de adsorção.
O mais importante método volumétrico de precipitação e, realmente, o único com
caráter mais ou menos amplo é a argentimetria. Este método se baseia no emprego de
solução padrão de nitrato de prata e na formação de sais (haletos, cianeto, tiocianato) de
prata pouco solúveis. A seguir estão apresentados alguns métodos argentimétricos.
5.5.1- Método de Mohr3
A solução neutra contendo íons cloreto é titulada com solução padrão de nitrato de
prata em presença de cromato de potássio como indicador.
O pH da solução deve estar entre 6,5 e 10,5. Se ácida, a solução pode ser
neutralizada por meio de adição de tetraborato de sódio, hidrogenocarbonato de sódio
(bicarbonato de sódio) ou carbonato de cálcio livres de cloreto. Se o pH estiver ácido há
formação de ácido crômico (H2CrO4), que é um ácido fraco e altera a solubilidade dos íons
cromatos, possibilitando a sua não precipitação e com isto o PE não seja evidenciado. Os
ácidos minerais podem ser eliminados mediante neutralização da maior parte com
hidróxido de amônio e adição de excesso de acetato de amônio. As soluções alcalinas são
neutralizadas com ácido nítrico livre de cloreto, em presença de fenolftaleína. No caso de
cloreto de amônio, se a solução for ácida, o pH deve ser ajustado entre 6,5 e 7,2. Para
valores de pH mais elevados, forma-se muita amônia livre, que aumenta a solubilidade do
cloreto de prata e do cromato de prata fazendo com que a mudança de coloração se
verifique tardiamente.
Quando a solução contém uma grande quantidade de eletrólitos, o cromato de prata
formado por um excesso local de reagente manifesta uma grande tendência a aglomerar.
Então, ele reage lentamente com o íon cloreto ainda presente, de sorte que o ponto final
pode ser observado prematuramente. Nestas condições, torna-se necessário agitar
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
vigorosamente perto do ponto de equivalência e adicionar nitrato de prata até que a
coloração vermelho-marrom não mais desapareça após agitação da mistura e repouso por
um curto período.
5.5.2- Método de Volhard3
O método consiste em adicionar solução contendo o íons halogenetos (cloreto,
brometo ou iodeto) um excesso de solução padrão de nitrato de prata e titular o excesso de
prata, em meio ácido, com solução padrão de tiocianato em presença de sulfato de amônio e
ferro (III)- FeNH4(SO4)2- como indicador. Normalmente acidifica-se o meio com ácido
nítrico com uma solução de ácido nítrico aproximadamente 0,3 M. É necessário remover o
cloreto de prata por filtração ou, então, adicionar nitrobenzeno para dificultar a reação do
precipitado com o tiocianato. No caso das determinações de brometo ou iodetos este
procedimento é desnecessário, pois os precipitados AgCl e AgBr possuem solubilidade
menor que o tioscianato de prata e desta forma não apresentam o problema de se
dissolverem. O indicador é usado na forma de uma solução saturada de sulfato de amônio e
ferro (III), cerca de 40%.
5.5.3- Método de Fajans2
O que caracteriza o Método de Fajans é a utilização de indicadores de adsorção.
Estes indicadores se adsorver no precipitado de prata formado e no primeiro excesso de
reagente desenvolverem cor. As substâncias utilizadas como indicadores de adsorção
podem ser ácidos, como a fluoresceína e a eosina, ou podem ser básicos, como a eosina e a
rodamina 6G. Estes indicadores são utilizados na forma de sais de sódio ou halogenados.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
O princípio deste metodo está baseado nas propriedades dos colóides. Quando há
formação de um precipitado este precipitado apresenta a tendência de adsorver íons iguais a
da sua composição. Esta camada de adsorção chama-se camada de adsorção primária.
Após, haverá uma segunda camada de adsorção, denominada de camanda de adsorção
secundária. Neste ponto entra em ação o indicador de adsorção. Após o ponto de
equivalência o íon em excesso se adsorveria, porém, o indicador é preferencialmente
adsorvido dando cor ao precipitado.
O íon indicador sempre deve possuir carga oposta a do íon do agente precipitante. O
precipitado formado deve separar-se na forma coloidal e o indicador somente deve se
adsorver após todo o composto a ser analisado estiver precipitado. Outro cuidado que deve
ser observado é a concentração do composto a ser analisado. Se a solução estiver muito
diluída, haverá pouco precipitado formado e desta forma a coloração desenvolvida será
pouca.
Uma desvantagem da utilização de indicadores de adsorção é a sensibilidade dos
haletos de prata à luz. Devido a isto, as titulações devem ser efetuadas com a menor
incidência de luz possível.
A fluoresceína pode ser empregada na determinação de íons cloreto, brometos e
iodeto, sendo sua alteração de cor no ponto de equivalência a passagem da cor verdeamarelo para rosa. Já a Rodamina 6G é utilizada na determinação de íons prata utilizandose íons brometo como reagente titulante. A alteração de cor no ponto de equivalência é
passando da cor rosa-alaranjado para violeta-avermelhado. Há indicadores que podem ser
utlizados na determinação de íons boratos, óxido de molibidênio e outros compostos.
