QBQ4020 – Bioquímica Metabólica
Início: 03/08/2015
Fim: 08/12/2015
Dias e horário: sextas-feiras das 19hs às 22h40
Local: sala 04 B6 inferior
Professor: Roberto K. Salinas
Contato: [email protected]; sala 1001 do bloco 10 inferior
Disciplina obrigatória para “Licenciatura Noturno” (sexto semestre) e “Química
Ambiental” (sexto semestre)
Pré-requisitos: Introdução à bioquímica e Química orgânica II
Bibliografia: Stryer e Lehninger
Mês
Agosto
Assunto
Hemoglobina e mioglobina, cooperatividade
Cinética enzimática, equação de Michaelis-Menten
Cinética enzimática
Mecanismos de catálise e tipos de inibição
28 Cinética
Membrana biológica
Compostos ricos em energia (ex. ATP, GTP)
Setembro
4
Transporte de membrana
Revisão de estruturas de carboidratos
Introdução ao metabolismo dos carboidratos
11 Semana da Pátria
18 Revisão para Prova 1
25 Semana da Química
Outubro
02 Prova 1
09 Via glicolítica
Ciclo de Krebs
16 Cadeia de transporte de elétrons e fosforilação oxidativa
23 Degradação e síntese de glicogênio
Neoglicogênese
Regulação, enzimas alostéricas
Integração do metabolismo de carboidratos, sinalização por
insulina e glucagon
30 Metabolismo de ácidos graxos
Fotossíntese
Novembro 06 Revisão
13 Prova 2 – não cai fotossíntese
20 Dia da consciência negra
27 Ação hormonal e integração metabólica/Biossíntese de
carboidratos em plantas e bactérias
Dezembro 04 Prova 3
Dezembro 11 Prova substitutiva (aberta)
Dezembro 15 Data máxima para cadastro de notas
Média Final = (P1 + P2 + P3)/3
Observação: 1) Os alunos que entregarem os exercícios obterão até 0.25 pontos na
média final caso necessitem.
Dia
7
14
21
1 2) A nota da prova substitutiva substitui a menor nota.
Introdução à Bioquímica
Roberto Salinas – [email protected]
Instituto de Química - USP
21 de outubro de 2015
1. Estrutura de proteínas
Proteínas são polímeros de aminoácidos conectados por ligações peptídicas. A ligação
peptídica ocorre devido a uma reação de condensação entre o grupo amino do
primeiro aminoácido e a extremidade carboxila do segundo aminoácido (Figura 1):
Figura 1: Reação de condensação entre Alanina e Serina formando um dipeptídeo Ala-Ser com
eliminação de uma molécula de H2O. As cargas líquidas do peptídeo e dos aminoácidos não estão
mostradas.
A ligação peptídica é planar devido à existência de delocalização de elétrons do
nitrogênio e do oxigênio da ligação amida. A planaridade da ligação peptídica possui
enormes consequências conformacionais. A primeira delas é que a conformação do
esqueleto polipeptídico pode ser descrita por apenas três ângulos diedrais: o ângulo ω
ao redor da ligação peptídica, e os ângulos φ e Ψ do carbono alfa. Os ângulos φ e Ψ
são correlacionados e nem todas as combinações são permitidas devido ao
impedimento esterico gerado entre a carbonila e o N-H da ligação peptídica, e entre as
cadeias laterais de aminoácidos vizinhos. O gráfico de correlação entre φ e Ψ é
chamado de Diagrama de Ramachandran em homenagem ao biofísico indiano que o
desenvolveu, e ainda é a principal ferramenta utilizada para avaliar a qualidade
estereoquímica das estruturas de proteínas (Figura 2).
2 Figura 2. Diagrama de Ramachandran. As regiões delimitadas pelas linhas verdes
demarcam combinações permitidas de Ψ e Φ. Pontos fora das regiões delimitadas
pelas linhas verdes indicam um aminoácido com algum problema ou correspondem a
glicinas.
Estruturas tridimensionais de proteínas com resolução atômica podem ser obtidas
experimentalmente através de cristalização e Difração de Raios-X, Ressonância
Magnética Nuclear em solução, e mais recentemente Crio-Microscopia Eletrônica.
Conservação e homologia de sequências
Proteínas são polímeros de aminoácidos. A sequência de aminoácidos de uma
proteína é chamada estrutura primária. A estrutura tridimensional de uma proteína é
determinada pela sequência de aminoácidos. A função biológica de uma proteína está
intimamente relacionada com a estrutura tridimensional.
A predição da estrutura tridimensional de uma proteína a partir da sequência de
aminoácidos não é tarefa simples, especialmente para moléculas com mais de 100 aa.
No entanto, a análise da estrutura primária pode oferecer pistas importantes sobre
topologia (presença de hélices transmembranares), estrutura secundária e grau de
desordem.
A relação evolutiva entre proteínas de diferentes organismos pode ser inferida a partir
de comparações entre sequências de aminoácidos. Famílias de proteínas que
compartilham a mesma estrutura e a mesma função podem ser facilmente
identificadas com base em similaridades entre sequências de aminoácidos. Membros
de uma mesma família são chamados de proteínas homólogas. A função de uma
3 proteína desconhecida pode eventualmente ser deduzida a partir da comparação de
sua sequência de aminoácidos com milhares de sequências de proteínas contidas nos
bancos de dados (por exemplo UNIPROT). Duas proteínas contendo ao menos 30%
de identidade de sequência geralmente compartilham a mesma estrutura e função.
Aminoácidos conservados são geralmente importantes para a função. Geralmente,
quando estes aminoácidos são alterados no decorrer da evolução as propriedades
físico-químicas são mantidas. Este fenômeno é chamado de mutação conservativa.
Por exemplo, a mutação de uma Ile por uma Val na mesma posição pode ser tolerável
pois trata-se de dois aminoácidos hidrofóbicos. Ou a mutação de um aspartato por um
um ácido glutâmico também pode ser tolerável pois trata-se de dois aminoácidos com
carga negativa na cadeia lateral. As alterações Ile – Val e Asp - Glu seriam duas
mutações conservativas (Figura 3).
Proteína A
Consenso
Proteína B
GVDEQRFRIEVIDDDVFEEDECFYIRLFN-------PSEGVK----------LAVPMIAT
G ++ R+ +IDDD+FEEDE F + L N
EG+
L P AT
GETQKEIRVGIIDDDIFEEDENFLVHLSNIRVSTEASDEGILEASRVSTLACLGSPSTAT
Figura 3. Exemplo de alinhamento de sequências de duas proteínas “A” e “B” de organismos
diferentes mas com a mesma função, detectar a concentração de Ca2+ intracelular. Note que “gaps”
foram introduzidos na sequência da proteína “A” para optimizar o alinhamento com a proteína “B”.
Função: proteínas que ligam Oxigênio (O2)
Proteínas assumem diversas funções, mas uma das funções mais comuns é a ligação
específica e reversível de outras moléculas. A molécula que se liga à proteína é
chamada de ligante. O ligante liga-se a um sítio de ligação, o qual deve ser
complementar em forma, tamanho, carga e hidrofilicidade ou hidrofobicidade ao
ligante (modelo chave-fechadura). A interação entre o ligante e a proteína é
específica, de forma que a proteína pode distinguir o ligante entre milhares de outras
moléculas parecidas. Exemplo: enzimas reconhecem especificamente o substrato.
No caso das enzimas, o sítio de ligação ao ligante é conhecido como sítio catalítico.
Enzimas frequentemente reconhecem um substrato de acordo com um modelo de
complementariedade espacial, chave-fechadura.
Proteínas são flexíveis, isto é, experimentam conformações distintas que se
interconvertem em diferentes escalas de tempo. A interação com um ligante
frequentemente induz uma mudança conformacional na proteína de forma a tornar o
sítio de ligação o mais complementar possível ao ligante, um processo conhecido
como “induced fit”. Alternativamente, a proteína em solução está em equilíbrio entre
distintas conformações sendo que apenas uma delas é espacialmente complementar ao
ligante. A interação com o ligante desloca o equilíbrio conformacional no sentido da
conformação complementar, um processo conhecido como “seleção de confôrmeros”
ou “modelo sequencial”.
A hemoglobina e a mioglobina foram as duas primeiras proteínas cuja estrutura
tridimensional foi determinada experimentalmente. Elas transportam oxigênio.
Proteínas não são capazes de coordenar oxigênio, para isso elas utilizam íons
metálicos como Fe(II). O Fe(II) normalmente liga-se à proteína na forma complexada
a um grupo prostético chamado Heme. O heme é uma protoporfirina que liga um íon
4 Fe2+ (Figura 4). O grupo Heme está ligado não covalentemente à proteína (veja PDB
1MBO).
Figura 4. Grupo Heme encontrado na mioglobina e na hemoglobina. O íon Fe2+ faz ligações de
coordenação com os quatro átomos de nitrogênio do anel. O Fe2+ ainda pode formar duas outras
ligações de coordenação que são formadas com as histidinas proximal e distal (veja PDB 1MBO).
A mioglobina consiste de um único polipeptídeo de 153 aminoácidos. O equilíbrio de
associação entre a mioglobina e uma molécula de O2 é descrito como:
𝐾𝑎 =
[!"]
! [!]
!
= !! (1)
!
onde P é a proteína, L é o ligante, e ka e kd são as constantes de velocidade de
associação (segunda ordem) e de dissociação (primeira ordem) do complexo. A fração
de sítios ocupados pelo ligante é dada por θ:
𝜃=
!"#çã! !" !"#$%í!"# !"#$%$&
!"#$%í!" !"!#$
=
[!"]
!" ![!]
!" ! [!]
= !" !
! ![!]
!"[!]
= !" ! !! =
[!]
! !!!
, (2)
em que [L] é a concentração de ligante total. O comportamento de θ em função de [L]
é uma hipérbole (Figura 5). É fácil observar que quando a proteína possui menor
afinidade (maior Kd) é necessário adicionar maior quantidade de ligante para atingir
saturação (Figura 5). Note que Kd (constante de dissociação) é equivalente à
concentração de ligantes necessária para saturar 50% dos sítios de ligação. Portanto o
valor de Kd é uma medida clara da afinidade de uma proteína pelo ligante.
5 isoterma de ligacao
1
0.9
theta (% sitios ocupados
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0
0
0.001 0.002 0.003 0.004 0.005 0.006 0.007 0.008 0.009
concentracao de ligante total (mol/l)
0.01
Figura 5. Simulação da curvas de ligação para um complexo ligante:proteína com
afinidade estimada por um Kd de 10 µM (azul) e 100 µM (vermelho).
Em água, o grupo heme liga-se ao monóxido de carbono (CO) com 20 mil vezes
maior afinidade do que ao oxigênio. Por outro lado, quando o heme está ligado na
mioglobina a diferença de afinidade cai para 200 vezes. Detalhes estruturais, em
particular a interação com a histidina distal explicam porque a afinidade do grupo
heme pelo CO diminui.
A hemoglobina possui estrutura quaternária, ela é um tetrâmero formado por duas
subunidades α e duas subunidades β. Ambas as subunidades α e β são homólogas à
mioglobina, mas mesmo assim apresentam baixa identidade de sequência (Figura 6).
1MBO:mioglobina
cadeia alfa
beta
beta
-VLSEGEWQLVLHVWAKVEADVAGHGQDILIRLFKSHPETLEKFDRFKHLKTEAEMKASE
-VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFD------LSHGSA
VHLTPEEKSAVTALWGKVNV--DEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNP
*: :
*
*.** .
* : * *:: .* *
* *
..
1MBO:mioglobina
cadeia alfa
beta
beta
DLKKHGVTVLTALGAILKKKGHHEAELKPLAQSHATKHKIPIKYLEFISEAIIHVLHSRH
QVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHL
KVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHF
.:* ** .* *:
: : .
: *:: *. * ::
:.::.. :: .* :
1MBO:mioglobina
cadeia alfa
beta
beta
PGDFGADAQGAMNKALELFRKDIAAKYKE
PAEFTPAVHASLDKFLASVSTVLTSKYRGKEFTPPVQAAYQKVVAGVANALAHKYH:*
.:.: :* : . . :: **:
Figura 6. Alinhamento de sequência da mioglobina (PDB 1MBO) com as cadeias
alfa e beta da hemoglobina (PDB 1HGA).
