UNIVERSIDADE DO ESTADO DO RIO DE JANEIRO
FACULDADE DE CIÊNCIAS MÉDICAS
DISCIPLINA DE FARMACOLOGIA I
TERAPIA ANTI-VIRAL DO HIV
MAIO/2012
GRUPO 6
Cícera Machado
João Batista
José Alves
Lílian Mohr
Lucas Nunes
Luiza Rivero Alcoforado
Luma Burguinhão
Marcella Bellini
Marcela Montojos
Maria Clara Zampirolli
Maria Eduarda La Roque
Marianna Cosentino
2
Sumário
Introdução..………………………………………………………………………….. 4
AIDS................................................................................................................ 4
Vírus HIV......................................................................................................... 5
Anti-retrovirais................................................................................................. 9
Inibidores nucleosídeos da transcriptase reversa................................ 9
Inibidores não-nucleosídeos da transcriptase reversa....................... 16
Inibidores da Protease........................................................................ 21
Inibidores da Fusão............................................................................ 25
Em fase de investigação.................................................................... 27
Tratamento............................................................................................. 27
Conclusão..............................................................................................29
Bibliografia.............................................................................................30
3
Introdução
Nos últimos anos, o grande avanço verificado no campo da farmacologia dos
anti-retrovirais (talvez a área de maior crescimento e desenvolvimento) mudou
a vida das pessoas vivendo com AIDS, possibilitando um aumento da
sobrevida e melhora da qualidade de vida. Apesar deste avanço, a facilidade
com que os vírus adquirem resistência, torna imprescindível o desenvolvimento
de novos medicamentos. Neste sentido, a Farmacêutica tem um papel
fundamental seja no estudo direto de novos fármacos ou na atenção
farmacêutica trabalhando a questão da adesão ao medicamento. A adesão ao
medicamento é essencial para a manutenção da baixa carga viral e menor
desenvolvimento de resistência. Através da genotipagem, hoje adotada apenas
para pacientes com falha terapêutica, é possível uma terapia de resgate mais
personalizada de acordo com o perfil de mutações apresentadas. A ampliação
do arsenal terapêutico aliado a politerapia baseada na genotipagem para todos
os pacientes com indicação de uso poderá quem sabe no futuro garantir o
controle farmacológico desta infecção por períodos maiores, auxiliando no
controle da epidemia.
AIDS
Aids, ou Síndrome da Imunodeficiência Adquirida, é uma doença infectocontagiosa causada pelo vírus HIV (Human Immunodeficiency Virus), que leva
à perda progressiva da imunidade. A doença – na verdade uma síndrome –
caracteriza-se por um conjunto de sinais e sintomas advindos da queda da taxa
dos linfócitos CD4, células muito importantes na defesa imunológica do
organismo. Quanto mais a moléstia progride, mais compromete o sistema
imunológico e, consequentemente, a capacidade de o portador defender-se de
infecções.
Na maioria dos casos, os sintomas iniciais podem ser tão leves que são
atribuídos a um mal estar passageiro. Quando se manifestam mais intensidade,
são os mesmos de várias outras viroses, mas podem variar de acordo com a
resposta imunológica de que cada indivíduo.
4
Os mais comuns são febre constante, manchas na pele (sarcoma de Kaposi),
calafrios, ínguas, dores de cabeça, de garganta e dores musculares, que
surgem de 2 a 4 semanas após a pessoa contrair o vírus.
Nas fases mais avançadas, é comum o aparecimento de doenças oportunistas
como tuberculose, pneumonia, meningite, toxoplasmose, candidíase, etc.
Existe um exame de sangue específico para o diagnóstico da AIDS, chamado
teste Elisa. Em média, ele começa a registrar que a pessoa está infectada 20
dias após o contato de risco. Se depois de três meses o resultado for negativo,
não há mais necessidade de repetir o exame, porque não houve infecção pelo
HIV.
O uso da camisinha nas relações sexuais é a forma mais eficaz de prevenção
da AIDS. Também é imprescindível usar somente seringas descartáveis.
Gestantes devem obrigatoriamente fazer o teste de HIV durante o pré-natal. Se
estiverem infectadas, é fundamental iniciar logo o tratamento a fim de evitar
que o vírus seja transmitido para o feto. Hoje, é perfeitamente possível para
uma mulher infectada engravidar e dar à luz um bebê livre do vírus.
Vírus HIV
Estrutura genômica
O HIV tem muitos genes que codificam proteínas estruturais.