5.6- Volumetria Complexação2, 3.
A volumetria de formação de complexos ou complexometria baseia-se em reações
entre um íon metálico e um ligante com formação de um complexo suficientemente estável.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
O caso mais simples é o de uma reação originando um complexo 1:1, como apresentado na
reação abaixo:
M+L
ML
Então, a constante de estabilidade é dada por
KML =
!ML"
!M"!L"
É possível construir uma curva de titulação representando pM, que pode ser
representado por - log[M], como uma fração da quantidade de agente complexante
adicionado. Os pontos da curva são achados com a equação
( !L " %
pM = log KML + log &&
##
' !ML" $
No ponto de equivalência, [M]eq = [L]eq e [ML]eq ) cM = concentração total do metal
em solução. Conseqüentemente,
[M]eq = ([ML]eq/KML)1/2 ) (cM/K ML)1/2
A figura a seguir representa as curvas de titulação de um metal M com um ligante L
correspondentes à formação de complexos 1:1 com diferentes estabilidades. A variação de
pM em torno do ponto de equivalência é tanto mais pronunciada quanto maior a
estabilidade do complexo: quando o salto é grande, a reação é apropriada para a titulação
complexométrica.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Figura 3- Curvas de titulação de um metal M com um ligante L correspondentes à
formação de complexos 1:1 de diferentes estabilidades1.
Quando um metal é capaz de unir-se a vários ligantes, a condição de equilíbrio é
determinada por uma série de constantes de estabilidade. Em uma curva de titulação, a
elevação de pM entre os vários estágios é, comumente, pequena. Para aplicar a formação de
uma seqüência de complexos na análise volumétrica, é preciso que se possa interromper a
titulação após a formação de um certo complexo MLn. Este complexo deve ser bastante
estável (pKn > 4) e sua estabilidade diferir apreciavelmente da do complexo seguinte MLn+1
(pKm – pKn+1 > 4). É raro que a titulação possa ser interrompida exatamente em um
complexo intermediário. Em geral a titulação deve ser conduzida até a formação do último
complexo, suposto, naturalmente, que seja bastante estável.
Durante muito tempo foi reduzido o número de métodos complexométricos. Os
mais importantes, já conhecidos em meados do século passado, eram o método de Liebig,
que titula cianeto com uma solução de nitrato de prata com formação de íon
dicianoargentato e a mercurimetria, que titula haletos com solução padrão de nitrato de
mercúrio (II) com formação de haletos de mercúrio (II) pouco ionizados. A limitação dos
métodos complexométricos devia-se, de um lado, à baixa estabilidade dos complexos
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
conhecidos e, de outro lado, ao fato de muitos metais se combinarem com os ligantes dando
complexos do tipo MLn, cuja formação ocorre em estágios. Em 1945, foi introduzido como
reagente volumétrico um poderoso agente complexante, o ácido etilenodiaminotetracético
(EDTA), capaz de formar quelatos com os metais pesados e alcalinoterrosos na proporção
1:1 e extraordinariamente estáveis. Com a utilização do EDTA ampliou-se enormemente o
campo da complexometria.
O EDTA pode ser obtido com alta pureza, na forma do ácido, ou na forma do sal
dissódico dihidratado. Ambos possuem alto peso molecular, porém, o sal possui a
vantagem de apresentar maior solubilidade em água.
O pH em uma titulação utilizando o EDTA é muito importante. O ácido
etilenodiaminotetracético é um ácido fraco, apresentando os seguintes pK: pK1= 2,00;
pK2= 2,66; pK3= 6,16 e pK4= 10,26. Desta forma, a dissociação dos prótons se dá com o
aumento do pH, e assim, aumenta o poder de complexação. Assim, melhora-se a
complexação de metais pelo EDTA realizando-se as titulações com pH em torno de 10.
Consideremos a titulação de íons Ca2+ pelo EDTA, apresentada no equilíbrio
abaixo, em pH 10.
Ca
2+
+ Y
4
_
CaY
2
_
Quando o ligante possui mais de um par de elétrons disponíveis para a
complexação, pode haver a formação de um anel com o metal. Se isto ocorrer, chamamos o
complexo de quelato e o ligante de quelante.
Algumas vezes há outras substâncias que competem na reação de complexação. A
presença destas substâncias podem ser proposital, com a intenção de complexar e eliminar
interferentes existentes no meio. Um exemplo disto é a adição de cianeto na determinação
da dureza da água. Os íons cianeto têm a função de complexar os demais metais existentes,
com exceção dos íons cálcio e magnésio, que ficam livres para a complexação do EDTA.
Estes reagentes complexantes que são adicionados à solução são denominados de agentes
mascarantes.