6 Apesar de diferenças na sequência de aminoácidos, as estruturas
tridimensionais da mioglobina (PDB 1MBO) e da hemoglobina (PDB 1HGA) são
muito parecidas. A hemoglobina possui quatro grupos heme, um em cada subunidade,
podendo transportar até 4 moléculas de O2.
A presença de quatro sítios de ligação e 4 subunidades afeta profundamente as
características da hemoglobina como molécula carreadora de oxigênio. A curva de
ligação do oxigênio na hemoglobina é sigmoide, indicando que oxigênio liga-se
cooperativamente à proteína (Figura 7). Em um processo cooperativo, a interação do
ligante com o primeiro sítio altera a afinidade do segundo sítio e assim por diante.
Como consequência da cooperatividade observada na curva de ligação a
oxigênio, a porcentagem de saturação da hemoglobina varia muito mais rapidamente
com a concentração de oxigênio do que no caso da mioglobina que não é cooperativa.
Por exemplo, no equilíbrio de ligação mostrado na Figura 7, nota-se que para ir de
20% a 80% de saturação seria necessário aumentar a concentração de ligante apenas
duas vezes, de 12.6 para 25.2 nM (Figura 7). Enquanto que no caso de um equilíbrio
não cooperativo como exemplificado na Figura 5, a concentração de ligante deveria
ser aumentada aproximadamente 10 vezes para aumentar a fração de sítios ocupados
de 20 a 80%. Portanto, uma proteína cooperativa é muito mais sensível a pequenas
alterações na concentração de ligante. Esta característica torna a hemoglobina capaz
de captar oxigênio nos pulmões, onde a pressão de O2 é maior, e liberar nos tecidos
onde a pressão de O2 é menor.
curva de Hill
4
v (numero de sitios ocupados
3.5
3
2.5
2
1.5
1
0.5
0
0
0.005
0.01
0.015
0.02
0.025
0.03
concentracao de ligante (mol/l)
0.035
0.04
Figura 7. Curva de Hill para uma proteína com 4 sítios de ligação e Kd 0.1 µM. A
concentração total de proteína é 1 mM.
7 A ligação cooperativa de um ligante com oxigênio em uma consequência da
interação entre as subunidades. A estrutura quaternária da hemoglobina envolve um
grande número de interações fracas (eletrostática, ligação de hidrogênio e van der
Waals) entre as subunidades alfa e beta. Estas interações são cruciais para modular a
afinidade dos diferentes sítios de ligação pelo oxigênio. Como ?
Monod, Wyman e Changeaux propuseram um modelo para explicar a
cooperatividade da hemoglobina. Este modelo, chamado de “concerted model”,
propõe que a hemoglobina está em equilíbrio entre um estado tenso (“T”) e outro
relaxado (“R”):
O O2 liga-se mais fortemente ao estado R deslocando o equilíbrio na direção da forma
“T”. Assim, quando O2 liga-se à primeira subunidades todas as outas subunidades
mudam de conformação para a forma “R” concomitantemente.
Exercícios para a aula 1
1. A proteína A interage com o ligante X com Kd 10-6 M. A proteína B interage
com o mesmo ligante mas com Kd 10-9 M. Qual proteína possui maior
afinidade pelo ligante X? Explique.
2. A proteína calcineurina liga-se à calmodulina com uma velocidade de
associação de 103 M-1s-1, e uma constante de dissociação, Kd, de 10 nM.
Calcule a velocidade de dissociação do complexo (koff), inclua as unidades
adequadas.
3. Desenhe a curva de desnaturação de uma proteína A em função da
temperatura. Assuma que a proteína A seja pequena, e portanto o equilíbrio de
desnaturação envolve apenas dois estados. Em seguida, suponha que um
mutante foi construído. Este mutante envolve a troca de dois resíduos de Ser
por Cys, de tal forma que as duas cisteínas estão próximas uma da outra na
estrutura tridimensional da proteína, e formam uma ponte dissulfeto
estabilizando a estrutura. Desenhe sobre o mesmo gráfico, a curva de
desnaturação do mutante.
4. Estudos de transporte de oxigênio em fêmeas grávidas mostraram que as
curvas de saturação com oxigênio do sangue do feto e do sangue materno são
diferentes. Os eritrócitos do feto contém uma variante estrutural da
hemoglobina (HbF), que consiste de duas subunidades α e duas γ (a2g2).
Enquanto que a hemoglobina da mãe (HbA) consiste de α2β2. Qual
hemoglobina possui maior afinidade pelo oxigênio nas condições nativas?
Explique.
8 between subunits.
6. Comparison of Fetal and Maternal Hemoglobins Studies of oxygen transport in pregnant mammals
show that the O2-saturation curves of fetal and maternal blood are markedly different when measured
under the same conditions. Fetal erythrocytes contain a structural variant of hemoglobin, HbF, consisting
of two a and two g subunits (a2g2), whereas maternal erythrocytes contain HbA (a2b2).
1.0
HbF
!BPG
v
0.5
HbA
!BPG
0
2
6
4
pO2 (kPa)
8
10
(a) Which hemoglobin has a higher affinity for oxygen under physiological conditions, HbA or HbF?
Explain.
5.
Depois
de
passar
alguns
dias
em
altitudes
muito elevadas,
a concentração de BPG
(b) What is the physiological significance of the different
O2 affinities?
nos When
eritrócitos
Qual será
o efeito
aumento
da and
concentração
de BPGOna-saturation
(c)
all theaumenta.
BPG is carefully
removed
fromdo
samples
of HbA
HbF, the measured
2
curva
de
saturação
da
hemoglobina?
Proponha
uma
explicação
para
porque
essanow has a
curves (and consequently the O2 affinities) are displaced to the left. However, HbA
adaptação
benéfica
para
o funcionamento
organismo
em altas
greater seria
affinity
for oxygen
than
does HbF. When do
BPG
is reintroduced,
the altitudes.
O2-saturation curves
returngráficos
to normal,para
as shown
in the graph.
What
is the effect of BPG on the O2 affinity of hemoglobin?
Desenhe
fundamentar
a sua
resposta.
How can the above information be used to explain the different O2 affinities of fetal and maternal
hemoglobin?
2. Cinética
enzimática
Enzimas são catalisadores em sistemas biológicos. As características mais notáveis
das enzimas são o seu poder catalítico e a sua especificidade. Quase a totalidade das
enzimas são proteínas, porém algumas moléculas de RNA também são capazes de
promover catalise. O exemplo mais conhecido de riboenzima é o RNA ribossomal.
Uma das funções mais importantes desempenhadas pelas proteínas é o
reconhecimento específico de ligantes através da formação de interações não
covalentes. Enzimas utilizam esta capacidade para interagir com o substrato (ligante)
posicionando-o de forma adequada para que reações químicas possam ocorrer.
Enzimas catalisam reações químicas porque estabilizam o estado de transição.
Elas catalisam apenas uma reação química, sendo que a ocorrência de reações
colaterais é rara em comparação com reações não catalisadas. A tabela 1 ilustra o
ganho em velocidade de reação promovido por enzimas selecionadas:
Tabela 1: Aumento de velocidade de reação proporcionado por certas enzimas
Enzima
Velocidade da reação
Velocidade da
não catalisada
reação catalisada
Anidrase carbonica
1.3×10-1 s-1
1×106 s-1
-6 -1
Triose fosfato isomerase
4.3×10 s
4.3×103 s-1
Carboxipeptidase A
3.0×10-9 s-1
578 s-1
Enzimas são altamente específicas para o substrato. Um exemplo desta especificidade
são as proteases, enzimas que catalisam a reação de clivagem da ligação peptídica. A
papaína é uma protease que não discrimina o aminoácido adjacente à ligação
peptídica, assim ela reconhece diferentes sequências de aminoácidos. A tripsina, por
outro lado, cliva somente ligações peptídicas adjacentes a lisinas ou argininas, que são
aminoácidos de cadeia lateral longa e carregados positivamente. Outro exemplo é a
DNA polimerase, uma enzima que catalisa a formação da ligação fosfodiéster para
9 formar uma nova dupla-fita de DNA. Ela é praticamente insensível a erros
incorporando menos de um nucleotídeo errado a cada mil. Dessa forma a DNA
polimerase é uma enzima altamente específica para a fita de DNA molde.
Equilíbrio
O sentido de uma reação química depende da variação da energia livre de Gibbs
envolvida (∆G = Gprodutos - Greagentes). Reações com ∆G negativo são espontâneas,
enquanto que reações com ∆G positivo não são espontâneas e precisam de energia
para ocorrer. Reações com ∆G = 0 estão no equilíbrio. O valor do ∆G depende apenas
dos estados inicial e final, independe do caminho.
A magnitude e o sinal do ∆G nada informam sobre a velocidade com que as reações
ocorrem. Por exemplo, a reação de oxidação de glicose (C6H6O6) a CO2 e H2O é
espontânea, mas não ocorre a taxas perceptíveis na ausência de enzimas. O ∆G de
combustão da glicose é o mesmo caso a reação seja catalisada ou não.
O grau de espontaneidade de uma reação química depende do valor do ∆G, o qual
depende, por sua vez, das concentrações de reagentes e produtos conforme a equação
3:
∆𝐺 = ∆𝐺° + 𝑅𝑇𝑙𝑛
!"#$%&#'
!"#$"%&"'
,
(3)
sendo que
∆𝐺° = −𝑅𝑇𝑙𝑛𝐾!" .
(4)
A Tabela 2 mostra a correlação entre constantes de equilíbrio e ∆G. Note como
reações com ∆G positivo não ocorrem na direção esperada, ao contrário de reações
com ∆G negativo (Tabela 2).
Tabela 2. Relação entre Keq e ∆G.
Keq
∆G (kJ/mol)
10-6
34.2
-3
10
17.1
1.00
0.00
1
10
-5.7
3
10
-17.1
6. Reações químicas que não são espontâneas (∆G0 > 0) podem tornar-se espontâneas
ajustando-se as concentrações de reagentes e produtos. Este aspecto é determinante
em condições celulares. Por exemplo, considere a reação de isomerização de
dihidroxicetona-fosfato (DHAP) em gliceraldeido-3-fosfato (GAP) que ocorre durante
a via glicolítica. A razão entre as concentrações de DHAP e GAP no equilíbrio é
0.0475 a 298 K e pH 7 (R = 8315 JK-1mol-1). Pergunta-se:
10 i)
ii)
A reação de isomerização de DHAP em GAP nas condições de equilíbrio é
espontânea?
A reação será espontânea quando as concentrações de DHAP e GAP forem
200 µM e 3 µM, respectivamente?
Enzimas aceleram reações químicas mas não alteram o equilíbrio da reação, que
é dado apenas em função da diferença de energia livre entre produtos e
reagentes (Figura 8).
A velocidade das reações químicas é expressa a partir de uma medida da variação da
concentração de produtos ou reagentes em função do tempo. Considere o equilíbrio
entre dihidroxicetona-fosfato (DHAP) e gliceraldeído-3-fosfato (GAP) ilustrado
abaixo:
Este equilíbrio possui velocidades no sentido da formação de gliceraldeido-3-fosfato
ou dihidroxicetona-fosfato. A velocidade da reação direta é expressa como:
𝑣=
![!"#$]
!"
= 𝑘[𝐷𝐻𝐴𝑃]
(5)
onde a constante de velocidade “k” é expressa em s-1. Reações que são diretamente
proporcionais à concentração do reagente são chamadas de “reações de primeira
ordem”. Reações bimoleculares e cuja velocidade é proporcional à concentração de
dois reagentes são chamadas “reações de segunda ordem”, neste caso a constante
cinética é expressa em M-1s-1.
Catálise enzimática
A velocidade de uma reação química é diretamente proporcional à energia de
ativação. Enzimas agem como catalisadores biológicos, e atuam para diminuir a
barreira de energia de ativação conforme mostrado na Figura 8, acelerando a reação.
