Bioquímica e Biologia Molecular
Bioquímica
1º Semestre - 2009/2010
2º ano - Mestrado Integrado em Engenharia Biomédica
Instituo Superior Técnico
Baseado nas aulas e no livro Principles of Biochemistry, Lehninger
Resumo por Inês Amorim
Índice
Evolução bioquímica ................................................................................................................. 4
Água e pH ................................................................................................................................. 9
Aminoácidos e proteínas......................................................................................................... 12
Catálise enzimática ............................................................................................................. 22
Glícidos ................................................................................................................................... 29
Lípidos .................................................................................................................................... 31
Metabolismo .......................................................................................................................... 35
Glicólise e Neoglucogénese ................................................................................................. 37
Ciclo de Krebs ..................................................................................................................... 39
Cadeia respiratória .............................................................................................................. 41
Outras vias metabólicas ...................................................................................................... 43
Fotossíntese............................................................................................................................ 47
2
Bioquímica
A bioquímica estuda a estrutura e função de biomoléculas e as reacções químicas envolvidas
nos processos biológicos.
Apesar da sua grande diversidade, todos os organismos vivos são constituídos por células e
partilham o mesmo tipo de moléculas e reacções.
De todos os elementos químicos, apenas uma pequena parte entra na composição das
biomoléculas. Os mais abundantes são H, O, C e N e existem vestígios de outros elementos
como Na, Ca, K, P ou S. Em concentrações ainda mais reduzidas, mas desempenhando funções
muito importantes, podemos encontrar Fe, Ni, Zn, I, Cu, etc.
Os diferentes átomos e moléculas reagem entre si e estabelecem diferentes ligações e
interacções:
Covalentes: têm origem na partilha de electrões e são as ligações mais fortes;
Não-covalentes: embora sejam mais fracas são responsáveis por muitas características
que permitem a manutenção da vida. Podem ser:
o Ligações iónicas;
o Interacções de Van der Waals;
o Ligações por pontes de hidrogénio;
o Ligações hidrofóbicas.
As biomoléculas englobam 4 tipos de macromoléculas, cuja natureza e função
aprofundaremos mais à frente, que são essenciais à vida:
Lípidos;
Glícidos;
Proteínas;
Ácidos nucleicos.
Todas estas moléculas e macromoléculas participam em reacções bioquímicas que
apresentam uma série de características:
Dão-se em ambiente aquoso;
Estão associadas a variações de energia. A sua forma mais vulgar de energia química é
o ATP;
Reacções bioquímicas diferentes localizam-se em diferentes partes da célula;
Estão frequentemente organizadas em vias metabólicas;
São reguladas de acordo com a necessidade de controlar a quantidade e a actividade
de enzimas do sistema.
3
Evolução bioquímica
A origem da vida na Terra pode ser explicada pela teoria da evolução pré-biótica, segundo a
qual a vida surgiu a partir de matéria inorgânica, inanimada.
Interacções entre substâncias químicas presentes na atmosfera primitiva levaram à formação
dos primeiros aminoácidos que gradualmente deram origem a moléculas mais complexas,
como as proteínas e o DNA. Estas, por sua vez, combinaram-se entre si e, eventualmente,
passaram a desempenhar diferentes funções e a organizar-se nas primeiras proto-células.
A atmosfera primitiva da Terra era provavelmente constituída por gases como hidrogénio,
azoto, amónia, metano, monóxido de carbono e dióxido de carbono, entre outros, não
apresentando oxigénio, ao contrário do que se verifica nos dias de hoje.
Há medida que a actividade interna da Terra foi diminuindo e esta passou a libertar menos
calor, a crosta começou a solidificar e a água a condensar. Na sequência das primeiras chuvas
surgiram lagos e, mais tarde, oceanos repletos de substâncias químicas que foram interagindo
entre si para formar novas moléculas. Os elementos presentes nesta sopa pré-biótica,
alimentados pela energia proveniente da radiação UV (não existia oxigénio na atmosfera, logo
não havia camada de ozono), de relâmpagos e do calor do interior da Terra, sofreram
inúmeras reacções que culminaram no aparecimento da vida.
A hipótese pré-biótica é apoiada pela experiência de Urey-Miller, na qual se provou que
alguns açúcares, bases e aminoácidos se formam naturalmente nas condições da Terra
primitiva. Num sistema fechado, Urey e Miller introduziram os principais gases atmosféricos
(hidrogénio, amónia, metano e água), submeteram-nos a descargas eléctricas e realizaram
ciclos de aquecimento e condensação de água, pretendendo simular as condições a que as
moléculas eram sujeitas no ambiente da atmosfera primitiva.
4
Uma semana depois do início da experiência os dois cientistas verificaram que algum do
carbono introduzido inicialmente no sistema fazia agora parte de compostos orgânicos.
Tinham-se formado açúcares (glicose, ribose e desoxirribose formam-se quando formaldeído,
CH2O, é exposto a radiação UV), lípidos, aminoácidos (glicina – mais abundante) e alguns
compostos dos ácidos nucleicos (adenina – a partir de 5 HCN e radiação UV). Formaram-se
ainda péptidos com cerca de 50 aminoácidos. Posteriormente, verificou-se que estes
compostos não se formavam quando a experiência era repetida num meio rico em oxigénio, o
que apoiada a hipótese de a vida ter tido origem em ambiente anóxico (sem O2).
Apesar das experiências a seu favor, a teoria da sopa pré-biótica tem alguns pontos fracos
como o facto de ser uma mistura diluída, já que nessas condições as moléculas têm tendência
a separar-se e não a serem sintetizadas. Contudo, a evaporação da água devido ao calor
interno da Terra pode ter levado ao aumento da concentração das substâncias presentes
nessa sopa e facilitado as reacções que levaram à formação de moléculas mais complexas.
Num meio cada vez mais concentrado, ter-se-ão formado espontaneamente coacervados a
partir de lípidos. Coacervados são aglomerados de moléculas sustentados por forças
hidrofóbicas do meio aquoso envolvente, cuja fronteira se assemelha a uma membrana. Estes
complexos são capazes de absorver e libertar moléculas, pelo que podem ter actuado como as
primeiras proto-células, embora ainda não fossem capazes de realizar processos metabólicos.
Pensa-se que a primeira “molécula da vida” a surgir terá sido o RNA pois este ácido nucleico
não necessita de enzimas ou primers para se replicar; pode, ele próprio, actuar como uma
enzima; e, ainda nos dias de hoje, se encontram organismos cujo material genético se
restringe a moléculas de RNA (ex. retro-vírus).
Num primordial Mundo do RNA terão sido produzidas moléculas de RNA com sequências
aleatórias. Alguns fragmentos ter-se-ão auto-replicado, num processo catalisado por ribozimas
- segmentos do próprio RNA. Os segmentos seleccionados terão catalisado a síntese de
péptidos específicos que, por sua vez, participaram na replicação do RNA. Deu-se então um
período de co-evolução do RNA e proteínas seguido da evolução dos sistemas primitivos de
tradução e do aparecimento no genoma de RNA. O papel catalítico e genético deste genoma
foi sendo separado ao longo do tempo, acabando o DNA por ser a fonte de material genético e
as proteínas os principais agentes catalíticos.
Estes e muitos outros factores contribuíram para a crescente complexidade do mundo prébiótico e para a evolução da vida.
5
Desde o aparecimento das primeiras células até à diversidade de organismos que se verifica
nos dias de hoje deu-se uma grande evolução. As primeiras células a surgir eram
quimioheterotróficas e utilizavam como fonte de carbono e energia moléculas sintetizadas
por processos não biológicos. O escasseamento de nutrientes favoreceu a evolução de seres
autotróficos capazes de sintetizar as suas próprias moléculas orgânicas, primeiro a partir de
compostos químicos e, mais tarde, a partir da radiação solar. A evolução de seres
fotossintéticos levou à libertação de 02 para a atmosfera terrestre e, apesar de este gás ser
tóxico para a maioria dos seres anaeróbios (os únicos existentes até esta altura), o aumento da
sua concentração favoreceu a evolução de seres aeróbios. Estes seres apresentavam uma
enorme vantagem em relação aos seres anaeróbios quando competiam num ambiente rico em
oxigénio e, devido à sua maior eficiência energética, tinham ainda maior potencialidade de se
desenvolverem para formas de vida mais complexas.
Relativamente à organização celular, as primeiras células tinham uma estrutura muito simples,
sem núcleo individualizado, organitos celulares ou sistema endomenbranar. Estas células
procarióticas foram sofrendo uma série de alterações até darem origem às células
eucarióticas, mais complexas e com um núcleo perfeitamente organizado e delimitado,
diversos organitos celulares e sistema endomenbranar.
Procarióticas
Eucarióticas
Animais
Membrana celular
Parede celular
Núcleo
Ribossomas
Complexo de Golgi e RE
Mitocôncrias
Vacúolo
Cloroplastos
Citoesqueleto
X
X
Não individualizado
X
-
X
X
X
X
X
- / muito pequeno
X
Vegetais
X
X
X
X
X
X
X
X
X
Actualmente considera-se que a evolução das células procarióticas em células eucarióticas
pode ser explicada pela teoria da endossimbiose.
6
O modelo endossimbiótico, desenvolvido por Lynn Margulis, defende que os seres eucariontes
terão resultado da evolução conjunta de vários organismos procariontes, os quais foram
estabelecendo associações simbióticas entre si. Este modelo admite que os sistemas
endomembranares e o núcleo resultaram de invaginações da membrana plasmática e que as
mitocôndrias e os cloroplastos, até há cerca de 2100 M.a., eram organismos autónomos. Nessa
altura, algumas células de maiores dimensões (células hospedeiras) terão capturado células
mais pequenas, como os ancestrais das mitocôndrias e dos cloroplastos. Alguns destes
ancestrais conseguiram sobreviver à digestão no interior da célula procariótica de maiores
dimensões, estabelecendo relações de simbiose. A íntima cooperação entre estas células terá
conduzido ao estabelecimento de uma relação simbiótica estável e permanente que trouxe
inúmeras vantagens, desde uma maior capacidade de metabolismo aeróbio (levado a cabo
pelas mitocôndrias) até a uma maior facilidade de obter nutrientes (produzidos pelo
endossimbionte autotrófico).