Genes retrovírus gerais:
gag : proteínas derivadas do gag sintetizam o capsídeo viral em forma
de cone (p24, i.e. proteína de 24 Quilo[[dáltons, CA) a proteína do
núcleocapsídeo (p17, NC) e um proteína da matriz (MA).
pol. :O gene pol codifica as proteínas enzimaticamente ativas do vírus.
A mais importante é a chamada transcriptase reversa (RT) que realiza a
única transcrição reversa do RNA viral em uma cadeia dupla de DNA. O
último é integrado ao genoma do hospedeiro, ou seja, em um
5
cromossomo de uma célula infectada de uma pessoa HIV-positiva pela
integrase (IN) pol-codificadora. Além disso, a pol codifica uma protease
viral específica (PR). Essa enzima cliva o gag e as proteínas derivadas
de gag e pol em pedaços funcionais.
env : env, abreviação para "envelope". As proteínas derivadas de env
são
uma
membrana
de
superfície
(gp120)
e
uma
proteína
transmembrana (gp41). Elas estão localizadas na parte externa da
partícula viral, formando um envelope viral o qual permite que o vírus se
anexe e incorpore às células-alvo para então iniciar o ciclo infeccioso. A
gp possui uma estrutura semelhante a uma maçaneta.
Genes específicos do HIV
tat. Um porção da estrutura do RNA do HIV é uma estrutura como um
grampo de cabelo que inicialmente impede que uma transcrição
completa ocorra. Parte do RNA é transcrita (ie. antes da parte do
grampo) e codifica a proteína tat. A tat liga-se à CdK9/CycT e a fosforila,
ajudando a alterar sua forma e a eliminar o efeito da estrutura de
grampo do RNA. Isso por si só aumenta a taxa de transcrição,
fornecendo um ciclo de retroalimentação positiva. Isto permite que o HIV
tenha uma resposta explosiva, uma vez que uma grande quantidade de
tat é produzida.
rev. A rev permite que fragmentos do mRNA do HIV que contém uma
unidade de resposta a rev (RRE) sejam exportados do núcleo ao
citoplasma. Na ausência da rev, a maquinaria de splicing do RNA no
núcleo rapidamente cliva o RNA. Na presença da rev, o RNA é
exportado do núcleo antes de ser clivado, num mecanismo de
retroalimentação positiva.
6
Ciclo Viral
O Vírus da Imunodeficiência Humana (VIH) pertence a um grupo de vírus
conhecidos como retrovírus. Estes vírus armazenam o seu material genético
como ARN (ácido ribonucleico), uma cadeia única de código genético. A maior
parte dos organismos tem ADN (Ácido desoxirribonucleico), uma cadeia dupla
de código genético. Quando chega à corrente sanguínea, o VIH ataca alguns
tipos
de
células,
sobretudo,
os
linfócitos
T.
O ciclo do vírus HIV inicia-se quando o próprio liga-se ao receptor CD4 na
superfície celular do linfócito T CD4, por meio da proteína de superfície gp 120,
presente no envelope viral. A proteína gp 120 sofre uma mudança
conformacional após a ligação e se liga a um co-receptor quimiocina (CCR5 ou
CXCR4) especifico na célula. Essa ligação libera a proteína transmembrana
gp41, que expõe o peptídeo de fusão para a membrana citoplasmática da
célula de defesa e produz então a fusão do envelope viral com a membrana
citoplasmática e a liberação do centro viral no citoplasma.
O genoma RNA é liberado e sofre ação da enzima transcriptase reversa,
tonando-se um cDNA de dupla fita. Este migra ao núcleo em associação com a
enzima integrase viral, integrando-se no genoma celular e tornando-se um
provirus. Novos RNA e mRNA genomicos virais são produzidos a partir do
provirus e exportados ao citoplasma, por meio da ação do gene rev, que
permite que fragmentos do mRNA do HIV que contém uma unidade de
7
resposta a rev (RRE) sejam exportados do núcleo ao citoplasma. O mRNA viral
é traduzido em proteínas virais. As proteínas do envelope viajam a membrana
plasmática, enquanto outras proteínas virais e o RNA genomicos são montados
em nucleocapsideos. Novas partículas virais brotam da célula, adquirindo seu
envelope lipídico e suas glicoproteinas do envelope no processo. Sendo assim,
saem da célula levando consigo todas as proteínas víricas e o ARN necessário
para formar partículas de vírus (os viriões).
Os novos viriões ainda estão imaturos quando entram na corrente sanguínea e,
nesta fase, são incapazes de infectar outras células, tendo de passar por um
processo de amadurecimento para se tornarem infecciosos.