A forma pela qual enzimas diminuem a barreira de energia é através da
estabilização do estado de transição. A primeira etapa de uma reação catalítica é a
formação de um complexo específico entre enzima e substrato. Dessa forma, a enzima
é capaz de orientar o(s) substrato(s) adequadamente para que a reação química ocorra.
11 G0
ET‡
Produtos
Reagentes
Coordenada de reação
Figura 8. Diagrama de energia livre em função da coordenada de reação para uma
reação na ausência (preto) e presença da enzima (vermelho). A enzima estabiliza a o
estado de transição, diminuindo a barreira de energia de ativação.
A velocidade das reações químicas depende da constante de velocidade e das
concentrações dos reagentes. Portanto, não é de estranhar que a velocidade de uma
reação enzimática, medida a partir da taxa de formação do produto “P”, aumente com
a concentração de enzima:
v = k[E]total .
(6)
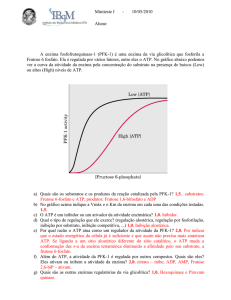
A velocidade da reação também aumenta com a concentração de substrato como seria
de esperar. O curioso é que a velocidade de reação aumenta linearmente para baixas
concentrações de substrato, mas deixa de aumentar em altas concentrações de
substrato e atingir um valor máximo (Vmax) (Figura 9). Com certeza, a velocidade de
uma reação não catalisada não apresenta este efeito de saturação.
0.12
0.1
v0
0.08
0.06
controle
maior Vmax
menor KM
0.04
0.02
0
0
0.2
0.4
0.6
[Substrato] (M)
0.8
1
Figura 9. Velocidade inicial (v0) em função da concentração de substrato (azul).
Comparação com enzimas diferentes apresentando menor KM (verde) ou maior Vmax
(vermelho).
Para compreendermos a origem deste efeito de saturação é preciso considerar as
condições em que as medidas foram realizadas. Medidas de velocidades de reações
enzimáticas são geralmente realizadas nos intervalos de tempo iniciais da reação, e
12 em condições de baixíssima concentração de enzima e um excesso de substrato.
Quando a enzima é misturada com um excesso de substrato, há um período inicial
durante o qual as concentrações de intermediários, de complexo enzima-substrato
(ES) aumentam até atingir valores estacionários. Uma vez que os intermediários
atingiram concentrações estacionarias, as velocidades de reação alteram-se muito
pouco com o tempo.
Leonor Michaelis e Maud Menten propuseram em 1913 um modelo simples para
explicar as características cinéticas observadas na Figura 9. O modelo de MichaelisMenten pressupõe que o complexo ES é um intermediário da reação catalítica. O
complexo enzima-substrato também costuma ser chamado de “complexo de
Michaelis”:
A reação catalítica é dividida em duas etapas. Inicialmente ocorre a interação entre
uma molécula da enzima e outra do substrato, formando o complexo “ES”. Este
primeiro equilíbrio é considerado rápido e reversível, e não envolve alterações das
ligações químicas. O complexo ES é mantido por interações não covalentes. As
reações químicas ocorrerão em uma segunda etapa, na qual o complexo ES se
decompõe em enzima livre e produto. Esta segunda etapa segue uma cinética de
primeira ordem, dependente da constante k2 ou kcat.
O complexo ES também pode ser formado a partir da reação reversa entre enzima e
produto. Mas como as medidas são realizadas nos instantes iniciais em que a
concentração de produto é muito pequena, a reação reversa pode ser ignorada. Esta
análise também assume que a decomposição de ES em enzima e produto livres é
rápida, e portanto pode ser ignorada. O esquema final é:
Com um pouquinho de álgebra e assumindo-se a condição de estado estacionário, é
possível chegar a uma equação simples para a velocidade inicial de reação em função
da concentração de substrato e das constantes cinéticas. Esta é a chamada “equação de
Michaelis-Menten”, e reproduz muito bem o comportamento ilustrado na Figura 9:
! [!] [!]
𝑣! = !!!!!!!!
!!
![!]
=
!!"# [!]
!! ![!]
.
(7)
A constante cinética k2 ou kcat é frequente chamada de “turnover”. Ela representa o
número máximo de moléculas de substrato convertidas em produto por unidade de
tempo. A constante kcat é uma constante de velocidade de primeira ordem. Na
presença de excesso de substrato, a velocidade inicial v0 atinge o valor máximo
(Vmax) que depende apenas da constante catalítica kcat.
13 𝑉!"# = 𝑘! [𝐸]!"!#$
(8)
KM é igual à concentração de substrato na qual a metade da velocidade máxima (v0 =
Vmax/2) é atingida. Dessa forma KM representa a concentração de substrato necessária
para se atingir uma taxa razoável de conversão de substratos em produtos. E na
situação particular em que k2 << k-1, KM representa a constante de dissociação do
complexo ES. As tabelas 3 e 4 apresentam os valores do turnover e de KM para
algumas enzimas.
0.12
0.1
v0
0.08
0.06
controle
maior Vmax
menor KM
0.04
0.02
0
0
0.2
0.4
0.6
[Substrato] (M)
0.8
1
A Figura 9 ilustra o efeito de variações de KM e Vmax sobre o comportamento
cinético de uma enzima.
Tabela 3. Turnover de algumas enzimas
Enzima
kcat (s-1)
Anidrase carbonica
6.0×105
Acetilcolinesterase
2.5×104
Lactato desidrogenase
1.0×103
Tabela 4. KM de algumas enzimas
Enzima
Substrato (s-1)
Anidrase carbonica
CO2
Lactose
β-galactosidase
Arginina-tRNA-sintetase
Arginina
KM (µM)
8.0×103
4.0×103
3
Normalmente, em condições fisiológicas, a velocidade de uma reação enzimática é
dada pela razão kcat/KM, que é uma constante de velocidade de segunda ordem
aparente:
𝑣! =
!!"#
!!
𝐸 [𝑆].
(9)
14 Em uma condição em que a concentração de substrato for muito menor do que o KM,
𝑣! =
!!"#
!!
[𝐸]!"!#$ [𝑆].
(10)
A razão kcat/KM depende tanto da eficiência da reação catalítica kcat, como da
afinidade entre enzima e substrato, KM. Esta razão é uma boa medida da
especificidade da interação entre enzima e substrato como mostra a Tabela 5. Neste
caso , fica evidente que a quimotripsina possui preferência por substratos volumosos
como a fenilalanina.
Tabela 5. Preferências da quimotripsina pelo substrato
Ester de amino ácido
kcat/KM (M-1s-1)
Glicina
1.3×10-1
Valina
2.0
Fenilalanina
1.0×105
É interessante imaginar o quão eficiente uma enzima pode ser. A resposta para essa
pergunta depende do limite da razão kcat/KM. Suponha que a decomposição do
complexo ES em enzima e produtos seja muito mais rápida do que a velocidade de
dissociação do complexo (k2 >> k-1), neste caso o limite é dado por k1, a constante de
velocidade de formação do complexo “ES”. O limite para k1 é a velocidade de difusão
das moléculas em solução, um número da ordem de 108 a 109 M-1s-1. Enzimas que
possuem valor kcat/KM nesta faixa são ditas “perfeitas”, pois atingiram a perfeição
catalítica (Tabela 6).
Tabela 6 Enzimas que atingiram perfeição catalítica
Enzima
kcat/KM (M-1s-1)
Acetilcolinesterase
1.6×108
Anidrase carbônica
8.3×107
Catalase
4.0×107
Representação gráfica dos dados
É muito útil linearizar a equação de Michaelis-Menten para determinar os valores de
kcat e KM, e também para examinar desvios da idealidade. Dessa forma, um gráfico de
1/v em função de 1/[S] obedeceria a uma equação de reta igual a:
!
!
!!
=!
!
!"# [!]
+!
!
!"#
.
(11)
A análise dessa equação indica que o ponto em que o gráfico cruza o eixo das
ordenadas equivale a 1/Vmax e que o coeficiente angular é dado por KM/Vmax.
Inibição
Enzimas podem ser inativadas pelo aumento da temperatura, ou pela interação não
covalente com inibidores, pequenas moléculas que competem com o substrato pelo
sítio ativo. Estes inibidores são chamados de “competitivos”. No caso de um inibidor
competitivo, o mecanismo de Michaelis-Menten teria que ser alterado para:
15 E não é difícil derivar a equação de Michaelis-Menten levando em conta o equilíbrio
entre a enzima e o inibidor competitivo:
𝑣=
!!"# [!]
!! !!
!
!!
![!]
.
12
Observa-se que o KM aparente aumentou por um fator de (1+[I]/KI), ou seja, a
afinidade aparente entre enzima e substrato diminuiu. Note que o inibidor competitivo
não afeta o Vmax, apenas KM.
Mecanismos
De onde vem o poder catalítico e a especificidade das enzimas? Nós já vimos que as
enzimas formam um complexo com o substrato através da formação de interações não
covalente, e que elas interagem melhor com o estado de transição. Como isto ocorre?
Estruturas cristalográficas e ensaios de mutação sítio-dirigida permitiram obter a
resposta em alguns casos.
Os mecanismos descritos abaixo trazem exemplos de um ou mais tipos de catálise:
a)catálise covalente: em que o sítio ativo contem um núcleofilo poderoso que liga-se
covalentemente a parte do substrato durante a catálise
b)catálise ácido-base: uma molécula que não seja a água assume o papel de
doador/aceptor de prótons
c)catálise por aproximação: enzimas facilitam proximidade entre os substratos
trazendo-os para uma mesma superfície de interação
d)catálise por íon metálico: íons metálicos agem cataliticamente, por exemplo ao
facilitar a formação de nucleófilos.
Proteases
Enzimas proteolíticas possuem a tarefa de acelerar a reação de hidrólise da ligação
peptídica.
16 Embora esta reação seja termodinamicamente favorável, ela é extremamente lenta na
ausência de um catalisador. Para promover a clivagem da ligação peptídica a enzima
deve facilitar um ataque nucleofílico no grupo carbonila da ligação peptídica. A
quimotripsina emprega um nucleofico forte capaz de fazer este ataque nucleofilico.
Este nucleofilo é o grupo hidroxil da cadeia lateral de uma serina extraordinariamente
reativa. A reatividade extraordinária da Ser195 vem do fato de que o grupo OH da
cadeia lateral forma uma ligação de hidrogênio com a His57. O grupo NH da His57
forma, por sua vez, uma ligação de hidrogênio com a cadeia lateral do Asp102. Este
arranjo, conhecido como “tríade catalítica”, estabiliza uma carga negativa na cadeia
lateral da Ser195, que se torna extraordinariamente ácida (Figura 10).
Figura 10. Tríade catalítica da quimotripsina formada pela Ser195, His57 e Asp102. A
rede de ligações de hidrogênio torna o grupo OH da Ser195 extremamente reativo e
estabiliza um oxiânion.
O mecanismo proposto para a reação catalítica envolve um ataque nucleofilico do
oxigênio da cadeia lateral da Ser195 sobre a carbonila do substrato, que assume
configuração teatraédrica intermediário e uma carga negativa no oxigênio da
carbonila. Esta carga negativa será estabilizada por grupos NH da proteína, que
formam o chamado “oxyanion hole”. O passo seguinte envolve a clivagem da ligação
peptídica ao mesmo tempo em que o próton ligado à His57 é transferido para o grupo
amino da cadeia polipeptídica livre. Como resultado, a enzima permaneceu acilada
por parte da cadeia polipeptídica. Portanto, este mecanismo envolve a formação de
um intermediário covalente, e a próxima etapa deve se a hidrólise desse
17 intermediário covalente. Na próxima etapa uma molécula de água ocupa o lugar do
grupo amino do substrato. A His57 age como uma base e abstrai um próton da água,
enquanto o grupo hidroxila ataca a carbonila do intermediário que colapsa liberando a
segunda metade do peptídeo e regenerando a enzima.