A dada altura, organismos unicelulares começaram a agrupar-se e a organizar-se em colónias.
As vantagens deste tipo de relações acabaram por prevalecer e surgiram os primeiros seres
multicelulares. Com o decorrer da evolução deu-se a diferenciação celular e a especialização
de grupos de células em determinadas funções.
Hoje em dia, considera-se que todos os organismos vivos pertencem a um dos domínios/ramos
da árvore da vida e que evoluíram a partir de um ancestral comum. Esses domínios são:
Eubacteria: seres procariontes que habitam os solos, meios aquosos ou tecidos de
outros organismos vivos ou em decomposição;
Archeabacteria: seres procariontes que vivem em ambientes extremos, tais como
lagos salgados, fundos oceânicos e meios extremamente quentes ou ácidos. Os ramos
bactéria e archea divergiram logo num estado primário da evolução;
Eukarya: seres eucariontes. Evoluiram a partir do ramo archea.
7
8
Água e pH
A água é a substância química predominante nos organismos vivos, perfazendo cerca de 70%
da sua massa. É um óptimo solvente para a maioria das moléculas polares e apresenta diversas
características que fazem dela uma molécula única, muitas delas derivadas do facto de a
molécula H2O formar um dipolo e ter tendência para formar ligações de hidrogénio.
Entre as principais características da água podemos destacar:
Molécula polar;
Bom solvente para moléculas polares;
Ponto de fusão e ebulição elevado;
Calor específico e calor latente de fusão/evaporação elevados;
Tensão superficial elevada;
Constante dieléctrica elevada.
Os átomos de oxigénio são mais electronegativos do que os átomos de hidrogénio, o que
resulta numa distribuição electrónica desigual nas moléculas de água: os electrões são mais
atraídos pelo núcleo do oxigénio, deixando a zona em torno de cada H com uma carga
parcialmente positiva e dando origem a um dipolo eléctrico. Consequentemente, verifica-se
uma atracção electrostática entre o oxigénio de uma molécula de água e o hidrogénio de
outra, a que se dá o nome de ligação hidrogénio.
Apesar de isoladamente cada ligação hidrogénio não ser muito energética, a uma escala
macroscópica estas interacções tornam-se bastante relevantes. No estado sólido, cada
molécula de água forma 4 ligações hidrogénio estáveis com moléculas vizinhas. Por outro lado,
no estado líquido essas ligações estão constantemente a formar-se e a ser quebradas, durando
cerca de 1 ns. No entanto, em qualquer dos casos, a quebra das ligações requer uma
quantidade considerável de energia, justificando o facto de os pontos de fusão e ebulição da
água serem elevados.
As ligações de hidrogénio podem também formar-se entre os átomos de hidrogénio da água e
átomos electronegativos (ex. oxigénio e azoto) de outras moléculas.
Uma das principais características da água é ser um bom solvente: dissolve moléculas polares
através da formação de ligações hidrogénio e sais através de interacções electrostáticas. Neste
último caso, a água rodeia os iões que compõem o sal e diminui as interacções electrostáticas
entres eles, contrariando a sua tendência para cristalizar e tornando-os solúveis.
9
Diz-se que uma substância é hidrofílica se se dissolve facilmente em água, tal como as
moléculas polares. Por outro lado, uma substância hidrofóbica é de difícil dissolução.
Compostos deste tipo, como a maioria as moléculas apolares, não formam ligações hidrogénio
e a sua presença força uma disposição da água em seu redor que é energeticamente
desfavorável. Numa tentativa de minimizar a superfície exposta à água as moléculas
hidrofóbicas tendem a formar agregados, não se dissolvendo.
Moléculas anfifílicas, que contém simultaneamente grupos polares e grupos não-polares,
tendem a dispor-se em aglomerados de modo a que as suas regiões hidrofóbicas não estejam
em contacto com a água, verificando-se assim a formação de micelas. Nas micelas, as
moléculas mantêm-se juntas não porque existam interacções entre si mas porque essa
configuração contribui para a estabilidade do sistema e para o aumento da sua entalpia. A
formação deste tipo de estruturas constitui o princípio base da formação de membranas
celulares.
Uma outra propriedade característica das moléculas de água é a sua tendência para sofrerem
ionização:
Como já é sabido, a esta reacção está associada uma constante K w, o produto iónico da água,
que relaciona as concentrações de OH- e H+ (ou H3O+ pois os iões hidrogénio não existem livres
- estabelecem logo ligação com as moléculas de água). A 25ºC, Kw=10-14.
Também já é conhecido o conceito de pH e de constante de dissociação, Ka, de um ácido, pelo
que serão apenas relembradas as respectivas fórmulas:
10
Para um qualquer ácido HA, quanto maior for a valor de Ka mais forte é o ácido.
A equação de Henderson-Hasselbach relaciona, para um qualquer ácido HA, a relação entre o
pKa, as concentrações existentes no equilíbrio e o pH da solução:
Em que pKa expressa a força relativa de um ácido: quanto mais baixo for o valor de pK a mais
forte é o ácido. Este valor corresponde ao valor de pH do ponto médio de uma curva de
titulação, ou seja, ao ponto para o qual
.
O pH do sangue ronda os 7.4 e pequenas variações, como 0.2, são suficientes para provocar a
morte de um indivíduo. No entanto, o pH noutros locais do organismo pode ter valores muito
diferentes (ex. pH do estômago pode variar entre 2.5 e 3.0) de acordo com as reacções que
ocorrem nos diferentes órgãos/tecidos. Apesar dos diferentes valores que se podem registar
nos mais variados tecidos, é necessário que, em cada caso, o pH se mantenha constante,
qualquer se que seja o seu valor ideal, para que as reacções metabólicas se dêem
correctamente. É para isso muito importante a existência de soluções tampão.
Alguns ácidos ou bases e os seus conjugados podem ser utilizados como soluções tampão na
medida que mantém o pH do meio relativamente constante face a pequenas variações das
concentrações de outros ácidos/bases da solução. Ácidos e bases fracas são os que melhor
desempenham esta função.
11
Aminoácidos e proteínas
Proteínas são as moléculas mais abundantes e mais versáteis de todos os organismos vivos.
Estão presentes em todos os locais das células e resultam da expressão de informação
genética. São formadas por polímeros de aminoácidos ligados através de ligações peptídicas,
surgem em dimensões variadas, desde pequenos péptidos até grandes cadeias, e
desempenham variadíssimas funções, desde mediar reacções químicas a transmitir impulsos
nervosos e a controlar o crescimento e diferenciação celular. Tanto podem ser estruturais
como participar num ciclo enzimático, e podem ser reactivas ou meramente mediadoras. Entre
os diferentes tipos de proteínas conhecidas podem destacar-se:
Enzimas (ex. amilase);
Hormonas (ex. insulina);
Proteínas transportadoras (ex. hemoglobina);
Proteínas de armazenamento (ex. mioglobina);
Proteínas estruturais (ex. colagénio);
Proteínas protectoras (ex. anticorpos);
Proteínas contrácteis (ex. actina e miosina);
…
Apesar da sua grande diversidade, as proteínas são formadas apenas por 20 tipos de
aminoácidos cuja natureza e sequência condiciona a estrutura final da cadeia polipeptídica e,
consequentemente, a função da proteína. Algumas das propriedades dos aminoácidos que
mais contribuem para a estrutura e função das proteínas são:
Estereoquímica (disposição espacial das moléculas);
Hidrofobilidade e polaridade relativas;
Propriedades das ligações hidrogénio;
Propriedades de ionização.
Cada aminoácido (aa) é constituído por um carbono central – carbono-α, que se liga a um
grupo amina (NH2), a um grupo carboxílico (COOH), a um hidrogénio e a um grupo lateral
variável, o grupo R ou grupo radical. Os grupos amina e carboxílico são ionizáveis e o grupo R,
que pode variar em estrutura, tamanho e carga eléctrica, é o responsável pelas características
que diferenciam os aminoácidos entre si. À excepção da glicina, cujo grupo R é apenas um
átomo de H, todos os aminoácidos apresentam um carbono-α quiral, isto é, um carbono que
estabelece quatro ligações todas diferentes.
Devido à sua configuração tetraédrica, dois aminoácidos com a mesma constituição química
podem apresentar imagens espelhadas, que representam uma classe de esteroisómeros - os
enateófilos. De um modo simples, podemos classificar cada isómero em L (levo) ou D (dextro)
conforme o seu grupo amina esteja orientado para a esquerda ou direita, respectivamente,
numa representação esquemática do aminoácido. (Esta classificação tem origem na
12
comparação dos aminoácidos com gliceraldeído, que também apresenta este tipo de
isómeros)
A treonina e a isoleucina, dois aminoácidos, apresentam dois carbonos quirais e, portanto,
podem apresentar 4 estereoisómeros.
Peptidoglicanos, constituintes das paredes celulares de células bacterianas, são formados por
D-aminoácidos. No entanto, essa é uma excepção pois, apesar de em laboratório a síntese de
aa dar origem a moléculas L e D, na maioria dos organismos vivos apenas se encontram L-aa.
Os aminoácidos podem ser classificados de acordo com vários critérios. A classificação mais
comum baseia-se na polaridade do grupo R e na sua carga eléctrica a valores de pH biológicos
(6-7):
Não polares: tendencialmente hidrofóbicos;
Polares: hidrofílicos, grupo R ionizável;
Carregados negativamente ou ácidos: o grupo R tem carga negativa, comportam-se
com um ácido;
Carregados positivamente ou básicos: o grupo R tem carga positiva, comportam-se
como uma base.