Outro reagente importante é complexante auxiliar. Este possui a função de impedir a
precipitação de metais no meio. Em geral, o próprio tampão exerce este papel. O tampão
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
mais difundido na complexometria utilizando-se EDTA é o tampão NH4OH/NH4Cl. Porém,
deve haver um cuidado na colocação do tampão, pois em uma maior quantidade de amônia
no meio, menor a inflexão da curva, dificultando a determinação do ponto final.
5.6.1- Indicadores Metalocrômicos
Os indicadores metalocrômicos são substâncias orgânicas que formam quelatos com
íons metálicos e desenvolvem uma cor diferenciada da sua forma livre.
Os indicadores mais utilizados nas titulações complexométricas são o Negro de
Eriocromo T (Erio T) e o Calcon. Suas estruturas estão apresentadas abaixo1:
OH
HO3S
OH
N=N
O2N
Erio T
OH
HO3S
OH
N=N
Calcon
As titulações realizadas como EDTA são geralmente em pH 10. Neste pH o
indicador Erio T apresenta a mudanção de coloração de violeta para azul no ponto de
equivalência.
O indicador deve ser adicionado por último, para evitar que não haja a formação de
um complexos estáveis com o indicador. Quando isto ocorre, dizemos que o indicador está
bloqueado, o que impossibilita a mudança de coloração no ponto de equivalência. Desta
forma, o complexo entre o metal e o indicador sempre deve ser menos estável que o
complexo entre o metal.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Poucos indicadores forma complexos estáveis na proporção 1:1. Entre elea estão o
EDTA, o que justifica sua larga utilização na complexometria, e a Trien
(trietilenotetramina).
5.7- Volumetria de Oxidação-redução1, 2, 3.
Este método está baseado nas reações de oxidação-redução. É
aplicado na
determinação de substâncias capazes de exibirem 02 ou mais estados de valência. Emprega
solução padrão de agente oxidante para determinação de substância redutora e solução
padrão de agente redutor para determinação de substâncias oxidantes.
A reação na volumetria de oxidação-redução deve ser quantitativa– o padrão só
deve reagir com a amostra e vice-versa e deve ser rápida– as que forem muito lentas podem
ser aceleradas pelo calor.
Podem ser classificados em dois grupos gerais, os métodos redutimétricos e os
métodos oxidimétricos.
Os métods redutimétricos empregam soluções padrões de agentes redutores, tais
como soluções de : Na2S2O3, FeSO4, SnCl2, As2O3, etc..., enquanto que os métodos
oxidimétricos empregam soluções padrões de agentes oxidantes, sendo os mais
importantes:
a)permanganimetria
b)dicromatometria
c)iodometria
d)cerimetria
O método oxidimétrico pode ser direto, onde a substância redutora é determinada
com solução padrão oxidante, exemplo: determinação de Fe2+ com K2Cr2O7, ou indireto,
que é empregado na determinação de substâncias oxidantes. A substância a determinar é
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
tratada com sol. padrão redutor auxiliar em excesso. O excesso de padrão redutor é
determinado com solução padrão oxidante. Pelo gasto de solução padrão oxidante, se
determina o quanto sobrou de padrão redutor e a diferença entre o que se colocou e o que
sobrou corresponde ao que reagiu com a substância oxidante a determinar. Exemplo:
determinação de MnO2 em pirolusita ( minério de manganês).
2MnO2 + As2O3 + H2SO4
2MnSO4 + As2O5 + 2H2O + As2O3 (excesso)
5As2O3 + 2MnO4- + nH+
+3
2Mn2+
+7
+ 5As2O5 + nH2O
+5
5x2=10 elétrons
2x5=10 elétrons
O Mn do MnO2 é reduzido a Mn2+ e o óxido arsenioso, As2O3, é oxidado a
pentóxido de arsênio, As2O5.
5.7.1- Determinação do Ponto Final
O ponto final da titulação redox pode ser realizada de formas diferenciadas. Pode
ser por:
a) indicação natural. Ex: MnO4-, então chamamos o reagente de auto indicador, pois ele
próprio funciona como indicador.
b) indicadores específicos, quando o indicador adicionado reage com alguma substância do
meio e desenvolve coloração. Ex: o amido interagindo com o ânion triiodeto, I3-, determina
o aparecimento de uma coloração azul ao meio reacional.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
c) métodos instrumentais, quando utiliza-se eletrodos que medem o potencial de oxidação:
interpretação gráfica
d) indicadores de oxidação-redução – Estes indicadores são agentes oxidantes ou redutores
fracos que apresentam na forma oxidada uma cor diferente daquela da forma reduzida. São
chamados de indicadores verdadeiros. Ex: difenilaminssulfonato de sódio, empregado na
determinação de Fe2+ com K2Cr2O7.
OBS: o indicador deve ter o E0 intermediário entre o E0 do padrão e o E0 da amostra. O
padrão oxida em primeiro lugar o mais redutor (amostra) e só depois vai oxidar o indicador
que na forma reduzida é incolor e na forma oxidada é violeta.
E0indicador= 0,85
E0amostra(Fe) = 0,77
E0dicromato = 1,38
O potencial do sistema no PE pode ser calculado a partir dos potenciais padrões, E0,
dos sistemas envolvidos, aplicando a equação de NERNST, apresentada abaixo.