As células CD4 não sobrevivem, habitualmente, à invasão do VIH.
Desintegram-se devido ao elevado número de vírus germinados ou porque o
sistema imunológico do corpo reconhece as proteínas de envelope vírico na
membrana da célula e destrói as células danificadas. Como as células CD4 são
elas próprias uma parte essencial do sistema imunológico, a sua destruição
pode provocar imunodeficiência profunda.
Depois do novo vírus deixar a célula, uma outra enzima vírica, a protease viral,
corta a molécula que contém as proteínas centrais do VIH. As proteínas
individuais libertadas são remontadas para formar um vírus estruturado e
maduro.
Este
vírus
pode
agora
infectar
outras
células.
O HIV destrói os linfócitos CD4 gradativamente (em média a contagem declina
80-100 células/ml/ano). A contagem relaciona-se inversamente com a
gravidade da doença. Para fins de tratamento com as drogas antirretrovirais
consideram-se os seguintes parâmetros:
Abaixo de 200 células/ml: Muito vulnerável, tratar imediatamente;
Entre 200 e 350 células/ml: Vulnerável, deve ser iniciado o tratamento
para evitar riscos;
8
Entre 350 e 500 células/ml: Pouco vulnerável, pode começar a critério
médico;
Acima de 500: Saudável, não precisa começar o tratamento.
Porém todos os pacientes com doença oportunista relacionada ao HIV devem
ser tratados mesmo com CD4 alto.
Anti-Retrovirais
Existem cinco tipos de drogas que influenciam no ciclo viral. Elas são
conhecidas como drogas antirretrovirais:
A) Inibidores nucleosídeos da transcriptase reversa (INTR)
Atuam pela inibição competitiva da transcriptase reversa do HIV-1 e também
podem ser incorporados na cadeia de DNA viral em crescimento, causando sua
interrupção.
9
Os inibidores exigem ativação intracitoplasmática em conseqüência da
fosforilação por enzimas celulares
Maioria exibe atividade contra HIV-1 e HIV-2
1) Zidovudina
A zidovudina (azidotimidina), AZT) é análogo da desoxitimidina.
É bem absorvida pelo intestino e distribui-se pela maioria dos tecidos e líquidos
corporais.
Sua ½ vida no soro é de 1 hora, já a ½ vida intracelular do composto fosforilado
é de 3,3 horas. No LCR os níveis do fármaco correspondem a 60-65% dos
níveis do soro.
Sofre excreção renal após glicuronidação no fígado e sua depuração encontrase diminuída em cerca de 50% dos pacientes urêmicos e a toxicidade do
fármaco pode aumentar em pacientes com insuficiência hepática avançada.
Esse fármaco diminui a taxa de progressão da doença clínica e prolonga a
sobrevida de pacientes infectados por HIV e sua eficácia também foi
demonstrada no tratamento da demência e trombocitopenia associada ao HIV.
Um esquema de zidovudina oral iniciado entre 14 e 34 semanas de gestação
(100mg, 5x ao dia), com zidovudina intravenosa durante o trabalho de parto
(2mg/kg durante 1 hora e, a seguir, 1mg/kg/h por infusão contínua) e xarope de
zidovudina administrado ao neonato desde o nascimento até 6 semanas de
idade (2mg/kg a cada 6h), diminui a taxa de transmissão vertical do HIV em
23%.
Em camundongos a zidovudina induz desenvolvimento de neoplasias epiteliais
vaginais , mas esse efeito não tem sido relatado em seres humanos.
A exemplo de outros INTR, a resistência pode limitar a eficiência clínica da
zidovudina, particularmente quando usado em monoterapia. A ocorrência de
uma ou duas mutações pode conferir resistência parcial, enquanto se observa
10
geralmente uma resistência de alto nível, em cepas com três ou mais
mutações.
Acredita-se que a terapia com outros agentes anti-retrovirais constitua-se a
melhor abordagem, pela maior potência e desenvolvimento tardio de
resistência.
O aparecimento de certas mutações que conferem menor susceptibilidade a
um fármaco parece aumentar a sensibilidade em cepas previamente
resistentes a zidovudina.
A interrupção da exposição a zidovudina pode permitir a reversão de isolados
de HIV resistente a zidovudina no fenótipo susceptível de tipo selvagem.