13
Há ainda uma outra de classe de aminoácidos, os aminoácidos aromáticos, que apresentam
anéis no seu grupo R, são hidrofóbicos e absorvem radiação ultravioleta ao contrário de todos
os restantes aa, que absorvem radiação na zona do infra-vermelho. A este grupo pertencem a
tirosina, o triptofano e a fenilalanina.
Os aminoácidos têm grupos ionizáveis (grupo amina, grupo carboxílico e, por vezes, também o
grupo R), pelo que podem encontrar-se sob a forma de catião ou anião. Excepto para valores
de pH extremos, coexistem várias formas ionizáveis de um aminoácido, predominando uma
delas, de acordo com a natureza ácida ou básica do meio.
Em condições fisiológicas, nomeadamente em meio aquoso, os aminoácidos designam-se
zwiteriões porque se apresentam na forma de dipolos iónicos, com ambos os grupos amina e
carboxílico ionizados. Estes grupos comportam-se como ácidos ou bases fracas,
respectivamente, pelo que é possível representar uma curva de titulação de um aminoácido.
Em aminoácidos com grupos R não ionizáveis destacam-se dois pontos na curva de titulação
que correspondem, cada um, ao pKa de um dos grupos amina ou carboxílico. O pKa do grupo
amina, NH3+ quando ionizado, localiza-se no ponto correspondente a 1.5 eq de OH- e tem um
valor básico, acima de 7. Já o pKa do grupo carboxílico, cuja forma ionizada é COO-, localiza-se
nos 0.5 eq de OH- e corresponde a um valor ácido, menor do que 7. Para aminoácidos com
grupos R ionizáveis distingue-se um outro pKa, num local que varia conforme a natureza ácida
ou básica do grupo. Em qualquer dos casos, o valor de pH para o qual se tem 1.0 eq de OH corresponde ao ponto isoeléctrico pI, i.e., ao ponto em que a carga global do aminoácido em
solução é neutra.
Para aminoácidos de grupo R não ionizável é possível calcular o valo de pI a partir dos valores
de pKa que estão associados a esse aa:
Onde
e
.
Quando o pH da solução é inferior ao pI de um dado aminoácido, este encontra-se em meio
ácido, fica carregado negativamente e migra para o cátodo. Já quando o pH do meio é superior
ao pI, diz-se que o aminoácido se encontra em meio básico, fica carregado positivamente e
migra para o ânodo.
14
A formação de péptidos dá-se por reacção de polimerização de condensação de aminoácidos.
A ligação peptídica forma-se entre o átomo de carbono (C) do grupo carboxílico e o átomo de
azoto (N) do grupo amina, com eliminação de água. As duas extremidades da cadeia assim
formada são designadas por "terminal amino" e " terminal carboxílico" ou N-terminus e Cterminus.
As ligações peptídicas são ligações covalentes muito estáveis, com 40% de carácter duplo (têm
comprimento de 0,133 nm, são mais curtas que uma ligação simples e mais longas que uma
ligação dupla), e têm uma disposição espacial dos seus átomos bem definida.
O carácter duplo parcial da ligação C-N inibe a rotação em torno da ligação peptídica e os
quatro átomos O, C, N e H definem um plano, o plano peptídico. Uma cadeia polipeptídica
pode então ser considerada como um conjunto de planos que podem rodar em torno das
ligações N-Cα (ângulo ) ou C-Cα (ângulo ).
15
Os ângulos de diedro, e , não podem assumir valores arbitrários pois certas configurações
da molécula são impossíveis devido à sobreposições de nuvens atómicas. Deste modo, estes
ângulos estão restritos a determinados valores que podem ser representados recorrendo a um
gráfico de Ramachandran:
Os diagramas de Ramachandran indicam quais as combinações dos ângulos de diedro
possíveis numa ligação peptídica e são semelhantes para todos os aminoácidos. Pequenas
variações podem ocorrer devido à complexidade dos grupos R envolvidos: as zonas favoráveis
tendem a diminuir quando a complexidade dos aa aumenta.
As ligações peptídicas são maioritariamente trans, ou seja, os grupos laterais de aminoácidos
consecutivos estão de lados opostos. As configurações cis, por norma, são mais desfavoráveis
energeticamente pelo que ocorrem numa razão de cerca de 1/1000 em relação às trans. A
excepção está no aminoácido prolina, em que a configuração cis e trans têm a mesma energia
– a sua razão cis/trans é 1/4. As ligações cis conferem instabilidade às proteínas, pelo que as
células desenvolveram mecanismos capazes de reverter as configurações das ligações.
Os aminoácidos ligam-se entre si em número variado, dando origem a cadeias de dimensões e
constituição diversa. Cadeias constituídas por até 10 resíduos são normalmente designadas
péptidos, ou oligopéptidos. Os polipéptidos apresentam geralmente entre 10 e 50 resíduos e
considera-se que as proteínas têm mais de 50 aminoácidos e pesos moleculares elevados
(expressa-se em Dalton – 1Da=1uma). No entanto, a distinção entre péptidos e proteínas não
se baseia unicamente na sua dimensão, mas sim nas respectivas funções biológicas de cada
molécula.
16
As proteínas apresentam 4 níveis de organização estrutural:
Estrutura primária: sequência linear de aminoácidos;
Estrutura secundária: arranjo local dos aminoácidos em cadeia - hélices- , folhas- ,
loops, turns, etc;
Estrutura terciária: arranjo 3D da molécula em solução;
Estrutura quaternária: organização da proteína em subunidades. Nem todas as
proteínas apresentam este tipo de estrutura.
Enquanto a estrutura primária é mantida por ligações covalentes, as restantes estruturas
devem-se a interacções mais fracas, mas não menos importantes, como ligações hidrogénio e
de Van der Waals.
A estrutura primária é a sequência linear de aminoácidos determinada pela informação
genética a partir da qual a proteína é transcrita. Os resíduos de aminoácidos ligam-se entre si
por ligação peptídicas e a cadeia apresenta um terminal carboxilo e um terminal amina. A
sequência de uma proteína lê-se a partir do terminal amina para o terminal carboxilo.
O estabelecimento de pontes de hidrogénio entre átomos de resíduos da mesma cadeia
estabiliza a estrutura da proteína e consegue-se através da variação dos ângulos e . A
estrutura resultante, designada estrutura secundária, resulta assim dos vários arranjos
espaciais de partes da cadeia linear de aminoácidos e depende da natureza destes últimos.
Identificam-se algumas estruturas frequentes - hélices, folhas, turns e loops, sendo as hélices-α
e as folhas-β as mais frequentes (mais de 60%).
Uma hélice-α tem uma forma cilíndrica, com o esqueleto da cadeia linear em hélice e os
grupos laterais em direcção ao exterior. Forma-se quando o oxigénio do grupo carboxílico
forma uma ligação de hidrogénio com o hidrogénio do grupo amina de um aminoácido quatro
resíduos a seguir, em direcção ao terminal-C. Ao longo da hélice, todos os átomos de oxigénio
do grupo carboxílico estabelecem este tipo de ligação na mesma direcção, o que confere
direccionalidade a este arranjo periódico. Os ângulos de diedro associados a este tipo de
ligação são φ~60º e ψ~45º/50º.
A cada ligação hidrogénio (estabelecida na mesma direcção) está associado um momento
dipolar, pelo que o próprio péptido acaba por apresentar um dipolo substancial – terminal-N
negativo e terminal-C positivo.
17
Cada espira da hélice representa, aproximadamente, 3.6 resíduos de aminoácidos e envolve 13
átomos – 3.613 helix. Cada resíduo estende-se por 1.5 Å, logo o passo da hélice (“altura” de
cada volta) tem 5.4 Å.
A solubilidade da hélice é determinada pela natureza das cadeias laterais porque os seus
grupos polares já estão envolvidos em ligações hidrogénio.
As folhas-β consistem em cadeias, de 5-8 resíduos, agrupadas lado a lado e cujas folhas
adjacentes se ligam por pontes de hidrogénio. A natureza das ligações estabelecidas leva as
folhas a formarem planos e a terem uma conformação final pregueada. Têm uma direcção
definida pelo que folhas adjacentes podem progredir na mesma direcção – paralelas, ou em
direcções opostas – anti-paralelas (menos estáveis). Em amba s as situações, cadeias laterais R
dispõem-se para um e outro lado da folha. Nas zonas de ligação, que fazem a continuação de
uma folha para a outra, podem existir turns e loops.
18
Turns são pequenas estruturas em forma de U, formadas por 3 ou 4 aminoácidos, que
redireccionam o esqueleto da cadeia para o seu interior, revertendo a sua direcção, e
permitem que as proteínas se tornem estruturas compactas. Em cada turn estabelece-se
apenas uma ligação hidrogénio - entre o oxigénio do grupo carboxílico do primeiro resíduo e o
hidrogénio do grupo amina do quarto aminoácido.
β-turns são das estruturas mais comuns e fazem a ligação entre cadeias anti-paralelas de
folhas-β. Muitas vezes contêm resíduos de glicina ou prolina e formam-se preferencialmente à
superfície da proteína, de modo a que os grupos que não estão envolvidos na ligação
hidrogénio possam interagir com a água.
Os loops fazem a ligação entre vários elementos da estrutura secundária das proteínas (ex.
ligam folhas-β paralelas). Ao contrário dos turns, podem ocorrer em várias formas e tamanhos.
No entanto, também se localizam à superfície das proteínas e, por isso, podem desempenhar
um papel importante no reconhecimento de processos biológicos (por exemplo, os antigénios
podem diferenciam-se entre si pelos loops que ligam as suas folhas-β).
Devido à sua estrutura não organizada, uma mutação que ocorra numa zona associada a um
loop tem tendência a ser menos grave do que se ocorresse numa folha ou numa hélice pois
não implica a destruição da estrutura proteica e, consequentemente, a perda de função.