Semelhante ao cálculo do potencial de uma pilha.
E = E0 +
0,0591/n log[oxidada]/[reduzida]
Podemo utilizar como exemplo a titulação de Fe2+ com Ce4+ , aonde uma espécie se
reduz e outra se oxida.
Ce4+ + Fe2+
Ce3+
+
Fe3+
Uma reação redox é considerada completa, quando no PE a razão entre as
concentrações da forma oxidada e da forma reduzida for * 103. Considerando a titulação de
Fe II com Ce IV:
Eeq. = E0Ce4+/Ce3+ + E0 Fe3+/Fe2+ = 1,45+ 0,77 = 1,11 V
2
2
1,11 = 0,77 + 0,0591 log [Fe3+]/[Fe2+] e fazendo [Fe3+]/[Fe2+] = X , teremos:
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
1,11 – 0,77 = 0,0591log X + log X = 0,34/0,0591 = 5,75 + X = 5,6 x 105 o que nos
indica uma reação completa!
Escolha do indicador:
Quanto maior for o salto potenciométrico mais fácil para escolher o indicador.
E
1,27V
1,11 V
Salto potenciométrico
0,95 V
99,9 100,0 100,1
volume do titulante em mL
Figura 4- Salto potenciométrico em uma análise volumétrica de oxidação-redução.
O indicador ideal é aquele que possui o valor de E0 intermediário entre o valor do E0
da amostra e o E0 da solução padrão e que possua cor na forma oxidada diferente da cor na
forma reduzida. A oxidação e a redução devem ser reversíveis. A primeira reação deve
ocorrer entre a amostra e o reagente titulante, após o térimino da reação (PE) a reação entre
o titulante e o indicador é necessária.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Um dos melhores indicadores é a FERROÍNA cujo E0 = 1,14 v. Antes de iniciar a
titulação de Fe2+ com Ce4+ predomina no erlenmeyer o Fe II, que forma com a FERROÍNA
um complexo de cor vermelha. Iniciada a titulação começa a se formar Fe III que reage
com a FERROÍNA retirada do complexo Fe II-FERROÍNA, formando um outro complexo
mais estável de Fe III-FERROÍNA de cor azul pálido (isto acontece porque a Kf do
complexo Fe II – FERROÍNA é menor que a Kf do complexo Fe III-FERROÍNA).
No PE há predominância de Fe III (mais estável) porém a mudança de cor vermelho
para azul pálido se dará num potencial de 1,12 V porque a cor vermelha é mais nítida do
que o azul. O erro cometido é muito pequeno uma vez que o Eeq. = 1,11 V .
OBS: para se fazer uma determinação por oxi-redução é necessário que a diferença
entre os E0 dos sistemas envolvidos seja igual ou superior 0,35 V.
5.7.2- Permanganometria
É o mais importante método oxidimétrico onde se emprega uma solução padrão de
KMnO4, agente oxidante forte.
Dependendo do pH do meio o íon MnO4- pode ser reduzido a Mn2+, MnO2, Mn3+ ou
MnO42-.
Com meio neutro, alcalino ou fracamente ácido, o produto da reação do MnO4- é o
MnO2, que após formado funciona como um catalizador da reação.
Em soluções com concentração de íons H+ 0,5M ou maior, o MnO4- se reduz a
Mn2+. Independente da reação que ocorra o íon MnO4- é um excelente agente oxidante.
MnO4- + 8H+
+ 5 e-
Mn2+ + 4H2O
E0 = 1,510 v
MnO4- + 4H+
+ 3 e-
MnO2 + 2H2O
E0 = 1,695 v
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Em geral a grande aplicação analítica do íon MnO4- é em meio fortemente ácido,
onde o produto de redução é o Mn2+. O ácido preferencialmente usado é o H2SO4 porque o
íon SO42- não sofre influência do MnO4-, embora em alguns casos se possa usar o HClO4.
Nas determinações de redutores tais como, As3+, Sb3+, H2O2, etc, pode se usar o HCl.
Nas determinações de Fe2+ não se pode usar HCl e HNO3 porque o Cl- é
parcialmente oxidado a Cl2 e o Fe2+ é parcialmente oxidado pelo NO3-. No primeiro caso
temos um erro para mais e no segundo caso um erro para menos na titulação.
Nas determinações de Fe2+ em minérios de ferro, emprega-se HCl para abrir a
amostra mas ele deve ser eliminado antes de se fazer a determinação do teor de Fe2+ ou
então se emprega a solução preventiva de ZIMMERMANN-REINHART. A eliminação do
HCl é feita por evaporação com H2SO4 ( ponto de ebulição igual a 3600 C).
5.7.3- Química do Iodo: Iodimetria e Iodometria
Iodimetria
É o método direto para determinar substâncias redutoras com solução padrão de
iodo.
I2 + 2e-
2I- E0 = 0,53 v
Na realidade, a maioria das titulações iodimétricas emprega uma solução de iodo em
iodeto de potássio e o que reage como oxidante é o íon triiodeto, I3-.