Este mecanismo não explica a especificidade da enzima. A quimiotripsina cliva
ligações peptídicas adjacentes a cadeias laterais grandes e volumosas, como
fenilalanina. O reconhecimento do substrato envolve a interação da cadeia lateral na
posição amino-terminal à ligação que será clivada, com um bolsão hidrofóbico. Este
bolso hidrofóbico possui, no caso da quimiotripsina, maior afinidade por cadeias
laterais aromáticas e volumosas.
A quimiotripsina é uma protease da classe das serino-proteases, pois utiliza a cadeia
lateral de uma Serina para fazer o ataque nucleofílico na carbonila. Outras classes de
proteases utilizam uma estratégia semelhante, as cisteíno-proteases, aspartilproteases e as metalo-proteases. Nestas três classes de proteases, o mecanismo de
catálise segue o mesmo padrão. Primeiro ocorre a ativação de um agente nucleofílico
capaz de realizar um ataque sobre a carbonila da ligação peptídica que será clivada.
No caso das cisteíno-proteases, esse agente será uma cisteína que assume a função da
serina. No caso das metaloproteases ou aspartil-proteases, o ataque nucleofílico é
efetuado por uma molécula de água reativa. Frequentemente, o grupo carbonila que
sofre o ataque também é polarizado, de forma a assumir menor densidade eletrônica.
O terceiro aspecto importante é a estabilização do intermediário teatraédrico através
da formação do “oxyanion hole”. No caso das metalo-proteases, um íon metálico, por
exemplo Zn2+, interage com uma molécula de água polarizando-a e tornando-a
altamente reativa.
Anidrase carbônica
A anidrase carbônica é uma enzima cuja função é acelerar a reação de hidratação de
CO2 formando o ânion bicarbonato:
As constantes k1 (pseudo-primeira ordem) e k-1 (primeira ordem) são 0.15 s-1 e 50 s-1,
respectivamente, na ausência de catalisador. Portanto o equilíbrio da reação é
deslocado no sentido da formação de CO2.
Anidrase carbônica acelera a reação de hidratação de CO2 para velocidades kcat = 106
s-1. A enzima possui um íon Zn2+ coordenado por três histidinas, o quarto ponto de
coordenação do Zn2+ é satisfeito por uma molécula de H2O.
O turnover da anidrase-carbônica é altamente dependente do pH, e apresenta uma
transição de um estado de baixa atividade em pH baixo para um estado de alta
atividade em pHs maiores que 9, sendo que o ponto médio dessa transição é o pH 7. O
18 grupo responsável por essa transição é uma molécula de água que está coordenada
pelo Zn2+. A interação com o Zn2+ faz com que o pKa da molécula de água diminuísse
de 15.7 para 7.0. Esta molécula de água é capaz de perder um próton facilmente
devido2/1/08
ao pKa1:11PM
mais baixo,
formando
um radical hidroxila
que é capaz de realizar um
2608T_ch06sm_S63-S77
Page S-66
ntt 102:WHQY028:Solutions
Manual:Ch-06:
ataque nucleofílico no CO2 gerando um íon bicarbonato.
Exercícios
1. Uma6
Chapter
enzima
Enzymesque catalisa a reação de conversão entre duas moléculas “X” e “Y” é
isolada de duas espécies de bactérias. As enzimas possuem o mesmo Vmax, mas
possuem
valores
KMPagediferentes.
A enzima
AManual:Ch-06:
possui from
KM =two
2.0bacterial
µM, enquanto
que enzymes
a
2608T_ch06sm_S63-S77
2/1/08 de
7:34AM
S-75 ntt 102:WHQY028:Solutions
z Y is
(c) An enzyme
that
catalyzes
the reaction
Xy
isolated
species. The
enzima
B
possui
K
=
0.5
µM.
A
gráfico
abaixo
mostra
a
cinética
de
duas
reações
have the same VM
max, but different Km values for the substrate X. Enzyme A has a Km of 2.0 !M,
efetuadas
com aB mesma
de below
enzima
e [X]
= 1 µM.
Qual carried
curva out
of 0.5 !M. The plot
shows
the kinetics
of reactions
while enzyme
has a Km concentração
with the asame
corresponde
qualconcentration
enzima? of each enzyme and with [X] ! 1 !M. Which curve corresponds to
which enzyme?
Chapter 6 Enzymes
S-75
(b) Using the Living Graph for Equation 6–30 and the kinetic parameters in (a), create a plot in
which both a and a! are 1.0. Now observe how the plot changes when a " 2.0; when a! " 3.0;
and when a " 2.0 and a! " 3.0.
(c) Using the Living Graphs for Equation 6–30 and the Lineweaver-Burk equation in Box 6–1, create
Lineweaver-Burk (double-reciprocal) plots for all the cases in (a) and (b). When a " 2.0, does
the x intercept move to the right or to the left? If a " 2.0 and a! " 3.0, does the x intercept
move to the right or to the left?
Answer
(a) When [S] increases from 0.2 to 0.4 mM, V0 increases by a factor of 1.96. When [S] " 10
mM, V0 " 50 mM s#1. When [S] increases from 100 to 200 mM, V0 increases by a factor of
1.048.
(b) When a " 2.0, the curve is shifted to the right as the Km is increased by a factor of 2.
When a! " 3.0, the asymptote of the curve (the Vmax) declines by a factor of 3. When
a " 2.0 and a! " 3.0, the curve briefly rises above the curve where both a and a! " 1.0,
due to a decline in Km. However, the asymptote is lower because Vmax declines by a
factor of 3.
(c) When a " 2.0, the x intercept moves
Timeto the right. When a " 2.0 and a! " 3.0, the x
intercept moves to the left.
[Y]
S-66
Answer
Data Analysis
Problem
2. O exame
da we
estrutura
de uma enzima permite formular
hipóteses
sobre a
0.25 V of an
. The
Michaelis-Menten
(a)23. Here
want to find the value of [S] when V0 !
Exploring and Engineering Lactate Dehydrogenase Examining
the structuremax
enzyme recontribuiçãoequation
de cada isaminoácidos
para a estrutura e para a atividade da enzima. Uma
sults in hypotheses
about the relationship between different amino acids in the protein’s structure and
the protein’s
One way toétest
these hypotheses
recombinant
DNA technology to para gerar
forma de testar
essasfunction.
hipóteses
usar
técnicasis todeuse DNA
recombinante
generate mutant versions of the V
enzyme
then examine"
the[S])
structure and function of these altered
Vand
0 !
max[S]/(K
m
mutantes, e examinar
os
efeitos
das
mutações
sobre
a
estrutura
e atividade da enzima.
forms. The technology used to do this is described in Chapter 9.
One
example
of
this
kind
of
analysis
is
the
work
of
Clarke
and
colleagues
on
the enzyme
A lactato desidrogenase
(LDH)
é uma
que or
catalisa a redução
delactate
piruvato com
so
V0 ! Vmax
when [S]/(K
[S])
! 0.25;
m "enzima
dehydrogenase,
published in 1989. Lactate
dehydrogenase (LDH) catalyzes the reduction of pyruvate
NADH lactato.
to form lactate
(seeesquema
Section 14.3). do
A schematic
of the enzyme’s
active
site
is
shown
below; piruvato
NADH para with
formar
Um
sítio ativo
da enzima,
contendo
[S]the!center:
0.33Km ! 0.33(0.0050 M) ! 1.7 # 10$3 M
the pyruvate is in
e NADH ligados, está mostrado abaixo:
(b) The Michaelis-Menten
equation can be rearranged to
Lactate dehydrogenase
102
V0/Vmax !Gln[S]/(Km " [S])
C
O equation
NH2
Substituting [S] ! %12% KmArg
into the
gives
Thr246
109
NH
H C
C
OH
3
C ! 0.5 K /1.5K
V0 /Vmax
m
m ! 0.33
+
NH
CH 3
H
OmC
HN
H
Similarly, substituting [S] ! 2K gives
His195
+
NH
C
Pyruvate
V0 /VHNmax ! O0.67
– O
H
H
And substituting [S]O!– O10Km gives
HN + NH
C
H
C
V0Asp
/V168
max ! 0.91
NH
H
N
H
H3C
H
CH3
C
CH2
(NADH)
Ile250
Arg171
(c) The upper curve corresponds to enzyme B ([X] is greater than the Km for this enzyme),
anddathereação
lower curve
corresponds
to enzymedeA.outras
When the
initial concentration
O mecanismo
é similar
ao mecanismo
reduções
por NADH.ofOsubstrate isenvolve
greater than
Km, the
rate ofdo
thepiruvato
reaction fortemente
is less sensitive
to the depletion of
estado de transição
o grupo
carbonil
polarizado:
substrate at early stages of the reaction and the rate remains approximately linear for a
longer time.
9. Applying the Michaelis-Menten Equation I A research group discovers a new version of happyase,
which they call happyase*, that catalyzes the chemical reaction
19 88z SAD
HAPPY y88
S-76
Chapter 6 Enzymes
The reaction mechanism is similar to many NADH reductions (Fig. 13–24); it is approximately the
reverse of steps 2 and 3 of Figure 14–7. The transition state involves a strongly polarized carbonyl
group of the pyruvate molecule as shown below:
CH3
A
OOC"
A
C
G G
O – O
!
Pergunta-se:
(a) A mutant form of LDH in which Arg109 is replaced with Gln shows only 5% of the pyruvate binding and 0.07% of the activity of wild-type enzyme. Provide a plausible explanation for the effects
i) Umofmutante
da LDH no qual a Arg109 foi substituída por Gln mostra apenas 5 %
this mutation.
de
a piruvato
e 0.07
% da
atividade
da with
enzima
selvagem.
Proponha
uma
(b)ligação
A mutant
form of LDH
in which
Arg171
is replaced
Lys shows
only 0.05%
of the wild-type
level ofpara
substrate
binding.
Why
is this dramatic effect surprising?
explicação
os efeitos
desta
mutação
171
(c)Você
In the
crystal
of LDH, da
theArg171
guanidinium
grouplisina
of Arg
and
the carboxyl
ii)
espera
questructure
a substituição
por uma
afete
a afinidade
da group of pyruvate
are
aligned
as
shown
in
a
co-planar
“forked”
configuration.
Based
on
this,
provide
a plausible
enzima pelo piruvato? Por quê?
171
explanation for the dramatic effect of substituting Arg with Lys.
(d)
A mutant
of LDH
in whichcatalisa
Ile250 is replaced
Gln shows
reduced binding of NADH. Pro3.
Uma
enzimaform
recém
descoberta
a reaçãowith
descrita
abaixo.
vide a plausible explanation for this result.
Clarke and colleagues also set out to engineer a mutant version of LDH that would bind and reduce oxaloacetate rather than pyruvate. They made a single substitution, replacing Gln102 with Arg;
the resulting enzyme would reduce oxaloacetate to malate and would no longer reduce pyruvate to
had therefore
converted
LDH
to enzima
malate dehydrogenase.
Olactate.
valor They
do turnover
encontrado
para
esta
é 600 s-1. Quando [E]T = 20×10-9 M
-1
e(e)
[T] Sketch
= 40 the
µM,
a velocidade
da reação
é voxaloacetate
. Calcule o KM para o
active
site of this mutant
LDH with
bound.
0 = 9.6 µMs
substrato.
(f) Provide a plausible explanation for why this mutant enzyme now “prefers” oxaloacetate instead
of pyruvate.
4.
hidrólise
pirofosfato
a ortofosfato
acoplada
importante
para a
(g)A The
authorsdewere
surprised that
substitutingé auma
largerreação
amino acid
in the active
site allowed
larger
substrate tode
bind.