As cadeias polipeptídicas podem ainda apresentar sequências com estrutura não tipificada,
chamas sequências random. Apesar do nome, estas sequências são bem definidas para cada
proteína e mantém-se mesmo após renaturação. Só não se encaixam é em nenhuma das
tipologias anteriores.
A função das proteínas está relacionada com a sua configuração tri-dimensional e esta
configuração, por sua vez, é especificada pela sequência de aminoácidos pelo que a natureza
dos vários resíduos favorece a adopção de diferentes tipos de estrutura secundária. Por
exemplo, o ácido glutâmico, a alanina e leucina dão sobretudo origem a hélices-α; a valina e a
isoleucina originam folhas-β; e a prolina, a glicina e a asparagina estão muitas vezes associadas
a turns.
Aos diferentes tipos de estruturas secundárias estão associados ângulos φ e ψ diferentes, pelo
que a cada tipo corresponde uma zona específica de um diagrama de Ramachandran.
19
A adopção de estruturas secundárias por parte das proteínas traz vantagens:
Permite uma compactação da estrutura, o que tende a minimizar o contacto das
cadeias laterais hidrofóbicas com a água;
As ligações por pontes de hidrogénio entre os grupos polares CO e NH das cadeias
principais (ligações essas que estabilizam as estruturas) compensam a energia gasta
para as desviar das interacções com a água.
A estrutura terciária das proteínas refere-se ao arranjo tridimensional dos resíduos de
aminoácidos e tem uma importância fundamental para a actividade biológica das proteínas.
Por exemplo, resíduos de aminoácidos muito distantes na cadeia linear podem aproximar-se
devido a enrolamentos e formar regiões indispensáveis ao funcionamento da proteína, como o
centro activo das enzimas.
Enquanto os elementos da estrutura secundária são estabilizados por ligações hidrogénio, a
estrutura terciária é influenciada essencialmente pela natureza dos grupos R dos aa e depende
de diversos tipos de ligações: iónicas, electrostáticas, pontes de hidrogénio, ligações
hidrofóbicas e covalentes. Podem ainda estabelecer-se ligações difusas que ajudam a
estabilizar a estrutura da proteína através de pontes cruzadas (crosslinks). As pontes mais
comuns são as pontes dissulfureto, formadas pela ligação S-S entre duas cistinas – aminoácido
cisteína após a oxidação do seu grupo HS, e que ocorrem maioritariamente em meio
extracelular.
20
A estabilização da estrutura final leva à dobra de elementos da estrutura secundária e, como é
maioritariamente devida a forças de interacção fracas, está em constante alteração
(mantendo, no entanto, uma conformação base), o que tem consequências importantes para a
sua função.
Por fim, a estrutura quaternária não está presente em todas as proteínas. Também é
influenciada pelos grupos R, é não linear e tem origem na associação entre várias cadeias
polipeptídicas, idênticas ou diferentes, através de ligações não covalentes. Permite o aumento
da estabilidade da proteína pela redução da relação superfície/volume, contribui para a
economia e eficiência energética e favorece a cooperatividade e a aproximação de centro
catalíticos – eficaz nas vias metabólicas/catalíticas.
A desnaturação de uma proteína é resultado da alteração das suas estruturas secundária,
terciária e quaternária, mas não da sua estrutura primária pois as ligações peptídicas só são
afectadas em condições extremas. A desnaturação leva à perda de função da proteína e pode
ser devida a agentes químicas, como o pH, ou físicos, como a temperatura.
Quando são repostas as condições fisiológicas a proteína adquire a sua conformação funcional
– ocorreu renaturação. Nalguns casos, nomeadamente quando a estrutura primária é alterada,
a proteína não é capaz de recuperar a sua função.
21
Catálise enzimática
Enzimas, uma classe particular de proteínas, são catalisadores bioquímicos cuja função é
baixar a energia de activação necessária a uma dada reacção e permitir que esta decorra a
uma velocidade muito mais elevada.
Em condições biologicamente favoráveis, muitas reacções comuns em bioquímica não são
favoráveis e só ocorrem devido à intervenção de enzimas.
Entre as características das enzimas podemos destacar:
São todas de natureza proteica, embora possam conter uma parte não proteica – o
cofactor;
Baixam a energia do estado transiente, diminuindo a energia de activação necessária a
uma reacção - aceleram a sua velocidade (velocidade até 1016x superior!);
Não modificam a constante de equilíbrio K;
Encontram-se intactas no final da reacção;
Têm elevada especificidade;
A sua actividade pode ser sujeita a controlo.
De acordo com o tipo de reacção que catalizam, as enzimas têm diferentes classificações:
No.
1
2
3
Classe
Oxidorredutases
Transferases
Hidrolases
4
Liases
5
Isomerares
6
Ligases
Tipo de reacção catalizada
Transferência de electrões (iões híbridos ou átomos H)
Transferência de grupos
Hidrólises
Adição de grupos para formar ligações duplas ou formação de
ligações duplas para remoção de grupos
Transferência de grupos entre uma mesma molécula para
formar isómeros
Formação de ligações C-C, C-S, C-O ou C-N por reacções de
condensação acopladas a clivagem de ATP
As enzimas são divididas em classes, subclasses, sub-subclasses e números de série. Na sua
nomenclatura internacional (EC) indicam-se 5 números que correspondem, respectivamente, a
cada uma das subdivisões mencionadas. Ex: Phosphoglucomutase (EC 5.4.2.2)
22
As enzimas podem ter especificidade para um determinado tipo de substrato e/ou de ligação,
apresentando diversos níveis de especificidade. Às enzimas pouco específicas costuma dar-se o
nome de enzimas promíscuas.
Em qualquer enzima, o substrato liga-se ao centro activo onde a reacção é catalisada. O centro
activo, constituído pelo conjunto de aminoácidos que entram em contacto com o substrato,
compreende o local de fixação, que se combina com o substrato por ligações fracas, e o centro
catalítico, que actua sobre o substrato levando-o a sofrer a reacção química.
A interacção enzima-substrato pode dar-se segundo dois modelos:
Chave-fechadura: sistema rígido, a configuração da enzima não é alterada aquando da
ligação do substrato;
Induced-fit: a enzima e o substrato alteram a sua conformação após a ligação.
23
Um grande número de enzimas necessita, para a sua acção catalítica, de um cofactor ou de
uma coenzima que promova a reacção.
Os cofactores são elementos químicos, como iões metálicos ou grupos prostéticos (grupos
orgânicos de natureza não proteica), e as coenzimas são moléculas, orgânicas ou metaloorgânicas, mais complexas e de natureza não proteica.
A reacção entre uma enzima (E) e um substrato (S) de modo a dar um produto (P) pode ser
representada por:
(Numa fase inicial, assume-se que a reacção inversa à formação do produto P é negligenciável porque se
verifica [P]~0 )
A velocidade a que uma reacção enzimática ocorre é, em geral, seguida através da quantidade
de produto formado por unidade de tempo. Numa fase inicial, a variação de [P] em função do
tempo é linear, pelo que se pode calcular a velocidade inicial da reacção (v0). Já a variação da
velocidade com a concentração de substrato pode ser estimada a partir da equação de
Michaelis-Menten, que assume algumas condições:
Formação de um complexo ES enzima substrato;
Equilíbrio rápido entre o complexo ES e a enzima livre;
A dissociação de ES em E+P é mais lenta que a formação do complexo ES a partir de
E+S e que a reacção inversa, a dissociação de ES para voltar a dar E+S.
Equação de Michaelis-Menten:
Velocidade máxima:
Vmax é a velocidade máxima da reacção quando há saturação da concentração do substrato. É
uma constante e representa um valor teórico pois nunca é alcançada na realidade – implicaria
que todas as enzimas estivessem ligadas ao substrato.
Constante de Michaelis:
Km, constante de Michaelis, é a constante de equilíbrio para a dissociação do complexo E-S e é
definida como a concentração de substrato quando a velocidade de reacção é metade da
velocidade máxima. É característica de cada enzima e não depende da concentração do
substrato.
24
É ainda uma medida da afinidade da enzima em relação ao seu substrato, sendo 1/K mo valor
dessa medida. Deste modo, quanto menor for o valor de Km maior é a afinidade enzimasubstrato e menor é a quantidade de substrato necessária para atingir metade da velocidade
máxima.
Pode ainda definir-se um último parâmetro, o turnover number, que se representa por Kcat e é
uma medida da actividade catalítica máxima de uma enzima. O Kcat é definido como o número
de moléculas de substrato que são convertidas em produto por molécula de enzima e unidade
de tempo quando a enzima está saturada de substrato.
O quociente
dá um índice da eficiência catalítica de uma enzima.
Os gráficos seguintes representam a curva de Michaelis para diferentes enzimas e para
diferentes concentrações de substrato, evidenciando os valores relativos de Km para cada caso.
A interpretação da cinética enzimática de Michaelis não estabelece uma relação linear entre as
grandezas em estudo. Na cinética de Lineweaver-Burk representam-se as variáveis 1/v e 1/[S],
em vez de v e [S], obtendo-se uma relação linear mais fácil de interpretar.
Equação de Lineweaver-Burk:
A partir desta equação e da sua representação gráfica é possível retirar facilmente os valores
de Km e vmax:
Quando a recta corta o eixo das ordenadas retira-se o valor de vmax:
Quando a recta corta o eixo das abcissas retira-se o valor de Km:
25
A velocidade das reacções é afectada por uma série de factores, tais como a temperatura e o
pH do meio, ou a presença de activadores ou inibidores.
A actividade enzimática tende a aumentar com a elevação da temperatura de forma
exponencial, até que se atinge um determinado valor de T a que a enzima desnatura. A relação
da velocidade com o pH é um pouco mais complexa, na medida que o pH do meio influencia
vários aspectos das enzimas, desde o seu estado de ionização à ionização dos aminoácidos do
centro activo e, em casos extremos, à desnaturação. Cada enzima tem assim uma gama de
valores de T e pH para os quais a sua actividade é optimizada.