I3- + 2S2O32-
Química Analítica Quantitativa
3I- + S4O62-
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
O iodo e o íon I3- são agentes oxidantes muito mais fracos que o MnO4-, o Cr2O72- e
o Ce4+ ( na forma de Ce(SO4)2. O iodo e o I3- se prestam para determinar substâncias muito
redutoras tais como Sn2+, SO32-, HSO3-, S2O32- e o H2S mesmo em soluções ácidas.
Para determinar redutores mais fracos como As3+ e Sb3+, a análise deve ser feita em
soluções neutras e para isso se acrescenta bicarbonato de sódio, NaHCO3 que consome os
íons H+ liberados na reação.
Na iodimetria a utilização de indicadores pode ser dispensada, pois a espécie I- é
incolor, porém, quando na forma I2 apresenta coloração castanho-amarelado. Desta forma,
na primeira gota que o reagente titulante não sofra redução, ocorrerá o surgimento de cor na
solução, indicando o ponto final da titulação. Caso queira-se evidenciar o ponto de
equivalência, pode ser utilizado uma solução de amido (indicador específico). Em presença
de I- o I2 forma o íon I3- que com o amido resulta num complexo de cor azul intensa. Não
pode ser usado em sol. muito ácida para evitar a hidrólise do amido. Um inconveniente da
sua utilização é que somente pode ser adicionado próximo ao ponto de equivalência. Outra
alternativa é o uso do amidoglicolato de sódio. É um pó não higroscópico, facilmente
solúvel em água quente e estável durante meses. Com excesso de iodo é verde e no ponto
de equivalência quando o iodo passa a iodeto mostra cor azul. Este indicador pode ser
adicionado desde o início da titulação.
Iodometria:
É um método indireto para determinar substâncias oxidantes. Consiste em tratar as
substâncias oxidantes com I- e titular o I2 liberado com solução padrão de S2O32-.
Aplicações:
Determinação de MnO4- e Cr2O72- em meio fortemente ácido
Determinação de BrO3- e IO3- em meio ácido
Determinação de IO- e ClO3- em meio fracamente ácido
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
Alguns cuidados são necessários para evitar erros na iodometria. Abaixo estão
apresentados as principais causas de erro neste tipo de análise volumétrica.
Oxidação do I- pelo O2 do ar:
No método direto:
2I- +
I2 + redutor
2I- + O2
subst. oxidada
I2
Neste caso o erro será para menos porque parte do redutor será oxidado pelo I2
proveniente da oxidação do I- pelo O2.
No método indireto:
2I- + oxidante
I2 + S2O322I- +
O2
I2
2II2
Neste caso o erro será para mais porque será necessário um maior consumo de
S2O32- para reduzir o I2 proveniente da oxidação do I- pelo O2.
A oxidação do I- é bastante lenta, mas é catalisada pela ação da luz direta, aumento
de acidez e presença de íon cuproso (Cu+) e NO2- e de monóxido de nitrogênio.
Para realizar a análise volumétrica de forma a evitar os erro apresentados, alguns
cuidados devem ser tomados, como por exemplo:
-titular sob luz indireta
-usar frascos âmbar para guardar as soluções
-quando for necessário esperar para continuar a titulação, guardar o erlenmeyer no escuro
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
-tampar os frascos
-na presença de NO adicionar uréia, ácido sulfanílico e corrente de CO2.
Volatilização do iodo.
Pode ser evitada quando se acrescentar I- formando o íon complexo I3- que é fixo.
Outra forma de diminuir, ou mesmo evitar a volatilização do iodo e não prolongar o tempo
das titulações e fazê-las em temperatura ambiente, sempre em erlenmeyer. Também podese diminuir a volatilização do iodo tampando-se a boca do erlenmeyer.
pH do meio
O pH do meio pode trazer interferências neste tipo de análises volumétricas. O pH
muito baixo favorece a oxidação do I-, porém, em pH acima de 8 ocorre hidrólise do iodo
formando I- e hipoiodito, que é estruturalmente instável.
I2
+ 2OH-
I- + IO- (ânion hipoiodito) + H2O
3IO-
2I- + IO3-
OBS: o potencial normal de redução do I2/2I- não é afetado pelo pH desde que ele não seja
menor que 8.
A análise também pode ser realizada utilizando-se CCl4 no meio reacional. No
tetracloreto de carbono o iodo é 85 vezes mais solúvel que na H2O. Sua cor no CCl4 é
violeta.
2I- + amostra oxidante
I2
+ S2O32-
I2
2I-
(determinação indireta)
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
No início da determinação do iodo liberado numa determinação indireta,
adicionando CCl4 no erlenmeyer se observará a cor violeta na camada orgânica. Após
adicionar S2O32-, o I2 vai se reduzindo a I- e a cor vai diminuindo a intensidade na camada
orgânica. No ponto final o CCl4 é incolor porque não forma composto colorido com o
iodeto.