O efeito adverso mais comum é a mielosupressão, resultando em neutropenia
ou anemia. Podem ocorrer intolerância gastro-intestinal, cefaléias e insônia,
que tendem a regredir com a terapia. Efeitos menos freqüentes incluem
trombocitopenia, hiperpigmentação das unhas e miopatia e a administração de
doses muito altas pode causar ansiedade, confusão e tremor. Foram relatados
casos raros de acidose lática e hepatomegalia grave com esteatose.
O tratamento com zidovudina deve ser suspenso na presença de níveis
rapidamente crescentes de aminotransferase, hepatomegalia progressiva ou
acidose metabólica ou lática de causa desconhecida.
Pode ocorrer aumento de seus níveis séricos com a administração
concomitante de probenicida, fenitoína, metadona, fluconazol, atovaquona,
ácido valpróico e lamivudina, através da inibição do metabolismo de primeira
passagem ou da diminuição da depuração. A zidovudina pode diminuir os
níveis de fenitoína, exigindo monitorização. A toxicidade hematológica pode
estar
aumentada
durante
a
co-administração
de
outros
agentes
mielodepressores, como ganciclovir e agentes citotóxicos.
2) Didanosina
A didanosina (ddI) é um análogo da desoxiadenosina.
11
Em pH ácido, a hidrólise da ligação glicosídica entre o açúcar e a base da ddI
inativa o composto. A formulação original , que consistia em pó tamponado, foi
substituída por comprimidos tamponados mastigáveis e dispersíveis com maior
biodisponibilidade
(30-40%).
Por
esses
comprimidos
conterem
tanto
fenilalanina quanto sódio, deve-se ter cautela em pacientes com fenilcetonúria
ou com dietas restritas em sódio.
A ASC da ddI encontra-se reduzida em 55% se o fármaco for ingerido dentro
de 2 horas após refeição. As concentrações séricas máximas são em média
1ug/ml após uma dose de 300mg. As concentrações no LCR são de
aproximadamente 20% das concentrações séricas.
A ½ vida de eliminação 0,6-1,5 hora, porém a ½ vida intracelular do composto
ativado é de 12-24 horas.
O fármaco é eliminado por filtração glomerular e secreção tubular. Por
conseguinte, é necessária uma redução da dose para a depuração diminuída
da creatinina, após hemodiálise ou diálise peritoneal ambulatorial contínua e
em caso de baixo peso corporal
Deve ser tomada com estomago vazio e devido a formulação tamponada, deve
ser administrada pelo menos 2 horas após administração de fármacos que
necessitam de acidez para sua absorção ótima (por exemplo: cetoconazol,
itraconazol,dapsona). A fluoroquinolonas e as tetraciclinas devem ser
administradas pelo menos 2 horas antes ou depois da ddI, a fim de evitar
concentrações plasmáticas diminuídas do antibiótico, devido à quelação. A coadministração com ganciclovir resulta em aumento da ASC da ddI e diminuição
da ASC do ganciclovir, enquanto co-administração com metadona resulta em
diminuição dos níveis séricos de ddI.
A resistência à ddI (devido a mutação), pode restaurar, em parte, a
susceptibilidade a zidovudina, mas pode conferir resistência cruzada ao
abacavir, zalcitabina e lamivudina.
Sua principal toxicidade clínica consiste no desenvolvimento de pancreatite
dose-dependente. Outros efeitos adversos relatados incluem neuropatia distal
12
periférica dolorosa, diarréia, hepatite, ulceração do esôfago, miocardiopatia e
toxicidade do sistema nervoso central (cefaléia, irritabilidade, insônia). A
hiperuricemia assintomática pode precipitar crises de gota em indivíduos
susceptíveis. A observação de alterações da retina e neurite óptica em
paciente em uso de ddI indica utilidade de exames periódicos da retina. À
semelhança de outros INTR, existem raros relatos de acidose lática ou
hepatomegalia grave com esteatose.
3) Lamivudina
A lamivudina (3TC) é um análogo da citocina com atividade in vitro contra o
HIV-1, que é sinérgico com uma variedade de análogos de nucleosídeos antiretrovirais – incluindo zidovudina e estavudina – contra cepas de HIV-1 tanto
sensíveis quanto resistentes à zidovudina.
Sua biodisponibilidade oral ultrapassa 80% e não depende da presença de
alimentos. Seus níveis séricos máximos são de 1,5 +- 0,5ug/ml.
A ½ vida de eliminação média é de 2,5 horas, enquando a intracelular é de
10,5-15,5 horas em linhagens infectadas por HIV-1 e de 17-19 horas em
linhagens infectadas por HBV.