A regulação alostérica, alteração da conformação proteica e das propriedades de um dos
locais de ligação (pode ser o centro activo) de um enzima pela associação de uma molécula
ligante a outro local de ligação do mesmo enzima (sem ser o centro activo), inclui mecanismos
tanto de activação como de inibição enzimática e é muito importante pois intervém no
controlo das sequências centrais do metabolismo.
26
Inibidores são compostos que, ao se ligarem a uma enzima, reduzem a sua actividade e a
velocidade da reacção catalisada. Os chamados inibidores reversíveis não provocam alterações
irreversíveis nas enzimas em que actuam. Existem diversos tipos de inibidores que provocam a
diminuição da velocidade da reacção actuando de modo diferente sobre a enzima:
Inibidores competitivos: são quimicamente semelhantes ao substrato e ligam-se ao centro
activo das enzimas impedindo a ligação enzima-substrato. Têm semelhanças estruturais
com o substrato para se conseguirem associar mas não reagem com o enzima e, portanto,
não dão origem a nenhum produto. Uma inibição deste tipo diminui à medida que se
aumenta a concentração de substrato - para concentrações mais elevadas é mais provável
que o enzima se ligue ao substrato que ao inibidor, mantendo-se assim o valor de vmax.
Apenas Km é alterado, verificando-se o seu aumento;
Inibidores não competitivos: ligam-se a locais da enzima que não o seu centro activo
antes ou depois da formação do complexo ES, pelo que podem formar-se três tipos de
complexos: ES, ESI (enzima-substrato-inibidor) ou EI. Como continuam a permitir a
ligação do substrato o valor de Km mantém-se inalterado. No entanto, como apenas o
complexo ES é funcional verifica-se a diminuição de vmax;
27
Inibidores anti-competitivos: ligam-se a locais da enzima que não o seu centro activo
apenas após a formação do complexo ES. Há a formação de dois complexos, ES e ESI,
sendo o complexo ES o único com actividade catalítica. Como o inibidor apenas se liga
ao complexo ES, à medida que a concentração de substrato aumenta a inibição é mais
acentuada. Verifica-se a diminuição do Km e de vmax na mesma proporção.
28
Glícidos
Glícidos ou hidratos de carbono, as moléculas mais abundantes na Terra, existem sob a forma
de monómeros – monossacarídeos, ou polímeros – dissacarídeos/dímeros e polissacarídeos.
São compostos de carbono, oxigénio e hidrogénio [(CH2O)n], e podem conter outros elementos
como azoto e enxofre. Desempenham diversas funções, desde energéticas (glucose e
sacarose) a armazenamento (amido e glicogénio são polímeros de reserva energética nas
células vegetais e animais, respectivamente) e estruturais (celulose e quitina são constituintes
da parede celular de células vegetais e fungos, respectivamente).
Alguns glícidos apresentam grupos aldeído (H-C=O) ou cetona (C=O). Se o grupo carbonilo
(C=O) se localizar no final da cadeia carbonada (inclusive num grupo aldeído), então o glícido é
uma aldose. Caso contrário, é uma quetose.
Os hidratos de carbono têm estereoisómeros cuja classificação (comparação com
gliceraldeído) segundo uma representação no plano de baseia na posição do grupo OH do
carbono mais afastado do grupo aldeído ou cetona: D - lado direito; L - lado esquerdo. Ao
contrário do que acontece nos aminoácidos, em que a forma mais comum é L-aminoácido, no
caso dos glícidos, estes apresentam-se maioritariamente sob a forma .
Pentoses e hexoses (glícidos com 5 e 6 carbonos, respectivamente) têm tendência a assumir
uma forma cíclica, principalmente quando se encontram em meio aquoso. No processo de
ciclização há eliminação de uma molécula de água quando o grupo carbonilo reage com um
grupo OH distante e o glícido passa a formar um anel. Também nestes casos se verifica a
existência de isómeros, desta vez relacionados com a posição do grupo OH correspondente ao
carbono-1: se este se localizar abaixo do anel tem a designação α, se se localizar acima do anel
designa-se β.
29
A formação de polissacarídeos dá-se pelo do estabelecimento de ligações glicosídicas. Estas
reacções de condensação resultam numa ligação covalente e na libertação de uma molécula
de água e têm liberdade de rotação. De acordo com o número de carbonos implicados na
ligação e da configuração α ou β de cada monómero, as ligações têm diferentes classificações.
Cada monómero pode estabelecer várias ligações glicosídicas em simultâneo.
Os polissacarídeos podem ser classificados de acordo com o tipo de monómeros e com o tipo
de cadeia que formam:
Homopolissacarídeos: constituídos por apenas um tipo de monómeros;
Heteropolissacarídeos: constituídos por diversos tipos de monómeros;
Não-ramificados: cadeia simples;
Ramificados: cadeia ramificada. Alguns monómeros estabelecem mais do que uma
ligação.
Para além de ligações glicosídicas os glícidos podem estabelecer outros tipos de ligações. Por
exemplo, podem estabelecer-se pontes de hidrogénio entre monómeros adjacentes e os
peptidoglicanos formam cadeias ligadas entre si por aminoácidos.
30
Lípidos
Lípidos são compostos de carbono e hidrogénio (cadeias alifáticas, formados por –CH2– ),
geralmente com mais de 8 carbonos, caracterizados por uma baixa solubilidade em água e
elevada solubilidade em solventes não-polares (como o benzeno e o clorofórmio).
Desempenham funções variadas destacando-se os papéis de reserva energética, precursores
hormonais, constituintes das membranas biológicas, cofactores, etc. A sua composição
química é variada e dividem-se em várias classes, entre as quais se destacam os ácidos gordos
e os fosfolípidos.
Os ácidos gordos são ácidos carboxílicos (têm um grupo COOH) com cadeias de
hidrocarbonetos formadas por um número variado de carbonos (entre 4 e 36). Raramente
ocorrem livres na natureza e podem ser considerados como a unidade fundamental da classe
dos lípidos.
O grupo carboxílico, que tem um pKa entre 4-5, em condições fisiológicas encontra-se ionizado
e constitui a extremidade polar do ácido gordo. A cadeia hidrocarbonada é apolar e as ligações
covalentes entre os átomos de carbono são maioritariamente simples, mas também podem
apresentar carácter duplo. Se todas ligações C-C forem simples, o ácido gordo diz-se saturado.
Caso existam ligações duplas, estamos na presença de um ácido gordo insaturado –
monoinsaturado se tiver apenas uma ligação dupla ou polinsaturado se estas forem em maior
número. O nível de saturação dos ácidos gordos está relacionado com o seu ponto de fusão.
Regra geral, quanto mais insaturado o lípido for menor será a sua temperatura de fusão.
Na nomenclatura dos ácidos gordos indica-se o número de átomos de carbono da cadeia
hidrocarbonada e o nº de ligações duplas, separados por “:”. Em índice superior e antecedido
do símbolo Δ, indica-se a posição das ligações duplas. Assume-se que essas ligações são cis,
excepto se especificado em contrário, e numeram-se os carbonos a partir do grupo carboxilíco.
Ex: C 18:3 Δ9,12,15.
Todos os ácidos gordos sintetizados pelo organismo humano são do tipo cis, ou seja, nas suas
ligações duplas C=C os átomos de hidrogénio ligados aos átomos de carbono encontram-se
orientados para o mesmo lado. Esta insaturação em cis altera a conformação das caudas
carbonadas - deixam de ser lineares e passam a estar “dobradas”, formando-se um ângulo de
~30º na zona da ligação dupla. Este facto é muito importante quando os ácidos gordos se
encontram em membranas ou agregados pois confere flexibilidade às estruturas.
Ácidos gordos insaturados em trans são produzidos por algumas bactérias presentes no
sistema digestivo dos ruminantes e são obtidos pelos humanos através do consumo de
produtos lácteos, carne animal ou pelo consumo de produtos que sofreram hidrogenação. A
natureza da ligação trans não altera a conformação das caudas carbonadas do mesmo modo
que a ligação cis e está associada ao aumento dos níveis de LDL (“mau colesterol”) e
diminuição de HDL (“bom colesterol”), pelo que é recomendável a sua ingestão moderada.
31
A partir de ácidos gordos podem formar-se diferentes tipos de lípidos, sendo os triglicéridos
dos mais simples. Os triglicéridos constituem uma grande fonte de reserva energética
(encontram-se armazenados nas células adiposas) e são formados a partir de uma molécula de
glicerol associada a três ácidos gordos. Se esses ácidos gordos forem iguais o triglicérido diz-se
simples.
A hidrólise de um triglicérido/diglicérido é uma forma de obter energia metabólica. A reacção
de degradação leva à formação de um diglicérido/monoglicérido e à libertação de um ácido
gordo, energia e uma molécula de água. Posteriormente o ácido gordo pode ser degradado
fornecendo grandes quantidades de energia.
32
Os fosfolípidos pertencem a uma outra classe de lípidos. Estas moléculas são os principais
constituintes das membranas biológicas e apresentam duas zonas com polaridade distintas –
são moléculas anfipáticas, ou anfifílicas.
Têm uma cabeça polar hidrofílica formada por um grupo fosfato ligado a uma molécula de
glicerol e, por vezes, a um outro grupo variável. Ao glicerol ligam-se dois ácidos gordos, que
podem ser diferentes entre si, e que constituem a cauda apolar hidrofóbica.
Os lípidos são moléculas hidrofóbicas mas muitas vezes são constituídas por uma zona polar
que tem afinidade com a água, como é o caso dos fosfolípidos. Deste modo, quando se
encontram em meio aquoso estas moléculas tendem a associar-se de modo a que as regiões
polares estejam em contacto com a água e as regiões apolares não. Os agregados que se
formam só são estáveis em ambiente aquoso e, dependendo do tipo de lípidos, podem
originar micelas – estruturas esféricas constituídas por ácidos gordos, ou lipossomas –
bicamadas de forma cilíndrica formadas por fosfolípidos. Este princípio está na base da
formação das membranas biológicas.