Provide
a plausible explanation
for thisa result.
deslocar
o equilíbrio
reações
biossintéticas,
por exemplo
síntese de DNA. Esta
reação de hidrólise é catalisada por uma pirofosfatase que possui massa de 120 kDa e
consisteAnswer
de seis subunidades idênticas. Para esta enzima, a unidade de atividade
(a)
the wild-type
the substrate
is held innecessária
place by a hydrogen
bond and an ionenzimática éIndefinida
comoenzyme,
a quantidade
de enzima
109 para hidrolisar 10
dipole interaction between the charged
side
chain
of
Arg
and
the
polar
carbonyl of
µmol de pirofosfato
em 15 minutos a 37 0C. A enzima purificada possui Vmaxthe= polarized
2800
pyruvate. During catalysis, the charged Arg109 side chain also stabilizes
unidades por carbonyl
miligrama
de enzima.
transition
state. In the mutant, the binding is reduced to just a hydrogen bond,
substrate binding is weaker, and ionic stabilization of the transition state is lost, reducing
catalytic
activity. são hidrolisados por segundo por miligrama de enzima
a) Quantos mols
de substrato
quando a(b)
concentração
substrato
for muito
do que
o Km?
Because Lysdo
and
Arg are roughly
the maior
same size
and have
a similar positive charge, they
probably have very similar properties. Furthermore, because pyruvate binds to Arg171 by
(presumably)
an ativos
ionic interaction,
an Arg
to Lys
would
probably
little
b) Quantos mols
de sítios
existem em
1 mg
demutation
enzima?
Assuma
quehave
cada
effect on substrate binding.
subunidade possui um sítio ativo.
(c) The “forked” arrangement aligns two positively charged groups of Arg residues with the
negatively charged oxygens of pyruvate and facilitates two combined hydrogen-bond and
c) Qual é o turnover
da enzima?
ion-dipole interactions. When Lys is present, only one such combined hydrogen-bond
and ion-dipole interaction is possible, thus reducing the strength of the interaction. The
5. Suponha que
uma enzima
mutanteis ligue-se
com o substrato com uma afinidade
positioning
of the substrate
less precise.
250
100x maior
do que
a enzima
selvagem.
Qual
é oofefeito
mutação
sobre iso not
(d) Ile
interacts
hydrophobically
with
the ring
NADH.desta
This type
of interaction
turnover (kcatpossible
) considerando-se
que a side
interação
with the hydrophilic
chain ofcom
Gln. o estado de transição não foi
afetada?
6. Para uma enzima que segue a equação de Michaelis-Menten, qual é o valor de Vmax
considerando-se que v0=1µM/min a 1/10 KM?
20 7. A penicilina é hidrolisada por uma enzima beta-lactamase que está presente em
algumas cepas de bactérias resistentes a antibióticos. A massa desta enzima é 29.6
kDa. A quantidade de enzima hidrolisada em 1 minuto em uma solução de 10 ml
contendo 10-9 gramas de beta-lactamase purificada foi medida em função da
concentração de penicilina (tabela abaixo). Assuma que a concentração de penicilina
não se altera apreciavelmente durante o ensaio.
Quantidade hidrolisada
(nanomols)
0.11
0.25
0.34
0.45
0.58
0.61
[Penicilina] µM
1
3
5
10
30
50
a) Faça um gráfico de V0 em função de [S] e de 1/V0 em função de 1/[S]. Responda: a
beta-lactamase parece seguir a cinética de Michaelis-Menten? Neste caso, qual é o
valor do KM? Observação: abaixo é dada a equação da reta do segundo gráfico:
𝑦 = 4.6×10! + 8.8×10!" b) Qual é o valor do Vmax?
c) Qual é o turnover da enzima nestas condições experimentais? Assuma a presença
de um sítio ativo por molécula de enzima.
11
5.5
x 10
5
0.01
4
0.008
1/v0 (s/mol)
3.5
0.006
3
2.5
2
0.004
0
v (nanomol/segundo)
4.5
1.5
1
0.002
0.5
0
0
10
20
30
[S]*1e6 (M)
40
50
0
−2
0
2
4
−1
1/[S] M
6
8
10
5
x 10
8. A cinética de uma enzima foi medida em função da concentração de substrato na
presença e ausência de 2 mM do inibidor “I”.
[S] (µM)
3
5
10
30
90
Velocidade (µmol/minuto)
Sem inibidor
Com inibidor
10.4
4.1
14.5
6.4
22.5
11.3
33.8
22.6
40.5
33.8
a) Quais são os valores de Vmax e KM na ausência e presença de inibidor? São
dadas as seguintes quações de reta:
21 Azul (sem inibidor): 𝑦 = 13𝑥 + 1.3×10!
Vermelho (com inibidor): 𝑦 = 40𝑥 + 1.3×10!
45
6
15
x 10
40
35
1/v0 (s/mol)
10
25
20
0
v (micromol/minuto)
30
15
5
10
5
0
0
10
20
30
40
50
[S]*1e6 (M)
60
70
80
90
0
−2
−1.5
−1
−0.5
0
0.5
1
−1
1/[S] M
1.5
2
2.5
3
3.5
5
x 10
b) Qual é o tipo de inibição?
c) Qual é a constante de afinidade do inibidor?
d) Se [S] = 10 µM e [I] = 2 mM, qual é a fração de moléculas de enzima ligadas
ao substrato? E com o inibidor?
e) Se [S] = 30 µM, qual é a fração de moléculas de enzima que estão ligadas com
o substrato na presença e na ausência de 2 mM de inibidor? Compare esta
razão com a razão entre as velocidades nas mesmas condições.
3. Metabolismo
Fermentação alcoólica
Em aulas anteriores vimos como a hidrólise de ATP formando ADP e fosfato
inorgânico libera uma grande quantidade de energia (Figura 11) que será utilizada
para promover processos celulares que requerem o fornecimento de energia. Estes
processo podem ser: i) contração muscular e movimentos celulares, ii) transporte
ativo de íons e moléculas através da membrana, iii) síntese de aminoácidos,
nucleotídeos, proteínas, DNA e outros compostos necessários à célula.
Como exemplo de transporte ativo, lembrem-se daquele promovido pela bomba de
Na+/K+ para a manutenção do gradiente iônico através da membrana plasmática
(aulas anteriores).
22 ∆𝐺° = −30.5 𝑘𝑗. 𝑚𝑜𝑙 !!
6
NH2
C
N
HC
N
O-
O-
O-
-O-P-O-P-O-P-O-CH
2
O
O
C
N
C
CH
Adenina
N
O
O
H
H
H
HO
H
Ribose
OH
AMP
ADP
ATP
ATP = Adenosina 5'-trifosfato
Figura 11. Reação de hidrólise de ATP formando ADP e fosfato inorgânico (Pi). A
variação de energia livre nas condições padrão (ΔG0) está indicada. Esta reação possui
um ΔG ainda mais
negativoTIPOS
nas condições
celulares devido às concentrações fora do
ALGUNS
DE ENZIMAS
equilíbrio de ADP e ATP (veja a equação 3, pág. 10)!! Estrutura dos compostos
fosforilados ATP, ADP e AMP.
Quinases: São enzimas que catalisam a transferência de um grupo fosfato de um
composto de alta energia (em geral do ATP) para um aceptor.
Isomerases: São enzimas
que catalisam
depara
isomerização
defundamentais:
função.
O metabolismo
celularreações
é voltado
dois aspectos
i) produção
ATP para fornecer
energia para
os processos celulares
e ii) síntese
de de baixa
Mutases: São de
isomerases
que catalisam
a transferência
de grupos
fosfatos
intermediários
de
biossíntese
(ác.
graxos,
lipídeos,
hormônios,
aminoácidos,
energia de uma posição para a outra, dentro da mesma molécula.
nucleotídeos, etc). O metabolismo degradativo é chamado de catabolismo, enquanto
Desidrogenases:
enzimas
que catalisam
de óxido-redução,
que oSão
metabolismo
biosintético
é conhecidoreações
como anabolismo.
Organismos quepor
+
energiado
livre
a partir dapara
luz do
sol coenzima,
são chamados
de fótotropicos,
que
transferência deobtém
hidrogênio
substrato
uma
geralmente
NAD enquanto
ou FAD.
Essas reações, na maioria dos casos são reversíveis.
Aldolases: São enzimas que cindem açúcares fosforilados, dando origem a
diidroxiacetona-fosfato
e a outro açúcar, com 3 átomos de carbono a menos que o 23 substrato original.
Fosfatases: São enzimas que catalisam reações de hidrólise de ésteres de fosfato.
organismos que obtém energia livre através da oxidação de moléculas (exemplo
açúcares) são chamados de quimiotrópicos.
Células clivam e oxidam moléculas glicose para produzir energia na
forma de moléculas de ATP e intermediários para as reações de biossíntese. A
principal via para obtenção de ATP é o metabolismo de glicose que se inicia através
da quebra da glicose pela via glicolítica, na qual uma molécula de glicose é clivada
em duas moléculas de piruvato (ácido pirúvico) com a concomitante formação de
duas moléculas de ATP.
A descoberta da via glicolítica (ou glicólise) está intimamente relacionada ao
estudo da fermentação alcóolica promovida por leveduras. De fato, uma parte da
bioquímica tal qual conhecemos hoje originou-se a partir do estudo da fermentação
alcoólica durante o final do século XIX e a primeira metade do século XX. Nessa
época, sabia-se que leveduras eram capazes de fermentar açúcar para produzir etanol
(Figura 12), e percebeu-se que tanto o extrato de levedura como as células vivas
possuíam a mesma atividade. Uma fração do extrato de leveduras que era não
dializável e sensível à temperatura foi chamada de zymase, enquanto que outra fração
que era dializável e insensível à temperatura foi chamada de cozymase. Mais tarde
descobriu-se que zymase e cozymase eram, respectivamente, uma mistura de enzimas
e de co-fatores como NAD+, ATP, ADP e íons metálicos.
Pesquisas realizadas para compreender este fenômeno acabaram conduzindo à
descoberta da sequência de reações de envolvidas na quebra anaeróbica de glicose
(hexose; açúcar de 6 carbonos) em etanol e gás carbônico, e à identificação e
isolamento das enzimas que catalisam estas reações. Estes estudos, realizados
simultaneamente tanto em leveduras como em extratos de músculo, mostraram que a
glicólise é uma via comum a todos os organismos, de procariotos às nossas células
humanas.
A glicólise quebra duas moléculas de glicose (uma hexose) em duas moléculas
de piruvato (ácido pirúvico). O catabolismo anaeróbico do piruvato em células de
levedura leva à formação de etanol, enquanto que em células musculares leva à
formação de ácido lático (Figura 13). O ácido lático é formado no músculo quando a
quantidade de oxigênio disponível é insuficiente para promover o metabolismo
aeróbico (por exemplo, uma situação de exercício físico intenso). O ácido lático
formado no músculo pode ser convertido novamente em glicose nas células do fígado
pelo chamado “ciclo de Cori”.
A sequência de reações enzimáticas da via glicolítica tal como a conhecemos
hoje pode ser observada na Figura 14.
24 C
C
C
C
cally, yeasts convert it to ethanol; but muscles convert it to lactate (Figure 3): ‘. . . a muscle resting in
nitrogen’, Meyerhof explains, ‘produces lactic acid
steadily; in oxygen no lactic acid accumulates’12
([157] p. 1415). Indeed, there have been many parallels and interconnexions in the research on the
two kinds of eukaryotic cell, those of yeasts and
hexose
those of muscles.13
C
C
Figure 2. Path of carbon atoms in the conversion of
glucose to ethanol and carbon dioxide. triose
Each carbon atom
of a glucose molecule is numbered to show its fate during
fermentation
CH3
CH2
OH CO2
C
C
C
Notes on some of the most exceptional
investigators
C
C
C
By 1940, the complete pathway of glycolysis
had been
elicited, largely by a few remarkable
triose
biochemists, of whom five were of Jewish origin,
as was an astonishing number of other outstanding
CO2 CH3
CH2
OH
12 Lactic acid is formed in muscle when the amount of oxygen is limiting; this occurs during great muscular activity and is a major
12. Destino
dos átomos
carbono
da formed
glicose
durante
a fermentação
em by liver cells;
factorFigura
limiting achievements
by athletes
and ballet de
dancers.
Lactic acid
in muscle
is converted
back into glucose
this cycle
of glucoseBaseado
→ lactate →em
glucose
is the ‘Cori
cycle’Yeast
[48]. 20: 509-543.
leveduras.