5.7.4- Oxidações com o dicromato de Potássio ( Cr2O72-/2Cr3+ E0 = 1,33 v )
Dicromatometria:
Apesar do não ser agente oxidante tão poderoso como o KMnO4, apresenta diversas
vantagens sobre ele. São elas:
pode ser obtido puro, é estável até o ponto de fusão e por isso é um padrão primário
solução de concentração exata é obtida através da pesagem exata e dissolução em
balão volumétrico
solução estável indefinidamente desde que não sofra evaporação
não é reduzido pelo HCl desde que a concentração do ácido não seja maior que 2M
não sofre redução pela ação da luz
A grande aplicação é direta, na determinação de Fe2+ no minério, em meio ácido.
Cr2O72-
+
6Fe2+
+ 14H+
2Cr3+
+
6Fe3+
+ 7 H2O
A determinação do ponto final pode ser realizada através de indicadores internos,
como difenilamina e difenilssulfonato de sódio (neste caso se acrescenta H3PO4 para
baixar o potencial Fe3+/Fe2+), ou utilizando métodos instrumentais potenciométricos.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6- PARTE EXPERIMENTAL1
6.1- Aula Sobre Preparo de Soluções
1. Preparar uma solução de cloreto de sódio (100 mL) a uma concentração de 10 g.L-1 do
sal.
,
Em um balão volumétrico de 100 mL colocar cerca de 20-30 mL de água
destilada/deionizada.
,
Adicionar o sal pesado e dissolve-lo.
,
Preencher até a marca.
,
Secar o gargalo com o auxílio de um bastão de vidro e papel absorvente.
,
Fechar o balão volumétrico com uma tampa e homogeinizar.
2. Preparar uma solução de hidróxido de sódio a uma concentração de 0,1 M (100 mL).
Seguir os procedimentos citados anteriormente.
3. Preparar uma solução (50mL) de ácido clorídrico (HCl) 0,1 N utilizando-se uma
solução de ácido clorídrico concentrado a 37%. Utilizar os procedimentos citados
anteriormente.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.2- Aula Sobre Calibração de Material Volumétrico
1. Medir a temperatura da água
2. Verificar o valor da densidade de acordo com a tabela abaixo
3. Pesar o aparelho volumétrico em balança analítica seco
4. Colocar água até a marca de aferição
5. Pesa-se novamente o material sem entrar em contato com as mãos.
6. Por diferença sabe-se a massa de água contida no material
7. Calcular o verdadeiro volume do material volumétrico pela equação d=m/V.
Tabela 1- Valores da densidade da água em relação a temperatura ambiente.
T°C
d (g/mL)
T°C
d (g/mL)
T°C
d (g/mL)
0
0,999841
10
0,999700
20
0,998203
1
0,999900
11
0,999605
21
0,997992
2
0,999941
12
0,999498
22
0,997770
3
0,999965
13
0,999377
23
0,997538
4
0,999973
14
0,999244
24
0,997296
5
0,999965
15
0,999099
25
0,997044
6
0,999941
16
0,998943
26
0,996783
7
0,999902
17
0,998774
27
0,996512
8
0,999849
18
0,998595
28
0,996232
9
0,999781
19
0,998405
29
0,995944
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.3- Aula Sobre Gravimetria (determinação de íons cloretos)
1. Pipeta-se uma alíquota da solução original, ou previamente diluída, para dentro de um
béquer.
2. Adiciona-se 1 mL de HNO3 1:1 (evita a precipitação de óxidos, carbonatos, ...).
3. Lentamente adicionam-se cerca de 20 mL de AgNO3 (C= 40 g.L-1) a frio.
4. Todas as operações subseqüentes devem ser realizadas na ausência de luz.
5. A suspensão deve ser aquecida até quase a ebulição, agitando-a durante dois minutos.
6. Deixar o precipitado depositar fora do aquecimento. Após testar completa pptação dos
íons cloretos adicionando-se gotas de AgNO3 sobre a solução.
7. Deixar em repouso por cerca de 1 a 2 horas.
8. Filtra-se o precipitado por decantação, lavando-se o precipitado com uma solução de
HNO3 0,01M (para remover excesso de íons Ag+ adsorvidos, e para evitar peptização
do AgCl).
9. Passa-se o precipitado, com o papel filtro, para uma estufa a 110°C por cerca de 1 hora.
10. A peso constante determinar a massa total de AgCl.
11. Calcular a massa de Cloreto contida neste precipitado e fazer relação com o volume da
alíquota utilizada.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.4- Aula Sobre Volumetria de Neutralização (Padronização de uma solução de HCl)
1. Preparar em um balão volumétrico de 100 mL uma solução de ácido clorídrico 0,1 N.
2. Retirar uma alíquota de 10 ou 25 mL para um erlenmeyer.
3. Adicionar água destilada até volume desejado.
4. Adicionar gotas de fenolftaleína (indicador ácido-base pH 8,0- 8,5).
5. Colocar em bureta limpa e ambientalizada solução de NaOH 0,100 N padronizada.
6. Titular a solução até o aparecimento da coloração rósea permanente.
7. Calcular a concentração da solução de ácido clorídrico e rotulá-la.
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.5- Aula Sobre Determinação da Concentração de Ácido Acético no Vinagre
1. Dilua o vinagre 10 vezes.
,
Com auxílio de uma pipeta volumétrica transferir 10 mL de vinagre para um balão de
100 mL.