A maior parte é eliminada de modo inalterado na urina. Deve-se reduzir a dose
em pacientes com insuficiência renal ou baixo peso corporal.
A terapia com lamivudina leva rapidamente à seleção de mutantes HIV M184V
resistentes, conferindo alto nível de resistência à lamivudina e uma redução na
sensibilidade ao abacavir, didanosina e zalcitabina.
A lamivudina , como outros agentes anti-retrovirais é utilizada de preferência
em terapias de combinação quem suprimem por completo a replicação viral,
reduzindo a geração de mutantes resistentes. A mutação M184V pode
restaurar a sensibilidade fenotípica à zidovudina, indicando que esses dois
fármacos
administrados
em
esquemas
de
combinação
podem
ser
particularmente benéficos. Todavia foram isoladas cepas de HIV-1 resistentes
tanto à lamivudina quando a zidovudina.
13
Recentemente foi aprovada para o tratamento da infecção crônica da hepatite
B. A terapia crônica com lamivudina em pacientes com hepatite pode ser
limitada pelo aparecimento de cepas de HBV resistentes a lamivudina
Efeitos colaterais potenciais consistem em cefaléia, insônia, fadiga e
desconforto gastrointestinal (condições tipicamente LEVES).
A ASC aumenta quando o fármaco é administrado concomitantemente com
tremetropim-sulfametoxazol. Os níveis máximos de zidovudina aumentam
quando o fármaco é administrado com lamivudina, embora não se acredite que
esse efeito tenha significado clínico.
4) Zalcitabina
A zalcitabina (ddC) é um análogo da citosina que possui atividade anti-HIV-1
sinérgica com uma variedade de agentes anti-retrovirais contra cepas de HIV-1
tanto sensíveis quanto resistentes à zidovudina. A resistência parece ser rara,
particularmente nos esquemas de combinação.
Sua atividade como monoterapia é menor do que quando o fármaco é
combinado com a zidovudina. Por isso costuma ser prescrita em esquemas de
combinação.
Possui ½ vida intracelular relativamente longa de 10 horas (apesar de sua ½
vida de eliminação de 2 horas e biodisponibilidade oral de mais de 80%). Seus
níveis plasmáticos diminuem em25-39% quando o fármaco é administrado com
alimentos
ou
com
antiácidos.
Suas
concentrações
no
LCR
são
aproximadamente 20% daquelas obtidas no plasma.
É necessário reduzir-se a dose em pacientes com insuficiência renal.
Efeitos adversos: Seu uso associa-se a uma neuropatia periférica dosedependente, que pode limitar o tratamento em 10-20% dos pacientes e que
parece ser lentamente reversível se o tratamento for interrompido de
imediatamente (a redução da depuração renal causada pela anfotericina B,
pelo foscarnet e aminoglicosídios pode aumentar o risco de neuropatia).
Ulcerações orais e esofágicas constituem outra toxicidade relatada, assim
14
como pancreatite (que ocorre menos freqüentemente do que com uso de
didanosina). Pode-se verificar a presença de cefaléia, náusea, exantema e
artralgias, que tendem a ser leves e regredir durante a terapia. Raramente
ocorreram miocardiopatia e acidose lática com hepatomegalia grave e
esteatose.
Quando administrada em combinação com probenicida ou cimetidina há
aumento da ASC e quando administrada concomitantemente com antiácidos ou
metoclopramida há diminuição da biodisponibilidade.
5) Estavudina
A estavudina é um análogo da timidina e possui alta biodisponibilidade oral
(>86%) que não depende da presença de alimentos.
Sua ½ vida plasmática é de 1,22 hora e intracelular é de 3,5 horas, e as
concentrações no LCR correspondem a 55% dos níveis plasmáticos.
A excreção do fármaco ocorre por secreção tubular ativa e filtração glomerular.
Sua dose deve ser reduzida em pacientes com insuficiência renal, em
pacientes submetidos a hemodiálise e em indivíduos de baixo peso corporal.
Resistência clinicamente significativa tem sido rara.
A principal toxicidade que limita sua dose é a neuropatia sensorial periférica
dose-dependente (a freqüência da neuropatia pode aumentar quando se
administra a estavudina com outros fármaco que induzem neuropatia como
ddC e ddI). Há regressão dos sintomas com sua interrupção. Outros efeitos
adversos incluem pancreatite, artralgias e elevação das aminotransferases
séricas. Raramente foi observada ocorrência de acidose lática e de
hepatomegalia grave com esteatose. A zidovudina pode reduzir sua
fosforilação, por isso não se deve administrar os dois juntos.