Barnett
(2003)
13 Valuable reviews of the history of research on glycolysis are to be found in the four editions of Harden’s Alcoholic Fermentation
[94,95,96,98], Dorothy Needham’s Machina Carnis [177], Fruton’s Proteins, Enzymes, Genes [80] and volume 31 of Comprehensive
Biochemistry by Florkin and Stotz [74].
Figura 13. Catabolismo do piruvato a etanol (levedura) ou ácido lático no músculo
em condições anaeróbicas. Retirado de Barnett (2003) Yeast 20: 509-543.
Figure 3. The catabolism of pyruvate to ethanol by yeasts, or to lactic acid by muscle
Copyright © 2003 John Wiley & Sons, Ltd.
Yeast 2003; 20: 509–543.
25 8
HOCH2
HO
O
GLICÓLISE
OH
OH
Glicose
OH
ATP
ATP
ADP
ADP
P -OCH2
O
HO
OH
Glicose 6-fosfato
OH
OH
P -O-CH2 O
CH2OH
HO
OH
Frutose 6-fosfato
HO
ATP
ATP
ADP
P -O-CH2 O
ADP
CH2O- P
HO OH
Frutose 1,6 bisfosfato
HO
HC=O
H2C-O- P
C=O
Diidroxiacetona
fosfato
HC-OH
H2C-OH
H2C-O- P
Gliceraldeído
3-fosfato
Pi
NAD+
Pi
NAD+
NADH
NADH
O=C-O- P
HC-OH
1,3 Bisfosfoglicerato
H2C-O- P
ADP
ADP
ATP
ATP
O=C-OHC-OH
3-Fosfoglicerato
H2C-O- P
COOHC-O- P
2-Fosfoglicerato
H2C-OH
H2O
COOC-O- P
CH2
Fosfoenolpiruvato
ADP
ADP
ATP
C=O
HC-OH
CH3
ATP
COO-
COO-
NAD+
NADH
CH3
Lactato
Piruvato
NAD+
NADH
Figura 14. Sequência de reações que compõem a glicólise e a fermentação láctica.
26 Todas as reações ocorrem no citosol das células eucarióticas. A glicose não
atravessa a membrana celular, entra na célula com auxílio de uma proteína carreadora
específica. Imediatamente, é possível notar que: i) a glicólise envolve duas etapas de
fosforilação por ATP, formando ADP e fosfato inorgânico (Pi); ii) cada molécula de
glicose contendo 6 átomos de carbono é convertida em duas moléculas de ácido
pirúvico, mais oxidado; iii) o processo global envolve a formação de 4 moléculas de
ATP, ou seja, produção líquida de duas moléculas de ATP; iv) ocorre uma reação de
oxido-redução quando gliceraldeído-3-fosfato é convertido em 1,3-bifosfoglicerato,
os elétrons são transferidos para a coenzima NAD+ formando NADH.
Vamos analisar estas reações em mais detalhe.
Reação 1: Fosforilação de glicose por ATP formando glicose-6-fosfato e ADP. Essa
reação é catalisada pela hexoquinase.
Figura 15. Reação de fosforilação da glicose por ATP formando glicose-6-fosfato e
ADP. Essa reação é catalisada pela hexoquinase. A variação de energia livre
envolvida é ΔG0 = -16.7 kJ.mol-1.
É interessante notar que já no início do século XX era sabido que glicose-6fosfato (hexose fosfato) não era fermentada ou hidrolisada por leveduras. A
explicação para essa observação é que as células não possuem proteínas de membrana
transportadores de glicose-6-fosfato (ou frutose 1,6 bifosfato). Portanto, o açúcar
fosforilado é incapaz de entrar ou sair da célula.1
A enzima que catalisa esta reação é chamada hexoquinase. Quinases são
enzimas que catalisam a adição de um grupo fosforil do ATP para outra molécula.
A reação de fosforilação da glicose formando glicose-6-fosfato ilustra um dos
aspectos principais do metabolismo: reações endergônicas (que requerem o
fornecimento de energia livre para ocorrer) são tornadas espontâneas através do
acoplamento com outra reação altamente exergônica (que libera uma grande
1
Note que a célula possui transportadores de glicose (exemplo GLUT1, GLUT2, etc).
Estes transportadores são específicos para glicose.
27 quantidade de energia livre). A reação entre glicose e fosfato inorgânico (PO4-3)
formando glicose-6-fosfato consome uma grande quantidade de energia e não
ocorreria caso não fosse acoplada à reação de hidrólise de ATP:
Glicose + Pi à glicose-6-fosfato + H2O
ATP + H2O à ADP + Pi
ΔG0 = + 13.8 kJ.mol-1
ΔG0 = - 30.5 kJ.mol-1
Glicose + ATP à glicose-6-fosfato + ADP ΔG0 = - 16.7 kJ.mol-1
Evidentemente estas duas reações não ocorrem isoladamente em água, mas
uma enzima, a hexoquinase aproxima os reagentes de propicia um ambiente adequado
para a reação ocorrer. Da mesma forma, a formação de ATP somente é possível
porque está acoplada a outra reação mais exergônica (ver abaixo):
Fosfoenolpiruvato + H2O à piruvato + Pi
ADP + Pi à ATP + H2O
ΔG0 = - 61.9 kJ.mol-1
ΔG0 = + 30.5 kJ.mol-1
Fosfoenolpiruvato + ADP à ATP + piruvato
ΔG0 = - 31.4 kJ.mol-1
Logicamente que se as reações de hidrólise de fosfoenolpiruvato e fosforilação
de ATP ocorressem isoladamente em água uma não influenciaria a outra. A enzima
piruvato kinase liga fosfoenolpiruvato e ADP no seu sítio ativo proporcionando
condições para que a reação acoplada ocorra.
Reação 2. Isomerização da glicose-6-fosfato em frutose-6-fosfato, catalisada pela
enzima fosfoglico isomerase (Figura 16).
Figura 16. Reação de isomerização de glicose-6-fofato, uma aldose, em frutose-6fosfato, uma cetose. Essa reação é catalisada pela enzima phosphohexose isomerase.
A variação de energia livre envolvida é ΔG0 = +1.7 kJ.mol-1, o que indica que a
reação é reversível.
Reação 3. Fosforilação da frutose-6-fosfato por ATP formando frutose 1,6 bifosfato.
Essa reação é catalisada pela enzima fosfofrutoquinase 1 (PFK1, do inglês
phosphofructo kinase).
28 Figura 17. Reação de fosforilação da frutose-6-fosfato catalisada pela enzima
fosfofrutoquinase 1 (PFK1) com o consumo de uma molécula de ATP. A variação de
energia livre envolvida é ΔG0 = -14.2 kJ.mol-1.
A variação de energia livre envolvida na reação de fosforilação de frutose-6fosfato catalisada pela fosfofrutoquinase-1 é altamente negativa, indicando que esta é
uma reação praticamente irreversível. Reações irreversíveis costumam ser pontos de
regulação no metabolismo. A fosfofrutoquinse-1 é uma enzima alostérica e está
sujeita à regulação a partir de uma série de fatores alostericos como ATP/ADP e
frutose-2,6-bifosfato. A figura 18 mostra a estrutura da PFK1 em complexo com os
produtos da reação. A estrutura consiste de duas cadeias polipeptídicas cada qual
contendo um sítio ativo e um sítio para ligação do efetor alostérico ADP.
Figura 18. Estrutura tridimensional da PFK1 (PDB PFK1) em complexo com os
produtos da reação, frutose 1,6 bifosfato e ADP no sítio ativo, e o ativador alosterico
ADP. Esta estrutura mostra duas subunidades da enzima, que é tetramérica.
Reação 4. A quarta reação corresponde à clivagem da frutose 1,6 bifosfato em dois
compostos de 3 átomos de carbono: gliceraldeido 3-fosfato e di-hidroxi-acetona
fosfato (Figura 19):
29 Figura 19. Reação de clivagem da frutose 1,6 bifosfato em dois compostos de 3
átomos de carbono, gliceraldeído 3-fosfato e dihidroxiacetona-fosfato. Essa reação é
catalisada pela enzima aldolase. A variação de energia livre envolvida é ΔG0 = +23.8
kJ.mol-1.
Apesar de que a variação de energia livre envolvida é positiva, nas condições
celulares os produtos da reação são rapidamente consumidos o que faz com que o ΔG
da reação seja menor e ela torne-se praticamente reversível. Apenas o gliceraldeído-3fosfato será utilizado para as próximas etapas da glicólise. Com a finalidade de não
perder um composto de 3 carbonos, as células produzem uma isomerase (triosefosfato isomerase – TPI ou TIM), que é capaz de catalisar a reação de isomerização
de uma cetose (dihidroxiacetona fosfato) em uma aldose (gliceraldeído-3-fosfato)
(Figura 20):
Figura 20. Reação de isomerização entre gliceraldeído-3-fosfato e dihidroxi-acetonafosfato. A variação de energia livre envolvida é ΔG0 = +7.5 kJ.mol-1.
Apesar de que o ΔG0 da reação é positivo, nas condições celulares esta reação
também é praticamente reversível. Esta etapa conclui a conversão de uma molécula de
glicose em duas moléculas de gliceraldeído-3-fosfato, um composto de 3 átomos de
carbono e mais oxidado do que a glicose. Para tanto foi necessário a hidrólise de duas
moléculas de ATP formando ADP. As próximas etapas envolvem a oxidação de
gliceraldeído-3-fosfato em piruvato, um ácido carboxílico e portanto um composto
mais oxidado. A conversão de gliceraldeído-3-fosfato envolverá perda de elétrons
(oxidação) e a formação de duas moléculas de ATP (quatro moléculas no computo
geral).
Reação 5. A reação de oxidação do gliceraldeído-3-fosfato formando 1,3-bifosfoglicerato (1,3-BPG) é catalisada pela gliceraldeído-3-fosfato desidrogenase. Esta
reação depende da presença de uma coenzima, NAD+. Historicamente, NAD+ foi
30 chamado de co-fermento ou co-enzima. O papel do NAD como carreador de
hidrogênio foi sugerido em torno de 1920, mas a natureza química da coenzima
somente foi elucidada nos anos 30. NAD (Figura 21) é um dinucleotídeo de
nicotinamida e adenina. Ele funciona como um carreador de átomos de hidrogênio
em uma ampla série de reações de oxido-redução ao aceitar um íon hidreto (H-; um
próton e 2 elétrons). NAD e NADH são solúveis, e podem mover-se de uma enzima
para outra.
Reações de oxido-redução:
Figura 21. Estrutura da coenzima nicotinamida adenina dinucleotideo (NAD) nas
formas oxidada (NAD) e reduzida (NADH).
Um grande número de enzimas utiliza NAD como co-enzima carreadora de
elétrons, catalisando reações nas quais NAD aceita um íon H- do substrato (oxidação)
ou doa um íon H- ao substrato (redução). Estas enzimas são chamadas de
oxidoredutases ou desidrogenases. A gliceraldeído-3-fosfato desidrogenase utiliza a
coenzima NAD+ para receber um íon H- do gliceraldeído-3-fosfato que é oxidado a 3fosfoglicerato formando NADH. O domínio responsável por ligar NAD+ ou NADH é
chamado de Rossmann fold. A coenzima NAD (e NADP) é fracamente associada à
enzima e pode difundir-se de uma enzima para a outra funcionando como um
composto carreador de elétrons solúvel. O NADH formado como resultado da ação da
gliceraldeído-3-fosfato desidrogenase difunde-se até encontrar a álcool desidrogenase
que promoverá a redução de acetaldeído à etanol durante a fermentação alcoólica:
A reação catalisada pela gliceraldeído-3-fosfato desidrogenase pode ser
compreendida como a soma de dois processos (Figura 22):
31 Figura 22. Reação de oxidação e adição de ortofosfato catalisada pela gliceraldeído3-fosfato desidrogenase.
Reação 6. A próxima reação envolve a transferência de um grupo fosforil do 1,3bifosfoglicerato (1,3 BPG) para ADP formando ATP e 3-fosfoglicerato. Essa reação é
catalisada pela enzima fosfoglicerato quinase (Figura 23).