As membranas biológicas são compostas por lípidos, maioritariamente fosfolípidos,
glicolípidos e colesterol, e proteínas, que conferem especificidade. A função das membranas
passa pela individualização das células e compartimentalização celular, criação de uma
barreira selectiva, localização de sistemas enzimáticos, de transporte e ainda de locais de
reconhecimento específico (hormonas, antigénios, etc).
As membranas são uma bicamada fosfolipídica assimétrica intercalada por outras moléculas,
tanto de natureza lipídica (colesterol) como de natureza proteica (proteínas transportadoras).
Os fosfolípidos têm liberdade para sofrer rotações, movimentos laterais e, por vezes, até
trocas entre camadas. A fluidez das membranas é afectada pelo nível de saturação das cadeias
carbonadas (quanto mais insaturadas forem maior será a fluidez) e pela presença de
colesterol.
À superfície das membranas destacam-se zonas mais densas, as jangadas lipídicas, que são
mais ricas em colesterol e ácidos gordos saturados. Estas jangadas são menos fluidas que as
zonas adjacentes e são ricas em proteínas com determinadas funções. São bem definidas mas
podem deslocar-se ao longo da membrana.
33
O colesterol é um tipo de lípido formado por um conjunto rígido de anéis associado a uma
pequena cadeia carbonada. É maioritariamente hidrofóbico mas apresenta um grupo
hidróxido OH polar, pelo que é uma molécula anfipática. É produzido apenas por animais (no
fígado), intercala-se nas membranas plasmáticas regulando a sua fluidez, e é um precursor de
hormonas como a testosterona e o estrogénio.
34
Metabolismo
Entende-se por metabolismo o conjunto de todas as reacções químicas que se dão dentro das
células. Inclui:
Catabolismo: processos oxidativos e exergónicos nos quais há libertação de energia
pela degradação de produtos complexos em produtos mais simples.
Ex: glicólise produz glicose a partir de glicogénio;
Anabolismo: processos redutivos endergónicos que requerem o uso de energia para
formar produtos complexos a partir de moléculas mais simples.
Ex: glicogénese armazena excesso de glicose sob a forma de glicogénio. Lipogénese
armazena glicose e aminoácidos sob a forma de lípidos.
Enquanto as vias catabólicas convergem para poucos produtos finais as vias anabólicas
divergem para a síntese de muitas biomoléculas diferentes. Por exemplo, o catabolismo de
lípidos, glícidos e proteínas origina um intermediário comum - acetil-CoA, que é utilizado na
respiração celular. Este intermediário, por processos anabólicos, pode depois dar origem às
mais variadas moléculas, desde fosfolípidos a triglicéridos, hormonas esteróides e vitaminas.
A maioria dos processos metabólicos inclui uma série de reacções e está organizada em vias
metabólicas reguladas por enzimas. Os enzimas intervenientes podem encontrar-se isolados,
em complexos multienzimáticos ou formar sistemas associados a membranas. A localização
de todo o complexo numa zona específica da célula, como uma região da membrana, permite
que o processo seja mais eficiente.
Uma via metabólica tem início num substrato específico e termina num determinado produto.
Durante todo o processo dão-se vários passos, cada um catalisado por um enzima específico, e
os produtos de uma reacção tornam-se substratos das seguintes, pelo que se designam
intermediários. O facto de as vias estarem divididas em várias reacções permite não só que se
obtenham produtos que de forma espontânea não seria possível (acoplamento de reacções
termodinamicamente favoráveis a processos desfavoráveis), como também que se regule o
calor/energia libertado e se mantenha a temperatura da célula dentro de valores fisiológicos.
Embora as vias anabólicas e catabólicas de degradação e síntese dos mesmos compostos
sejam semelhantes, é necessário que apresentem alguns passos diferentes. Só assim se obtêm
mecanismos de regulação que permitem favorecer uma das vias e inibir a outra, e também só
deste modo é que ambas as vias podem ocorrer em simultâneo e de forma espontânea, já que
se não existisse qualquer diferença entre elas as leis do equilíbrio termodinâmico ditariam que
as vias apenas se dessem num sentido.
As vias metabólicas principais da maioria dos organismos apresentam muitas semelhanças, o
que sugere que todas as formas de vida descendem de um ancestral comum. A molécula de
35
ATP, por exemplo, é o intermediário energético mais utilizado por todos os seres vivos embora
outras moléculas, como o NAD ou NADP, também desempenhem o mesmo papel.
O ATP, adenosina trifosfato, armazena energia nas ligações dos seus grupos fosfato e a sua
síntese ou degradação está acoplada a muitos processos exergónicos ou endergónicos,
respectivamente. Através do ciclo do ATP, a energia conseguida durante a fotossíntese ou
outros processos catabólicos é transportada para os processos celulares que necessitam de
energia.
Para além do ATP as células têm outros co-factores que actuam como intermediários
energéticos. FAD, NAD, e NADP, cujas formas reduzidas são, respectivamente, FADH, NADH e
NADPH, são transportadores de electrões e armazenam energia sob a forma de electrões num
alto nível energético.
A síntese de ATP dá-se por 3 processos:
Glicólise, ou outras vias metabólicas que levem à formação de acetil-CoA
(metabolismo glicídico, lipídico ou proteico);
Ciclo de Krebs, ou ciclo do ácido cítrico;
Cadeia respiratória.
36
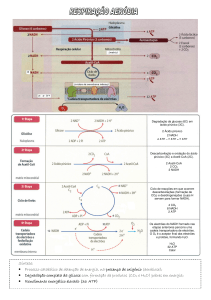
Glicólise e Neoglucogénese
A glicólise é o processo no qual uma molécula de glicose é degrada, através de 10 reacções
catalisadas enzimaticamente, e forma 2 moléculas de ácido pirúvico, um composto de 3
carbonos. É comum a todos os organismos, não utiliza oxigénio e dá-se no citoplasma.
Pode dividir-se em 3 fases:
Fase 1 (fase de activação): fosforilação da glicose com consumo de 2 ATP;
o Reacção 1: fosforilação da glicose em glicose-6-fosfato com gasto de 1 ATP;
o Reacção 2: isomerização da glicose-6-fosfato em frutose-6-fosfato;
o Reacção 3: fosforilação da frutose-6-fosfato em frutose-1,6-fosfato com gasto
de 1 ATP;
Fase 2 (fase de clivagem): formação de 2 moléculas de gliceraldeído-3-fosfato;
o Reacção 4: cisão da frutose-1,6-fosfato em gliceraldeído-3-fosfato e no seu
isómero di-hidroxiacetona-fosfato;
o Reacção 5: isomerização de di-hidroxiacetona-fosfato em gliceraldeído-3fosfato;
Fase 3: oxidação de cada gliceraldeído-3-fosfato (GAP) em ácido pirúvico e formação,
no total, de 4 ATP e 2 NADH+H+. As reacções seguintes referem-se a cada molécula de
gliceraldeído-3-fosfato;
o Reacção 6: oxidação do gliceraldeído-3-fosfato em ácido 1,3-difosfoglicérico
com redução de NAD a NADH + H+;
o Reacção 7: conversão do 1,3-difosfoglicérico em ácido 3-fosfoglicérico com
síntese de 1 ATP;
o Reacção 8: isomerização do ácido 3-difosfoglicérico em ácido 2-fosfoglicérico;
o Reacção 9: desidratação do ácido 2-fosfoglicérico em fosfoenolpiruvado;
o Reacção 10: conversão de fosfoenolpiruvado em piruvado (ácido pirúvico) com
síntese de 1 ATP;
37
A equação global da glicólise é:
Reacção
1
Glucose glucose-6-fosfato
3
Frutose-6-fosfato frutose-1,6-fosfato
6
2 x ( gliceraldeído-3-fosfato ácido 1,3-difosfoglicérico )
7
2 x (1,3-difosfoglicérico ácido 3-fosfoglicérico )
10 2 x (fosfoenolpiruvado piruvado )
Balanço energético final
ATP
-1
-1
0
2
2
2
NADH+H+
0
0
2
0
0
2
O produto final da glicólise é o ácido pirúvico, que vai depois ser utilizado na respiração
celular. Em condições anaeróbicas (ausência de oxigénio) ocorre fermentação. Há vários tipos
de fermentação, cada um com produtos finais diferentes, mas em todos os casos o balanço
final de ATP é sempre de 2 moléculas.
Na fermentação alcoólica, que ocorre, por exemplo, nas leveduras, o produto final é o CO2 e o
etanol. Neste tipo de fermentação o ácido pirúvico proveniente da glicólise é descarboxilado
libertando CO2 e originando aldeído acético, um composto com 2C. Por acção do NADH
reduzido, também proveniente da fase da glicólise, forma-se etanol.
38
Na fermentação láctica, que se verifica, por exemplo, nos bacilos lácticos, o ácido pirúvico é
reduzido a ácido láctico. Após a glicólise, o piruvado combina-se com o H+ transportado pela
NADH e origina o ácido láctico.
Na respiração aeróbia (em presença de oxigénio) o ácido pirúvico formado durante a glicólise é
transportado para o interior das mitocôndrias, onde vai integrar o ciclo de Krebs 40
e a cadeia de transporte electrónico. No final de todo este processo o balanço teórico final de
ATP será de 36 nos organismos eucariotas e de 38 nas bactérias.
A neoglucogénese, ou gluconeogénese, é via metabólica que conduz à síntese de glicólise a
partir de ácido pirúvico. Ocorre principalmente no fígado e nos rins e, apesar de a maioria das
suas reacções serem inversas às da glicose, algumas, que por razões de ordem termodinâmica
não são reversíveis, variam. Nesses casos, as reacções são catalisadas por enzimas diferentes e
constituem os principais pontos de regulação das duas vias. Os passos alterados são:
(Reacção 10) conversão de piruvato em fosfoenolpiruvado;
(Reacção 3) conversão da frutose-1,6-fosfato em frutose-6-fosfato;
(Reacção 1) conversão da glicose-6-fosfato em glicose.