,
Levar à marca com água destilada/deionizada
,
Secar o gargalo
,
Fechar o balão volumétrico com uma tampa adequada
,
Homogeneizar a solução
2. Transferir com o auxílio de uma pipeta volumétrica uma alíquota desta solução para
um erlenmeyer.
3. Em uma bureta previamente ambientalizada colocar solução padronizada de NaOH
0,1M, aproximadamente.
4. Adicionar gotas de fenolftaleína no erlenmeyer.
5. Titular até o aparecimento da coloração róseo.
6. Fazer o cálculo da concentração.
Reação envolvida:
O
O
H3 C
C
+
NaOH
OH
Química Analítica Quantitativa
H3C
C
_
O Na+
+ H2O
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.6- Aula Sobre a Determinação da Concentração de Mg(OH)2 no Leite de Magnésia
1. Transfere-se por diferença uma amostra de Leite de Magnésia para um erlenmeyer.
2. Adiciona-se uma quantidade em excesso de HCl padronizado (a solução deve ficar
límpida).
3. Goteja-se 2 a 3 gotas do indicador Fenolftaleína.
4. Titula-se a solução com uma solução padrão de NaOH até o ponto de virada.
5. O indicador passa de incolor para rosa.
Reações envolvidas:
Mg(OH)2 + 2 HCl
HCl (excesso) + NaOH
Química Analítica Quantitativa
MgCl2 + 2H2O + HCl (excesso)
NaCl + H2 O
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.7- Aula Sobre Determinação do íon Carbonato por Um Ácido Forte (deslocamento)
1. Em um erlenmeyer transferir uma alíquota de 10 a 20 mL da solução contendo íons
carbonatos com o auxílio de uma pipeta volumétrica.
2. Em uma bureta ambientalizada colocar HCl padrão 0,1M e zerá-la.
3. Adicionar gotas do indicador indicado.
4. Titular a solução até ponto de equivalência indicada pela virada do indicador.
5. Realizar os cálculos apropriados para determinação da quantidade de íons
Carbonatos em solução (k1= 4,3. 10-7), assim [H+]= (4,3.10-7. M)1/2= X
CO2
3
_
+
+ H
HCO
_
3
pH= 8,3 (fenolftaleína, azul de timol,...)
OU
CO2
3
_
+ 2H
+
H CO
2 3
Química Analítica Quantitativa
pH= 3,7 (amarelo de metila, meltilorange,...)
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.8- Aula Sobre Determinação de íons Cloreto pelo Método de Mohr
1. Uma amostra contendo Cl- é recebida em um balão volumétrico de 100 mL e diluída até
a marca, se necessário.
2. Retirar uma alíquota com auxílio de uma pipeta volumétrica e passá-la para um
erlenmeyer.
3. Adicionar gotas do indicador cromato de potássio K2CrO4 (cor amarela).
4. Titular com solução de AgNO3 padrão 0,100 N até o aparecimento da coloração
vermelho-tijolo, devido precipitação do cromato de prata.
Reações envolvidas:
_
+
Ag
+
Ag+
2_
+ CrO
4
Cl
Química Analítica Quantitativa
AgCl
Ag2CrO4
(vermelho-tijolo
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.9- Aula Sobre Determinação de íons prata pelo método de Volhard (mét. direto)
1. Uma amostra de AgNO3 é recebida em um balão volumétrico de 100 mL e diluída
até a marca.
2. Retirar uma alíquota e passa-la para um erlenmeyer.
3. Adicionar 1 mL de solução saturada de sulfato férrico amoniacal Fe(NH4)(SO4)2.
4. Acidifica-se meio com HNO3.
5. Titula-se com tiocianato de potássio (KSCN) padrão até primeira aparição de cor
marrom-avermelhada persistindo sob forte agitação.
Reações envolvidas:
_
+
Ag + SCN
_
+3
Fe
+
SCN
Química Analítica Quantitativa
AgSCN
+2
Fe(SCN)
Vermelho
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.10- Aula Sobre Determinação de Íons Cloreto pelo Método de Volhard (mét.
Indireto)
1. Uma amostra de Cl- é recebida em um balão volumétrico de 100 mL e diluída até a
marca, se necessário.
2. Retirar uma alíquota e passá-la para um erlenmeyer.
3. Adicionar uma alíquota de AgNO3 padrão suficiente para precipitar todo o
halogeneto.
4. Adicionar Nitro-Benzeno (1 ml).
5. Adicionar 1 mL de solução saturada de sulfato férrico amoniacal Fe(NH4)(SO4)2.
6. Acidifica-se meio com HNO3 para evitar hidrólise do indicador e precipitação dos
íons metálicos.