Figura 23. Reação catalisada pela fosfoglicerato quinase. A variação de energia livre
dessa reação nas condições padrão é ΔG0 = -18.8 kcal.mol-1, no entanto nas condições
celulares a reação é reversível com ΔG = + 1.3 kcal.mol-1.
Reações 7 a 9: As próximas reações envolverão a conversão de 3-fosfoglicerato a
fosfoenolpiruvato, um composto com alto potencial de transferência de um grupo
fosforil, e finalmente a piruvato com a concomitante formação de uma molécula de
ATP (Figura 24):
32 Figura 24. Reações de isomerização de 3-fosfoglicerato a 2-fosfoglicerato (ΔG0 =
+4.6 kJ.mol-1); de desidratação de 2-fosfoglicerato formando uma dupla ligação e o
composto fosfoenolpiruvato (ΔG0 = +1.7 kJ.mol-1); e de transferência de um grupo
fosforil do fosfoenolpiruvato ao ADP formando ATP e piruvato (ΔG0 = -31.4 kJ.mol1
).
É interessante notar que a reação de transferência do grupo fosforil do
fosfoenolpiruvato ao ADP é extremamente favorável. Uma explicação para essa
observação é que a forma enólica do piruvato é altamente instável e converte-se
espontaneamente para a forma cetônica, piruvato.
A reação completa da quebra de glicose em duas moléculas de piruvato é:
Esta reação envolve uma variação de energia livre da ordem de ΔG0 = -73.1
kJ.mol , e resulta na formação de duas moléculas de ATP e na redução de duas
moléculas de NAD+. Esse processo poderia seguir indefinidamente não fosse o fato de
que a disponibilidade de NAD+ na célula é finita. Conforme já discutido acima, em
condições anaeróbicas, o piruvato é reduzido a lactato (células musculares) ou a
etanol (levedura) regenerando NAD+. Esse processo é conhecido como fermentação
(Figura 25):
-1
33 Figura 25. Reações enzimáticas que ocorrem nas células de levedura ou do músculo
durante a fermentação e levam à regeneração de NAD+.
A eficiência da glicólise expressa em termos de conversão de energia
armazenada na forma de glicose para moléculas de ATP é muito baixa. A
fermentação alcoólica gera um total de -235 kJ.mol-1 de glicose (-196 kJ.mol-1 para a
fermentação láctica) sendo que aproximadamente 61 kJ.mol-1 de energia livre são
necessários para a síntese de duas moléculas de ATP. Ou seja, apenas 1/3 da energia
liberada pela fermentação é utilizada para a síntese de ATP. O resto é dissipado na
forma de calor !
À medida em que as reações da fermentação foram sendo elucidadas, outros
bioquímicos voltaram sua atenção ao metabolismo aeróbico em levedura (após 1930).
Na presença de oxigênio, as células são capazes de oxidar a glicose completamente a
CO2 e H2O extraindo muito mais energia e formando um número muito maior de
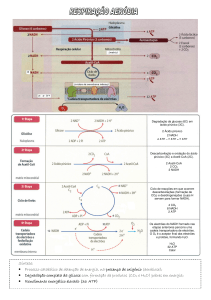
moléculas de ATP a partir de uma única molécula de glicose. Este processo é
conhecido por respiração celular, e envolve duas vias metabólicas que vamos discutir
em detalhes: o ciclo de Krebs ou ciclo do ácido cítrico, a cadeia de transporte de
elétrons e a fosforilação oxidativa.
Uma das principais diferenças entre a glicólise, o ciclo de Krebs e a
fosforilação oxidativa é que enquanto a primeira via metabólica ocorre no citoplasma
da célula, as duas outras ocorrem dentro da mitocôndria. Esta diferença de
compartimentos irá auxiliar a regulação das duas vias.
34 Glicólise exercícios
1. Complete o mapa metabólico mostrado na Figura 14 indicando quais são as
enzimas responsáveis por catalisar cada reação, os efetuadores alostericos e o
número de carbonos de cada metabolito.
2. Responda:
i)
Quais os passos irreversíveis que aparecem no mapa?
ii)
Quantas moléculas de piruvato se formam a partir de uma molécula de
glicose?
iii)
Qual hexose da origem a trioses?
iv)
Indicar as reações de oxido-redução que aparecem no mapa.
v)
Sabendo-se que a concentração celular de NAD+ é da ordem de 10-5M, é
possível estimar a quantidade de glicose que pode ser convertida a lactato?
vi)
Verificar no mapa os compostos que apresentam ligações do tipo:
a) fosfoenol
b) anidrido fosfórico
c) éster fosfórico
vii) Identificar no mapa as reações catalisadas pelas seguintes enzimas
a) quinase
b) mutase
c) isomerase
d) aldolase
e) desidrogenase
3) Considerando-se o número de moléculas de ATP consumidas e formadas,
estabelecer o saldo final de ATP na oxidação de uma molécula de glicose pela via
glicolítica
4) Qual a porcentagem de energia que a célula armazena, a partir de um mol de
glicose, pela sua degradação através da via glicolítica? Sabe-se que:
Glicose à Lactato
ΔG0’ = -195 kJ/mol
5) Citar os compostos que devem ser fornecidos à via glicolítica para:
a) iniciá-la (haver formação de lactato)
b) mantê-la em funcionamento
6) Indicar a função da via glicolítica
35 Ciclo de Krebs
O ciclo de Krebs é uma espécie de ponto de encontro metabólico da célula.
Todas as moléculas que podem ser convertidas em um grupo acetil ou em
intermediários do ciclo podem ser aproveitadas e oxidadas completamente a CO2
gerando elétrons (na forma de NADH ou FADH2) que depois serão transferido ao
oxigênio pela fosforilação oxidativa gerando água e promovendo a síntese de ATP
(vide infra). De forma geral, o ciclo de Krebs oxida o grupo acetil da Acetil-CoA em
duas moléculas de CO2 com a geração concomitante de três NADHs, um FADH2 e
um GTP.
O piruvato gerado pela via glicolítica é transportado para dentro da
mitocondria por um carreador específico. Na matriz mitocondrial, o piruvato será
descarboxilado oxidativamente pelo complexo piruvato desidrogenase gerando um
grupo acetil. Este grupo acetil é ativado por esterificação com a Coenzima-A,
formando o grupo acetil-CoA. Este é o ponto de partida para entrada no ciclo de
Krebs (Figura 26):
Figura 26. Destino do piruvato que entra na mitocôndria.
A primeira reação do ciclo de Krebs envolve a condensação de um grupo
acetil proveniente do piruvato, com oxaloacetato, formando citrato. Para que esta
reação possa ocorrer, o grupo acetil precisa ser ativado pela ligação à Coenzima A. A
elucidação da estrutura da Coenzima-A e a comprovação de que o grupo acetil precisa
ser ativado para reagir com oxaloacetato formando citrato foram fundamentais para a
elucidação das reações do ciclo de Krebs. O complexo piruvato desidrogenase
catalisa a reação de descarboxilação oxidativa do piruvato, transferindo um par de
elétrons para o NAD+ e gerando acetil-CoA (Figura 27):
36 Figura 27. Reação de descarboxilação oxidativa de piruvato formando NADH e um
radical acetil ativado através da ligação com a Coenzima A (acima). Estrutura
molecular da Coenzima A (abaixo). Esta reação é irreversível como indica a
descarboxilação.
O complexo piruvato desidrogenase é formado por três enzimas que
promovem a descarboxilação e oxidação do piruvato formando um grupo acetil, e
promovem ainda a reação de condensação entre o grupo acetil e o grupo sulfidril da
Coenzima-A gerando, através de uma ligação tio-éster, Acetil-CoA (Figura 28). Uma
das co-enzimas importantes nesse processo chama-se FAD, um grupo prostético
associado fortemente a uma das enzimas do complexo piruvato desidrogenase e que
aceita um um par de elétrons (dois prótons e dois elétrons) assumindo a forma
reduzida, FADH2 (Figura 29). As enzimas associadas a um grupo FAD são chamada
de flavoproteínas. Ao contrário do NAD, o FAD associa-se fortemente à enzima e é
incapaz de difundir-se livremente em solução.
Figura 28. Sequência de reações promovidas pelo complexo piruvato desidrogenase.
37 Figura 29. Estrutura do grupo prostético FAD na forma oxidada (esquerda) e na
forma reduzida, FADH2 (direita).
A formação de Acetil-CoA é o ponto de partida para a entrada de piruvato no
ciclo de Krebs. As reações seguintes envolvem a condensação de Acetil-CoA com
oxaloacetato formando citrato, um composto de 6 átomos de carbono. Essa reação é
catalisada pela enzima citrato sintase (Figura 30). Em seguida, o citrato sofre uma
reação de isomerização que altera a posição do radical OH. Essa reação de
isomerização que é realizada através de uma desidratação seguida de uma hidratação,
é catalisada pela enzima aconitase (Figura 30). A posição do radical OH no carbono-2
do isocitrato permite a próxima reação, que é a descarboxilação oxidativa do
isocitrato formando alfa-cetoglutarato. Os elétrons do átomo de carbono que é
oxidado são transferidos para NAD+ formando a coenzima reduzida NADH. Note que
nesta reação de descarboxilação, o átomo de carbono que é eliminado na forma de
CO2 não pertence ao grupo Acetil-CoA (Figura 30).
38 Figura 30. As três primeiras reações do ciclo de Krebs. A entrada de Acetil-CoA no
ciclo da-se pela condensação com oxaloacetato (um alfa-ceto ácido) formando citrato.
Em seguida o citrato sofre uma isomerização formando isocitrato, que é oxidado e
descarboxilado gerando alfa-cetoglutarato.
O alfa-cetoglutarato sofre uma reação de descarboxilação oxidativa catalisada
pelo complexo alfa-cetoglutarato desidrogenase, gerando succinil-CoA e outra
molécula de NADH. Note que succinil-CoA é um composto de 4 átomos de carbono
(Figura 31). A hidrólise de succinil-CoA a succinato libera uma grande quantidade de
energia livre, que é mais do que suficiente para permitir a fosforilação de GDP
formando GTP, o único nucleotídeo trifosfato formado pelo ciclo de Krebs (Figura
31).
Figura 31. Reações de descaboxilação oxidativa do alfa-cetoglutarato formando
succinil-CoA, e de hidrólise do succinil-CoA gerando succinato. Esta última reação é
acoplada à fosforilação de GDP gerando GTP.
39 Succinato é um composto de 4 átomos de carbono que deve ser oxidado a
oxaloacetato. A forma para fazer isso é primeiro oxidar succinato a fumarato
gerando FADH2, seguido de uma reação de hidratação que forma malato, este por sua
vez pode ser oxidado a oxaloacetato completando o ciclo de Krebs e gerando a
terceira molécula de NADH (Figura 32).
A enzima succinato desidrogenase distingui-se das outras enzimas do ciclo de
Krebs pois utiliza FAD como grupo aceptor de elétrons, além disso ela esta embebida
na membrana mitocondrial interna onde ocorrem as reações da cadeia de transporte de
elétrons. Dessa forma, a succinato desidrogenase faz o link entre o ciclo de Krebs e a
fosforilação oxidativa responsável pela formação de ATP.
Figura 32. Motivo de três reações necessárias para oxidar succinato a oxaloacetato.
Este motivo consiste de uma reação de oxidação catalisada pela enzima succinato
desidrogenase, seguido de uma reação de hidratação catalisada pela enzima fumarase
e outra oxidação que finalmente converte malato em oxaloacetato. Duas coenzimas
reduzidas são produzidas neste processo, FADH2 e NADH.
Olhando estas reações de forma global, vemos que elas perfazem um ciclo em
que uma unidade de dois carbonos entra (Acetil-CoA) e sai completamente oxidada
na forma de duas moléculas de CO2. Note que os átomos de carbono da Acetil-CoA
somente irão sair na forma de CO2 após a quarta volta no ciclo. A cada volta, são
formadas coenzimas reduzidas NADH e FADH2 (Figura 33). Seguramente, o ciclo
pode ser alimentado por unidades de Acetil-CoA provenientes da quebra de glicose
pela via glicolítica, ou provenientes de outras vias metabólicas como a oxidação de
ácidos graxos e a degradação de certos aminoácidos. Subprodutos do metabolismo
também podem alimentar o ciclo de Krebs, como succinato ou oxaloacetato
provenientes do metabolismo de aminoácidos, assim como o ciclo de Krebs também
fornecerá esqueletos de carbono para outras reações biossintéticas.