6) Abacavir
15
Abacavir é um análogo da guanosina que parece ser significamente mais eficaz
que os outros dessa classe. É bem absorvido após administração oral (>83%)
e não é afetado pela presença de alimento.
Sua ½ vida de eliminação é de 1,5 hora e seus níveis no LCR são
aproximadamente 1/3 dos níveis plasmáticos .
O fármaco é metabolizado pela álcool desidrogenase e glicuronosil transferase
em metabólitos inativos que são eliminados primariamente na urina.
Resistência de alto nível ao abacavir parece exigir ao menos duas ou três
mutações concomitantes, e por esse motivo pode se desenvolver lentamente
Em 2-5% dos pacientes foram relatadas reações de hipersensibilidade
(algumas vezes fatais). Os sintomas, que geralmente aparecem nas 6
primeiras semanas de terapia, afetam múltiplos sistemas de órgãos e incluem:
febre, mal-estar e queixas gastrointestinais. Pode haver ou não erupção
cutânea. Embora a síndrome tenda a regredir rapidamente com sua
suspensão, a reexposição ao abacavir resulta no reaparecimento dos sintomas
dentro de poucas horas e pode ser fatal. Outros efeitos adversos podem incluir
exantema (na ausência de hipersensibilidade), náuseas, vômitos, diarréia,
cefaléia e fadiga. Outros efeitos que parecem ser raros incluem: pancreatite,
hiperglicemia, hipertrigliceridemia e acidose lática.
Co-administração de álcool e abacavir pode resultar em aumento da ASC do
abacavir.
B) Inibidores Não-Nucleosídeos da Transcriptase Reversa (INNTR)
Os INNTR ligam-se diretamente a um sítio na transcriptase reversa viral
localizado próximo ao sítio de ligação dos INTR,porém distintos deles. A
contrário do grupo dos INTR,os INNTR não competem com trifosfatos de
nucleosídios
nem
necessitam
de
fosforilação
para
serem
ativos,e,normalmente,exibem atividade específica contra HIV-1.
A ligação do INNTR ao sítio ativo da enzima resulta em bloqueio das atividades
da DNA polimerase RNA-e DNA-dependente. Em geral, a resistência a um
16
INNTR está associada à mutação K103N,bem como à mutação Y181C/Imenos
crítica. O rápido aparecimento de resistência impede o uso de qualquer INNTR
como
monoterapia
para
tratamento
das
infecções por HIV. Não há resitência cruzada entre os INNTR e os INTR ou os
inibidores da protease.
1)Neverapina
Apresenta excelente biodisponibilidade oral(> 90%) e não depende da
presença de alimento. O fármaco é altamente lipofílico,e cerca de 60% ligamse às proteínas. Atinge níveis no líquido cefalorraquidiano ,que correspondem a
45% dos níveis alcançados no plasma.Esse fármaco é extensamente
metabolizado pela isoforma P450 CYP3A a metabólitos hidroxilados e,a seguir,
excretada primariamente na urina.
Indicação: A nevirapina é tipicamente utilizada como componente de um
esquema de combinação anti-retroviral. Além disso, recentemente, foi
constatado a eficácia de uma dose única de nevirapina (200mg) na prevenção
da transmissão do HIV da mãe para o recém-nascido ,quando administrada a
mulheres
no
início
do
trabalho
de
parto,seguida
de
uma
dose oral de 2mg/Kg administrada ao neonato dentro de 3 dias após o parto.
Efeitos adversos : Ocorreram erupções cutâneas graves e potencialmente
fatais durante a terapia com nevirapina,incluindo síndrome de Stevens-Johnson
e necrólise epidérmica tóxica. Vale ressaltar que a terapia com nevirapina deve
ser imediatamente interrompida em pacientes com exantema intenso,bem
como naqueles em que o exantema é acompanhado de sintomas
constitucionais. A ocorrência de exantema é verificada em cerca de 17% dos
pacientes,tipicamente nas 4-8 semanas de terapia,limitando a dose do fármaco
em 7% dos pacientes.Ao iniciar o tratamento, recomenda-se o escalonamento
da dose durante 14 diaspara diminuir a frequência do exantema.
Já foram constatados alguns casos de hepatite fulminante ,com ou sem
exantema.Recomenda-se a monitorização das provas de função hepática,com
interrupção
do
tratamento
com
nevirapina
em
casos
de
elevações
17
significativas.