A regulação da neoglucogénese está relacionada com a regulação da glicólise:
Quando a glicólise está em funcionamento, a gluconeogénese não está a realizar-se;
Quando o estado energético da célula é elevado a glicólise deve ser desligada e o
piruvato, entre outros, deve ser utilizado para a síntese e armazenamento de glucose;
Quando o estado energético da célula é baixo a glucose deve ser rapidamente
degradada de modo a fornecer energia;
Os passos regulados da glicólise são os mesmos passos que são regulados na direcção
oposta.
Ciclo de Krebs
O ciclo de Krebs, ou ciclo do ácido cítrico, dá-se na matriz mitocondrial e consiste na oxidação
de acetil-CoA a CO2 com libertação de energia sob a forma de electrões, que é armazenada em
NADH e FADH2, moléculas transportadoras de electrões.
A acetil-coenzima A é o produto de diversas vias metabólicas mas, como vimos anteriormente,
não é um produto final da glicólise. Para que o ácido pirúvico possa integrar o ciclo de Krebs é
necessário um passo preparatório em que o piruvato é convertido em acetil-CoA.
Na respiração aeróbia, após a etapa da glicólise o ácido pirúvico entra nas mitocôndrias e, ao
nível da matriz mitocondrial, sofre 3 reacções que culminam na formação de acetil-CoA:
39
1. Descarboxilação: é removido um carbono ao ácido pirúvico. Forma-se acetaldeído e
dióxido de carbono;
2. Oxidação: são removidos dois H ao acetaldeído. Forma-se ácido acético e reduz-se
NAD+ a NADH+H+;
3. Formação de acetil-CoA: o ácido acético combina-se com uma coenzima A e forma-se
acetilcoenzima A.
O acetil-CoA vai agora integrar o Ciclo de Krebs, que tem 8 passos:
Reacção 1: formação de citrato: condensação de acetil-CoA com o ácido oxaloacético.
Há transferência do grupo acetilo e libertação da coenzima A;
Reacções 2a e 2b: formação de ácido isocítrico pela transferência do grupo OH da
posição 3 para a posição 4;
Reacção 3: oxidação do isocitrato a ácido α-cetoglutárico com libertação de NADH ou
NADPH e CO2;
Reacção 4: oxidação do ácido α-cetoglutárico a succinil-CoA. Libertação de CO2 e
produção do cofactor NADH;
Reacção 5: hidrólise de succinil-CoA a ácido succínico com síntese de ATP;
Reacção 6: oxidação de ácido succínico a ácido fumárico com libertação de FADH 2;
Reacção 7: hidratação do ácido fumárico a ácido málico;
Reacção 8: oxidação do ácido malático a ácido oxaloacético.
40
Balanço do Ciclo de Krebs
ATP
NADH+H+
FADH2
CO2
Por acetil-CoA
1
3
1
2
Por ciclo
2
6
2
4
Cada acetil-CoA contribui para a produção de 12 ATP:
Balanço no final do Ciclo de Krebs
Glicólise
Obtenção de acetil-CoA
Ciclo de Krebs
Por molécula de glicose
ATP NADH+H+ FADH2
2
2
2
2
6
2
4
10
2
CO2
2
4
6
Cadeia respiratória
A cadeia respiratória, ou cadeia de transporte electrónico, é a etapa em que culminam todos
os processos oxidativos de degradação de glícidos, lípidos ou proteínas. Esta fase dá-se nas
cristas mitocondriais e permite a síntese de ATP utilizando a energia libertada durante a
redução de O2 a H2O, cujos electrões são doados pelo NADH e FADH2 provenientes da glicólise
e ciclo de Krebs.
Nas cristas mitocondriais encontra-se um conjunto de 3 complexos enzimáticos (I, III, IV),
ligados por dois transportadores electrónicos (CoQ e Citocromo C). O conjunto destas 8
enzimas, associadas a átomos metálicos (cofactores) capazes de aceitar e doar electrões,
forma a cadeia respiratória. Seis das proteínas que fazem parte desta cadeia encontram-se
fixas, enquanto as restantes duas são móveis.
Quando os cofactores NADH+H+ e FADH2 libertam hidrogénio para a cadeia respiratória dão
início ao processo de fosforilação oxidativa. Neste processo as reacções de oxidação e
fosforilação estão acopladas, o que permite a síntese de ATP:
Cada H fornecido à cadeia é separado em H+ + e-;
o Os protões são bombeados para o espaço intermembranar criando um
gradiente de pH e um potencial electroquímico transmembranar de H+. Por
cada NADH+H+ são bombeados 10 H+ e por cada FADH2 apenas 6 H+ - o FADH2
liberta os seus protões apenas no segundo transportador;
o Os electrões vão passar de transportador electrónico em transportador
electrónico até serem entregues ao oxigénio;
Os electrões são fornecidos ao O2, dando origem a iões;
Os iões oxigénio atraem H+ e forma-se água;
O potencial de H+ provoca a difusão destes iões de novo para o interior da matriz
mitocondrial e permite a síntese de ATP via ATPsintase;
o O complexo V, ATPsintase, é formado por três partes. A corrente criada pela
passagem de H+ provoca a rotação de duas dessas partes, o rotor e o haste, e
41
o
activa os sítios activos da terceira parte, o bastão, onde é formado ATP a partir
de ADP+Pi;
Cada NADH origina 3 ATP e cada FADH2, como só leva ao bombeamento de 6
H+, permite apenas a síntese de 2 ATP.
Nº de moléculas
NADH+H+ 10
FADH2
2
Balanço final
Glicólise
Obtenção de acetil-CoA
Ciclo de Krebs
Balanço intermédio
Cadeia Respiratória
Por molécula de glicose
42
ATP por molécula
3
2
ATP
2
2
4
34
30** 36* 38
ATP formado
30
4
34
NADH+H+ FADH2
2
2
6
2
10*
2
-10
-2
0
0
CO2
2
4
6
6
* Nos eucarioentes um dos cofactores NADH é transportado por uma molécula que não
atravessa a membrana das mitocôndrias, o que leva a que no final da cadeia respiratória só se
tenham formado 36 ATP. Nas bactérias todos os NADH+H+ são aproveitados, pelo que a
contabilização total de ATP é 38.
** Na verdade, os valores aqui mencionados são apenas os esperados teoricamente. Na
prática, verifica-se que os organismos eucariontes produzem apenas 30 ATP por molécula de
glicose. Esta diferença deve-se ao facto de a célula utilizar o gradiente de protões criado
durante o transporte electrónico para outros propósitos que não apenas a actividade da
ATPsintase.
Outras vias metabólicas
Outras vias metabólicas que levam à produção de energia metabólica são a oxidação dos
ácidos gordos (as reservas de lípidos no corpo humano perfazem cerca de 85% do total de
energia disponível) e o catabolismo de proteínas.
Oxidação de ácidos gordos
Os triglicéridos podem ser degradados e dar origem a 3 ácidos gordos, que entram no ciclo de
oxidação dos ácidos gordos, e a uma molécula de glicerol, que se torna um substrato da
glicólise. As enzimas responsáveis pela quebra das ligações dos ácidos gordos são as lipases.
Para poderem entrar no ciclo de oxidação os ácidos gordos livres têm que ser activados, isto é,
têm que ser convertidos em acil-CoA gordo e transportados para as mitocôndrias, onde se vai
dar todo o processo de extracção de energia. A activação dos ácidos gordos dá-se no
citoplasma, pela enzima acil-CoA gordo sintetase e com gasto de 2 ATP, e o seu transporte
está dependente da bomba de carnitina, onde o grupo acil-CoA é substituído por acil-carnitina
para poder entrar na mitocôndria. Uma vez dentro do organelo a acil-carnitina volta a ser
convertida em acil-CoA.
43
A oxidação de ácidos gordos (activados) ocorre no interior das mitocôndrias e envolve três
etapas:
1ª etapa – β-oxidação: uma cadeia de ácidos gordos é oxidada dando origem a
resíduos de acetil-CoA;
2ª etapa: acetil-CoA entram no ciclo de Krebs e são oxidadas a CO2;
3º etapa: os electrões provenientes das oxidações anteriores entram na cadeia
respiratória e permitem a síntese de ATP.
A β-oxidação é um ciclo em espiral e engloba 4 reacções:
Reacção 1, oxidação: remoção de H dos carbonos α e β (primeiros dois carbonos
ligados a hidrogénios), formação de uma ligação dupla C=C em trans e redução de FAD
a FADH2;
Reacção 2, hidratação: adição de H20 à ligação dupla acabada de formar, processo
catalisado pela enzima enoil-CoA hidratase. Ao carbono α é adicionado um H e ao
carbono β é adicionado um grupo OH, formando um grupo hidroxilo (COH).
A enzima enoil-CoA hidratase só actua em ligações duplas do tipo trans.
Consequentemente, ácidos gordos insaturados, que contém ligações duplas do tipo
cis, requerem a acção um outra enzima - uma isomerase, que converte as ligações cis
em trans;
Reacção 3, segunda oxidação: oxidação do grupo hidroxilo, formação de um grupo
cetona no carbono β e redução de NAD+ a NADH;
Reacção 4, clivagem: a ligação entre Cα e Cβ é clivada dando origem a uma molécula
de acetil-CoA e a um acil-CoA gordo com menos dois carbonos que entra de novo no
ciclo de β-oxidação.