40 C2
acetil-CoA
NADH
C4
oxaloacetato
condensação
C6
citrato
C6
isocitrato
oxidação
Ciclo de Krebs
C4
Malato
C4
Fumarato
FADH2
descarboxilação
oxidativa
C5
α-cetoglutarato
hidratação
H 2O
isomerização
descarboxilação
oxidativa
oxidação
hidrólise
C4
Succinil-CoA
CO2
NADH
CO2
NADH
C4
Succinato
GTP
Figura 33. Visão global do ciclo de Krebs. O tipo de cada reação está indicado em
vermelho. Os compostos que depois serão úteis para a síntese de ATP estão indicados
em azul.
A reação global do ciclo de Krebs é mostrada abaixo. Note que a oxidação
completa de uma molécula de glicose a CO2 pela via glicolítica e pelo ciclo de Krebs
leva à formação de duas moléculas de ATP, duas moléculas de GTP, 6 moléculas de
NADH e duas moléculas de FADH2.
Qual é o destino dos elétrons de alta energia contidos em NADH e FADH2?
Esses elétrons serão transferidos para a cadeia de transporte de elétrons e depois
finalmente transferidos para uma molécula de oxigênio que será reduzida formando
41 água. Um gradiente de prótons através da membrana interna da mitocôndria é gerado
à medida em que os elétrons são transportados pela cadeia de transporte de elétrons.
Esse gradiente será utilizado para promover a síntese de ATP.
Exercícios sobre o ciclo de Krebs
1. Desenhe o ciclo de Krebs, indique as enzimas que catalisam cada reação.
Comece a partir de Acetil-CoA
2. Na oxidação de uma molécula de acetil-CoA no ciclo de Krebs, indicar a
enzima que catalisa a reação onde há produção ou consumo de:
a) CO2
b) GTP
c) NADH
d) FADH2
e) H2O
3.
4.
5.
6.
Indicar o composto rico em energia do ciclo de Krebs e a reação que o produz
Citar as vitaminas que participam do ciclo de Krebs
Indicar a localização do ciclo de Krebs
Na reação catalisada pela aconitase indicar o composto predominante no
equilíbrio
7. Esquematizar a reação catalisada pela piruvato carboxilase e citar o seu
efetuador alostérico
8. Citar as funções do ciclo de Krebs
9. Mostras as consequências metabólicas da ativação da piruvato carboxilase por
acetil-CoA
10. Descrever a regulação do ciclo de Krebs em função das relações ATP/ADP e
NAD+/NADH
11. Relacionar a velocidade da via glicolitica com a atividade da isocitrato
desidrogenase
42 Fosforilação oxidativa
Glicose é completamente oxidada a CO2 através das reações da via glicolítica
e do ciclo de Krebs. Os elétrons retirados da glicose são utilizados para reduzir O2
formando H2O culminando o fim do processo oxidativo. Fosforilação oxidativa é o
processo pelo qual ATP é formado como resultado de uma série de reações de
transferência de elétrons das coenzimas reduzidas NADH e FADH2 para O2 formando
H2O. As moléculas carreadoras de elétrons são proteínas de membrana localizadas na
membrana interna da mitocôndria e formam a cadeia de transporte de elétrons. O
fluxo de elétrons através dos carreadores provoca a saída de prótons da matriz
mitocondrial para o espaço inter-membrana. A distribuição desigual de H+ nos dois
lados da membrana interna gera um gradiente de pH e uma diferença de potencial
elétrico através da membrana interna. O retorno dos íons H+ para a membrana interna
através de uma proteína transportadora de H+ (ATP sintase) é altamente exergônico
(ΔG << 0). Essa variação de energia livre será suficiente para tornar
termodinamicamente favorável a reação de síntese de várias ATP a partir de ADP e
fosfato inorgânico pelo complexo ATP-sintase (Figura 34).
A mitocôndria possui duas membranas: membrana interna e membrana
externa. O compartimento interno, delimitado pela membrana interna, é chamado de
matriz mitocondrial. A matriz contém todas as enzimas do ciclo de Krebs e as
enzimas que catalisam a oxidação de ácidos graxos. A membrana interna contém 75%
de proteínas em massa. Todas as enzimas envolvidas na cadeia de transporte de
elétrons estão localizadas na membrana interna, bem como uma série de proteínas
transportadoras para permitir a entrada de metabólitos essenciais na matriz. Entre
estes metabolitos encontra-se NADH produzido no citosol durante a glicólise. A
membrana interna também contem um transportador de ADP/ATP, de forma que o
ATP produzido na matriz possa ser transportado onde ele será utilizado para fornecer
energia para outros processos celulares.
espaço inter-membrana
H+
I
NADH
NAD+
matriz
H+
II
FADH2
eFAD
III
H+
H+
IV
O2
H 2O
H+
ADP + Pi
ATP
Figura 34. Elétrons provenientes das coenzimas reduzidas NADH e FADH2
(provenientes do ciclo de Krebs) são transferidos para as proteínas carreadoras da
cadeia de transporte de elétrons através de uma série de reações de oxido-redução.
Estes elétrons são utilizados para reduzir O2 formando H2O. O fluxo de elétrons
43 através dos carreadores é acoplado ao transporte de prótons através da membrana,
gerando uma força próton-motriz que é suficiente para promover a síntese de ATP
pela enzima ATP-sintase.
Vamos ver detalhes sobre como funciona a cadeia de transporte de elétrons. A
direção do equilíbrio de uma reação química deve ser examinada de acordo com a
variação de energia livre envolvida na reação. No caso de reações de oxidoredução,
DG depende da diferença de potencial de oxidoredução da seguinte forma:
ΔG0 = -nFΔE0
onde DE é proporcional à afinidade por elétrons do substrato oxidado. Por exemplo,
considere as duas meias reações de oxidoredução:
(1)
(2)
2H+ +1/2 O2 + 2e- à H2O
NAD+ + H+ + 2e- à NADH
E0 = +0.82 V
E0 = -0.32V
Claramente, como o oxigênio possui maior potencial de redução ele irá oxidar o
NADH. Dessa forma, o NADH será doador e o O2 acceptor de elétrons e a reação
global deve ser escrita como:
No caso da transferência de elétrons de NADH e FADH2 para o O2, é possível
calcular a variação de energia livre através da diferença de potencial de redução
envolvida. Por exemplo, os potenciais de redução de oxigênio e NAD+ são dados por:
(1)
(2)
2H+ +1/2 O2 + 2e- à H2O
NADH à NAD+ + H+ + 2e-
E0 = +0.82 V
E0 = +0.32V
A soma das duas meias reações resulta:
(3)
H+ +1/2 O2 + NADH à NAD+ + H2O
ΔE0 = +0.82 +0.32 = +1.12V
Considerando-se que a variação de energia livre envolvida em uma reação de
oxido-redução é dada por:
∆𝐺 ! = −𝑛𝐹∆𝐸
onde n é o número de elétrons envolvido, F é a constante de Faraday e ΔE é a
diferença de potencial de redução, a variação de energia livre envolvida na reação de
redução de oxigênio por NADH é:
∆𝐺 ! = −𝑛𝐹∆𝐸 = −2 96.48 𝑘𝐽𝑚𝑜𝑙 !! 𝑉 !! 1.12𝑉 = −220.1 𝑘𝐽𝑚𝑜𝑙 !!
Considerando-se que para sintetizar uma molécula de ATP a partir de ADP e
Pi (PO4-3) são necessários 30.5 kJ/mol, se acoplada à síntese de ATP, a oxidação do
NADH fornecerá energia suficiente para a síntese de vários mols de ATP. Este
acoplamento é realizado pelos componentes da chamada cadeia de transporte de
elétrons, centros redox com afinidades crescentes por elétrons, ao invés de transferir
os elétrons diretamente para o O2 (Figura 34).
A quantidade de energia livre liberada a partir da transferência de elétrons
para o O2 é inicialmente utilizada para criar um gradiente de H+ através da membrana
44 interna da mitocôndria (Figura 34). A fim de calcular qual é a variação de energia
livre associada ao transporte de prótons para fora da matriz podemos fazer:
[!]!"#$
∆𝐺 = 𝑅𝑇𝑙𝑛 [!]
!"#$%&
+ 𝑛𝐹∆𝐸
(4)
Em condições típicas, o pH no espaço inter-membrana é 1.4 unidades menor do que
[!]!"#$
na matriz mitocondrial, portanto 𝑙𝑜𝑔 [!]
!"#$%&
= 1.4 , ΔE = 0.14V e Z=+1 (H+).
Considerando-se a temperatura como 310K, e substituindo-se os valores na equação
acima encontramos que ΔG para mover um H+ para fora da matriz é 21.8 kJ.mol-1.
Portanto a redução de O2 por uma molécula de NADH produzirá energia suficiente
para o transporte de vários íons H+ para fora da matriz mitocontrial. O retorno de H+
para dentro da matriz e a síntese de ATP são acoplados pela enzima ATPsintase
(Figura 35). A oxidação de um NADH resulta em 2.5 ATPs, enquanto que a oxidação
de um FADH2 resulta em 1.5 ATP.
F0
Axle
Stator
F1
doi:10.2210/rcsb_pdb/mom_2005_12
Figura 35. Estrutura da ATP-sintase.
Considerando-se que estes valores estão corretos podemos estimar a
quantidade de moléculas de ATP geradas a partir da oxidação completa de uma
molécula de glicose a CO2 e H2O: [8NADH/Glicose (Krebs) + 2NADH/Glicose
(glicólises)]x2.5 ATP/NADH + [2FADH2/Glicose]x1.5ATP/FADH2 + 2ATP/Glicose
(Krebs) + 2ATP/Glicose (glicólise) = 32 ATP/Glicose.
45 Exercícios sobre fosforilação oxidativa
1. Citar os compostos que fazem parte da cadeia de transporte de elétrons
2. Esquematizar a sequência dos complexos da cadeia, indicando os
transportadores de elétrons, os transportadores de prótons e elétrons, bem
como o composto doador dos elétrons
3. Citar a localização celular da cadeia de transporte de elétrons
4. Citar 3 inibidores da cadeia de transporte de elétrons, indicando os complexos
sobre os quais atuam
5. Os compostos seguintes, localizados na membrana interna da mitocôndria,
fazem parte de uma cadeia que transfere elétrons do NADH (E0’ = -0,315 V)
para o oxigênio molecular, O2 (E0’ = +0,815 V):
Coenzima Q (E0’ = +0,04 V)
Citocromo a-a3 (E0’ = +0,55 V)
Citocromo b (E0’ = +0,06 V)
Citocromo c (E0’ = +0,235 V)
Citocromo c1 (E0’ = +0,215 V)
Os potenciais de óxido-redução destes compostos dão alguma indicação sobre
a sequência que eles se encontram na cadeia de transporte de elétrons?
03. Uma suspensão de mitocôndrias foi incubada em diferentes condições,
medindo-se a formação de NAD+,, o consumo de oxigênio, a produção de
ATP, e a diferença de pH entre o interior e o exterior da organela. Os
resultados encontrados estão apresentados na tabela abaixo:
Tubo
Condição
NAD+
1
2
Com NADH
Com NADH e
droga A
Sem NADH
Com NADH e
pH externo
mantido
constante
Sem NADH
mas mantendo
ΔpH
+++
+++
3
4
5
+++
Consumo
de O2
+++
+++
Produção
de ATP
+++
ΔpH
1.4
0
0
0
+++
+++
1.4
a. Verificar se é possível:
a1. oxidação de NADH sem síntese de ATP.
a2. oxidação de NADH sem consumo de oxigênio.
a3. consumo de oxigênio sem síntese de ATP.
a4. consumo de oxigênio sem formação de gradiente de H+.
a5. consumo de oxigênio sem oxidação de NADH.
46