7. Titula-se com tiocianato de potássio (KSCN) padrão até primeira
aparição de cor marrom-avermelhada persistindo sob forte agitação.
Reações envolvidas:
+
Ag (excesso) + Cl
Ag+ + SCN
_
SCN
_
AgCl + Ag+ (excesso)
_
+3
+ Fe
AgSCN
+2
Fe(SCN)
Vermelho
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.11- Aula Sobre Determinação de íons Ca2+ no leite
Transferir três alíquotas com o auxílio de uma pipeta volumétrica para três
1.
erlenmeyers de 250 mL.
2.
Adicionar 10-15 mL de solução tampão pH= 10 (NH4OH/NH4Cl).
3.
Adicionar alguns cristais de KCN para mascarar íons Zn2+, Cu2+, e Fe3+ (para impedir
erro de análise e bloqueamento do indicador).
Adicionar uma pitada do indicador Erio T. Este deve ser adicionado após a adição de
4.
cianeto para não haver bloqueamento do indicador, ou seja, formação de um
complexo mais estável com íons metálicos os quais o EDTA não consegue deslocar,
impedindo a “virada” do indicador.
5.
Titular com uma solução de EDTA padrão 0,02M, utilizando uma bureta
ambientalizada previamente, até a mudança da coloração púrpura para azul.
Calcular o valor de Cálcio em g.L-1, ppm e %; considerando 1 mol de EDTA ragindo
6.
com 1 mol de íon cálcio.
Principais reações:
4
2+
Ca
+ Y
Y
4
_
_
_
CaIn
+
(púrpura)
Química Analítica Quantitativa
CaY
CaY
2
2
_
_
_
3
In
+
(azul)
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.12- Aula sobre a determinação de peróxido de hidrogênio na Água Oxigernada
(iodometria)
1. Dilui-se a amostra de Água Oxigenada em em um balão volumétrico de 100ml
2. Transfere-se uma alíquota de 25ml desta solução para um erlenmeyer de 250ml
3. Adiciona-se 10ml de H2SO4 2M
4. Adiciona-se 1-2g de iodeto de potássio ao erlenmeyer
5. Goteja-se 3gotas de molibdato de amônio 3%, como catalisador
6. Titula-se com solução padrão de tiossulfato de sódio.
7. Uiliza-se amido como indicador (azul para incolor)
Reação
_
H2 O2 + 2I + 2H+
Química Analítica Quantitativa
I2 +
2H2 O
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.13- Aula Sobre Determinação de Ácido Ascórbico por Oxidação-redução
(iodimetria)
1. Pesam-se três amostras de vitamina C em três erlenmeyers, ou pesa-se uma amostra
e dilua-a em um balão volumétrico.
2. Coloca-se uma solução padronizada de Iodo, aproximadamente 0,03M, em uma
bureta previamente ambientalizada.
3. Após a bureta pronta, dissolver o sólido, com agitação e o frasco fechado para evitar
oxidação pelo oxigênio do ar.
4. Caso tenha sido preparado uma solução da amostra, pipetar 25mL desta para um
erlenmeyer.
5. Titula-se com a solução de Iodo até o primeiro aparecimento da coloração do Iodo.
O ponto de equivalência pode ser evidenciado pela adição de solução de amido pelo
aparecimento da coloração azul.
OBS- O erlenmeyer deve estar fechado para evitar entrada de oxigênio em excesso.
Principal reação:
O
2 I 2 + C4 H6O4 (OH)C
COH
Química Analítica Quantitativa
_
+
4 I + 2 H + C4 H6O4 C
C
O
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
6.14- Aula sobre a Determinação de Fe3+ em medicamentos (oxi-redução)
1. Dissolve-se parte da amostra precisamente pesada em 100ml de água destilada
2. Adicionam-se 10ml de HCl 12M
3. Transferir 3g, aproximadamente de KI
4. Agitar fortemente e após deixar 5-10 minutos de repouso no escuro
5. Titula-se o iodo liberado com uma solução de tiossulfato padrão.
6. Pode-se utilizar amido como indicador
Reações envolvidas:
3+
Fe + 2I
_
2_
I 2 + 2S O
2 3
2+
Fe + I 2
_
_
2
S4O6 + 2I
Química Analítica Quantitativa
Prof. Daniel Arsand
UNIVERSIDADE DE CRUZ ALTA- UNICRUZ
7- BIBLIOGRAFIA
1 BACCAN, N. at all: Química Analítica Quantitativa Elementar. Campinas, SP, Ed.
Blucher, 1979.
2 VOGEL, A.: Análise Química Quantitativa. 6ª ed., Rio de Janeiro, RJ, Ed. LTC, 2000.
3 HARRIS, D.: Análise Química Quantitativa. 5ª ed., Rio de Janeiro, RJ, Ed. LTC, 2000.
4 OHLWEILLER, O.: Química Analítica Quantitativa. Rio de Janeiro, RJ, Ed. LTC, 1976.
Química Analítica Quantitativa
Prof. Daniel Arsand