44
A oxidação de ácidos gordos é o processo mais rentável para a obtenção de energia
metabólica. O número de carbonos de um ácido gordo determina o número de oxidações
necessárias e o número de acetil-CoA formadas, determinando consequentemente a
quantidade de ATP que pode ser sintetizada:
Balanço energético genérico
Activação do ácido gordo
-2 ATP
Β-oxidação
1 NADH + 1 FADH2 por ciclo – 5 ATP
Ciclo de Krebs (Acetil-CoA) 12 ATP por cada acetil-CoA
Deste modo, por cada ácido gordo com n carbonos (n par) temos:
Balanço energético final – n carbonos
Activação do ácido gordo
Β-oxidação
n/2-1 ciclos
Ciclo de Krebs (Acetil-CoA) n/2 moléculas
-2
5 x (n/2-1)
12 x (n/2)
Por exemplo:
Carbonos
12
14
16
18
Acetil-CoA
6
7
8
9
Β-oxidação
5
6
7
8
ATP total
167
196
225
254
Catabolismo proteico
O catabolismo de proteínas nos mamíferos dá-se principalmente no fígado e envolve:
Quebra das ligações peptídicas e degradação das proteínas em aminóacidos;
Deaminação dos aminoácidos (remoção de um grupo amina - NH2) com formação de
amónia (NH3) e de um ácido orgânico;
o A amónia entra no ciclo da ureia e produz ureia, que é eliminada pelo
organismo;
o O ácido orgânico formado pode entrar em diferentes fases da glicólise ou do
ciclo de Krebs, de acordo com a natureza do seu grupo R. Por exemplo, a
deaminação da alanina dá origem a piruvato mas o aspartato já origina
oxaloacetato.
Devido às diferentes moléculas que podem resultar do catabolismo das
protéinas não é possível estimar a quantidade de ATP formado de um modo
geral – cada caso é um caso.
45
46
Fotossíntese
Fotossíntese é o processo através do qual alguns seres vivos produzem matéria orgânica a
partir de matéria mineral utilizando energia luminosa. Os seres que realizam este processo
chamam-se fotoautotróficos e neles estão incluídas as plantas, as algas e algumas bactérias.
A fotossíntese dá-se ao nível dos cloroplastos. O prolongamento da sua membrana forma
tilacóides onde se encontram os pigmentos fotossintéticos, responsáveis pela absorção da luz,
e a cujo conjunto se dá o nome de grana. Estas estruturas estão inseridas no estroma, uma
matriz fluida.
Existem diversos pigmentos fotossintéticos, que apresentam diversas cores e se encontram em
diferentes seres vivos:
Tipo de pigmento
Clorofilas: a
b
c
d
Carotenóides: carotenos
xantofilas
Ficobilinas: Ficoeritrina
Ficocianina
Cor
Verde
Laranja
Amarela
Vermelha
Azul
Distribuição
Plantas, algas, algumas bactérias
Plantas, algas verdes
Algas castanhas, diatomácias
Algas vermelhas
Algas e plantas
Algas castanhas, diatomácias
Algas vermelhas, algumas bactérias
Os dois grandes grupos de pigmentos fotossintéticos que absorvem energia luminosa,
elevando electrões para níveis de energia mais altos, são as clorofilas e os carotenóides. Cada
um destes pigmentos absorve preferencialmente uma gama de radiações: as clorofilas a e b
absorvem principalmente radiações correspondentes às faixas azul-violeta e vermelhoalaranjado (apresentando assim cor verde), enquanto os carotenóides se restringem à faixa
azul-violeta. No entanto, apenas a clorofila a participa directamente nas reacções luminosas.
47
Todos os outros pigmentos apenas adicionam energia à clorofila ou dissipam energia que foi
absorvida em excesso.
Em 1930, Van Niel, estudando o comportamento de bactérias sulfurosas capazes de realizar a
fotossíntese, concluiu que o oxigénio libertado neste processo provinha da água e não do
dióxido de carbono. Marcou a água com oxigénio 18O e verificou que este era libertado na
fotossíntese e não integrava os compostos orgânicos formandos. Mais tarde, na experiência de
Calvin, através da marcação do dióxido de carbono com carbono radioactivo comprovou-se
que as substâncias formadas, de carácter radioactivo, derivavam do CO2 fornecido.
Gaffron, em 1951, através de uma experiência provou que a luz era necessária para iniciar o
processo fotossintético, o qual poderia depois continuar durante alguns segundos mesmo na
sua ausência. A fotossíntese compreende assim duas fases sucessivas:
Fase fotoquímica, ou fase luminosa/clara: dependente da luz. Dá-se a excitação dos
pigmentos fotossintéticos e a fotólise da água. É uma fase exergónica, produzindo-se
ATP, NADPH e 02;
Fase química, ou fase escura: também conhecida com Ciclo de Calvin. Esta fase
depende não da luz mas sim da presença de CO2 e é endergónica. A partir de dióxido
de carbono e ATP e electrões do NADPH, provenientes da fase fotoquímica, produz-se
glicose (C6H12O6).
Fase fotoquímica
A fase fotoquímica ocorre nos tilacóides e transforma a energia luminosa captada pelos
pigmentos fotossintéticos em energia química. Nesta fase intervêm dois fotossistemas conjuntos de pigmentos associados a proteínas que se encontram na membrana dos tilacóides
e providenciam a energia necessária para excitar os electrões da clorofila:
Fotossistema II (PSII) absorve comprimentos de onda correspondentes a 680 nm e
participa na síntese de ATP. Encontram-se na face da membrana dos tilacóides que
está virada para o interior da grana;
Fotossistema I (PSI) absorve comprimentos de onda correspondentes a 700 nm e
participa na síntese de NADPH. Encontra-se na face da membrana que está em
contacto com o estroma.
48
Nos fotossistemas fotões são absorvidos pelos pigmentos antena e a sua energia é canalizada
para o centro de reacção, composto por uma clorofila e por um aceitador primário de
electrões. A energia captada oxida a clorofila e o electrão libertado é removido através da
redução do aceitador primário de electrões, podendo então ser conduzido por dois caminhos:
Fotofosforilação acíclica: contribui para a síntese de ATP e de NADPH, em igual
quantidade, e utiliza os dois fotossistemas PSI e PSII. Os electrões do PSII são
removidos e substituídos por electrões provenientes da fotólise da água,
;
Fotofosforilação cíclica: contribui apenas para a síntese de ATP e só utiliza o PSI, cujos
electrões são reciclados.
Na fotofosforilação acíclica a clorofila a do centro de reacção do PSII é oxidada e os electrões
libertados vão ser transferidos para outras moléculas através de sucessivas reacções de
oxidação-redução, dando início ao transporte electrónico que permite a síntese de ATP por
quimiosmose. Electrões provenientes da fotólise da água reduzem a clorofila desse sistema,
deixando-a apta a recomeçar um novo ciclo, e é libertado O2.
Ao longo do transporte electrónico os electrões vão perdendo energia e ao chegar ao PSI, já
num baixo nível energético, reduzem a Cla+ aí existente. Pela incidência da luz solar, a Cla é
49
oxidada de novo e dá-se início a mais um transporte electrónico que, juntamente com os
protões libertados pela água, contribui para a redução de NADP+ a NADPH.
A fosforilação do ATP dá-se por quimiosmose, ou seja, a difusão de iões, neste caso H+,
permite a síntese de ATP aquando da passagem de 2H+ pela enzima ATP Sintetase. A difusão
destes protões é garantida pelo gradiente de concentração existente entre o estroma e o
interior do tilacóide, um meio de pH muito mais baixo. Uma vez que na fotólise da água se
libertam não só e- como também H+, este processo ajuda a assegurar a diferença de pH
necessária ao sucesso da fotossíntese.
Ao passarem para o estroma um dos H+ é utilizado na formação de NADPH enquanto o outro é
de novo enviado por transporte activo para o interior do tilacóide. A energia necessária a este
transporte provém da energia libertada durante o transporte electrónico de electrões no PSII e
durante a fotofosforilação cíclica.
50
A fotofosforilação cíclica só contribui para a sintetização de ATP. Um electrão sai do PSI em
direcção à zona de transporte activo de H+, voltando depois ao PSI onde recebe energia para
recomeçar o processo. Durante o seu percurso o e- liberta energia que vai ser utilizada no
transporte activo de H+ que cria o gradiente necessário à síntese de ATP.
Fase química
A fase química é também conhecida como Ciclo de Calvin e é onde se dá a produção de
açúcares. Este ciclo engloba 3 fases:
Fase 1, fixação de CO2: 3 moléculas de CO2 reagem com 3 moléculas de RuDP (ribulose
bifosfato), um composto com 5 carbonos e 2 fosfatos, dando origem a um composto
com 6C muito instável. A enzima que participa nesta reacção é a RuBP carboxilase, ou
rubisco. O composto instável divide-se rapidamente formando 6 moléculas de 3fosfoglicerado, que têm apenas 3C cada;
Fase 2, produção de açúcares: seguidamente, dá-se a fosforilação do 3-fosfoglicerado
em 1,3-bifosfoglicerado utilizando-se 6 ATP e a sua redução em gliceraldeído 3fosfato (PAGL) recorrendo-se a 6 NADPH. Formam-se então 6 PGAL, dos quais um
abandona o ciclo e é mobilizado para a síntese de moléculas orgânicas. Os restantes 5
PGAL permanecem no ciclo de Calvin;
Fase 3, regeneração de RuDP: 5 gliceraldeído 3-fosfato originam 3 moléculas de RuMP
(ribulose monofosfato), um composto com 5C mas apenas 1P. Recorrendo-se a 3 ATP
regeneram-se os 3 RuDP que serão utilizados para dar início a um novo ciclo.
Quando se fala no ciclo de Calvin consideram-se 3 ciclos como a unidade, pois apenas assim se
permite a formação e regeneração correcta de todos os elementos. O ATP e NADPH utilizados
neste processo provêm da fase fotoquímica e o ADP e NADP+ resultantes são reintegrados na
nessa fase, onde se regeneram. Durante este processo gasta-se mais ATP do que NADH o que
explica a necessidade da fotofosforilação cíclica.
51