Outros efeitos adversos associados à terapia com nevirapina,que são relatados
com frequência, consistem em febre,náusea ,cefaléia e sonolência.
Interações medicamentosas: A nevirapina induz o metabolismo de fármacos
pela CYP3A,e a co-administração resulta em diminuição dos níveis de
indinavir,ritonavir e saquinavir,bem como dos anticoncepcionais orais.
A
nevirapina
não deve
ser administrada
concomitantemente
com o
cetoconazol, devido ao fato de isso resultar num aumento da nevirapina e na
redução dos níveis de cetoconazol.
Ao ser co-administrada com inibidores do metabolismo da CPY3A,como a
cimetidina e os macrolídios, a nevirapina tem seus níveis aumentados, todavia,
ao ser administrada concomitantemente com indutores da CYP3A,como a
rifabutina e a rifampicina, seus níveis são reduzidos.
Portanto, a co-administração desses agentes deve ser feita com muita
cautela,e apenas se não houver outras alternativas satisfatórias.
2) Delarvidina
Possui biodisponibilidade oral de cerca de 85%,que é reduzida na presença de
antiácidos.O fármaco liga-se extensamente (cerca de 98%) às proteínas
plasmáticas.
Os
correspondem,em
níveis
média,
alcançados
a
apenas
no
líquido
0,4%
das
cefalorraquidiano
concentrações
plasmáticas,representando cerca de 20% da fração não-ligada às proteínas
plasmáticas.
A delavirdina é extensamente metabolizada a metabólitos inativos pelas
enzimas P450 CYP3A e CYP2D6. Todavia, o fármaco inibe o seu metabolismo
,ao inibir a CYP3A.Deve-se ter cautela ao administrar delavirdina a pacientes
com insuficiência hepática de grau significativo,visto que a experiência clínica
18
nessa situação é limitada.
Efeitos adversos: Ocorre erupção cutânea em cerca de 18% dos pacientes,que
aparece, tipicamente , no primeiro mês de terapia e que não impede nova
exposição.
Outros efeitos adversos observados em mais de 2% dos pacientes em uso de
delavirdina incluem cefaléia,fadiga,náusea,diarréia e aumento dos níveis
séricos de aminotransferases.
Interações medicamentosas: Observa-se uma redução das concentrações
plasmáticas
de
delavirdina
na
presença
de
antiácidos,
didanosina,
fenitoína,fenobarbital,carbamazepina,rifabutina,rifampicina,nelfinavir
e
saquinavir, enquanto que ocorre aumento das concentrações durante a
co-administração com claritromicina,fluoxetina e cetoconazol.
A
delavirdina,todavia,determina
uma
redução
dos
níveis
de
claritromicina,indinavir,nelfinavir,saquinavir,rifabutina,dapsona,alprazolam,mida
zolam,triazolam,nifedipina,cisaprida,quinidina,varfarina
e derivados do esporão do centeio.
A ASC da didanosina exibe uma diminuição de cerca de 20% na presença
de delavirdina.
Não
se
recomenda
a
co-administração
de
delavirdina
com
fenitoína,fenobarbital,carbamazepina,rifabutina,rifampicina e antagonistas dos
receptores H2.
A co-administração com antiácidos ou didanosina deve ter um intervalo
de pelo menos uma (1) hora.
Recomenda-se uma redução da dose de indinavir para 600mg,três vezes ao
dia, caso esse fármaco seja administrado concomitantemente com a
delavirdina.
Aconselha-se
a
monitorização
das
provas
de
função
19
hepática caso a delavirdina seja administrada com saquinavir.
Contra-indicação:
delavirdina,
Deve-se
uma
vez
evitar
que
foi
a
gravidez
constatado
durante
que
a
o
uso
delavirdina
de
é
teratogênica em ratos ,causando defeitos do septo ventricular e outras
malformações que não diferem das obtidas em seres humanos em exposição
ao fármaco.
3) Efavirenz
O efavirenz (antigamente DMP 266) pode ser administrado uma vez ao
dia, em virtude da sua meia-vida prolongada( 40-55 horas). É bem
absorvido
após
a
administração
oral(45%),e
sua
biodisponibilidade
aumenta em cerca de 65% após uma refeição rica em gordura.As
concentrações plasmáticas máximas do efavirenz são observadas dentro
de 3-5 horas após a administração de doses diárias,e a concentração
plasmática em estado de equilíbrio dinâmico é alcançada em 6-10 dias.
O efavirenz é metabolizado principalmente pela CYP3A4 e CYP2B6 a
metabólitos hidroxilados inativos,sendo o restante eliminado na forma
inalterada nas fezes. O fármaco liga-se intensamente à albumina (>
99%). Os níveis no líquido cefalorraquidiano variam de 0,3 a 1,2%
daqueles alcançados no plasma, e são aproximadamente três vezes
maiores
do
que
a
fração
livre
de
efavirenz
no
plasma.
Efeitos adversos: Embora o fármaco seja geralmente bem tolerado, os
principais efeitos adversos afetam o sistema nervoso central (tontura,
sonolência, insônia, cefaléia, confusão, amnésia, agitação, delírios, depressão,
pesadelos, euforia), e tendem a ocorrer nos primeiros dias de terapia,
regredindo durante o tratamento. A administração do fármaco ao deitar pode
ser útil.
Também foi relatada a ocorrência de erupção cutânea,no início da
terapia,em até 28% dos pacientes,e o exantema ,tipicamente, regride
20
com a continuação da medicação.
Outras reações adversas relatadas em mais de 2% dos pacientes incluem
náusea e vômitos, diarréia,cristalúria,elevações das enzimas hepáticas
e aumento de 10-20% dos níveis séricos de colesterol total.
Interações medicamentosas: O efavirenz induz a CYP3A4,com consequente
indução de seu próprio metabolismo,bem como alteração do metabolismo
de muitos outros fármacos.
Devido ao potencial de interações farmacológicas graves, os fármacos
que utilizam a mesma via do efavirenz não devem ser administrados
concomitantemente,e esses fármacos incluem:cisaprida, midazolam, triazolam
e derivados do esporão do centeio. Além disso, deve-se ter cautela com o uso
concomitante de varfarina,fenobarbital, fenitoína, rifampicina e eritromicina.
Embora a experiência clínica seja limitada, recomenda-se muita cautela
em pacientes com comprometimento hepático grave.
Os níveis de ritonavir e nelfinavir podem aumentar na presença de
efavirenz,enquanto
claritromicina
são
que
os
níveis
de
significativamente
amprenavir,saquinavir,indinavir
reduzidos
na
e
co-administração.
As doses de indinavir (até 1.000mg a cada 8horas) e de rifabutina(em
50%) devem ser aumentadas na administração concomitante com efavirenz.
Contra-indicação: A gravidez deve ser evitada em mulheres tratadas com
efavirenz, uma vez que foram observadas altas taxas de anormalidades
fetais em fêmeas grávidas de macacos expostas ao efavirenz em doses
aproximadamente equivalentes à dose de 600mg/dia administrada a seres
humanos.
C) Inibidores da Protease (IPs)
A protease é responsável pela clivagem das poliproteinas, que são moléculas
precursoras, produzindo as proteínas estruturais finais do cerne do vírion
maduro. Ao impedir esse processo, são formadas partículas virais imaturas e
não infecciosas. Devido a ocorrência de alterações genotípicas específicas que
21
conferem resistência fenotípica a esses agentes, é contra-indicada a
monoterapia.
Amprenavir
É rapidamente absorvido pelo trato gatrointestinal e pode ser ingerido com ou
sem alimento, evitando-se refeições ricas em gorduras, uma vez que diminuem
sua absorção. Sua meia-vida plasmática é relativamente longa (6-10,6 h). É
metabolizado no fígado, devendo ser utilizado com cautela no contexto da
insuficiência hepática.
Os efeitos adversos mais comuns são náuseas, diarreia, vômitos, parestesias
periorais, depressão e exantema.
Atazanavir
É um novo IP azapeptídico, cujo perfil permite a administração de uma dose
única ao dia. Sua biodisponibilidade oral é de cerca de 60 a 68% e deve ser
ingerido com alimentos. O fármaco necessita de um meio ácido para sua
absorção e apresenta solubilidade aquosa dependente do pH, recomendandose assim, um intervalo de pelo menos 12 h
para a ingestão de agentes
redutores de ácido. Consegue penetrar tanto no LCR quanto no líquido
seminal. Sua meia-vida plasmática é de 6 a 7 h, aumentando para cerca de 11
h
quando é administrado juntamente com o ritonavir. A principal via de
eliminação é biliar e não deve sem administrado em pacientes com
insuficiência hepática.
Os efeitos adversos mais comuns são náuseas, vômitos, diarreia, dor
abdominal, cefaleia, neuropatia periférica e exantema. Pode ocorrer também
hiperbilirrubinemia indireta com icterícia franca devido a inibição da enzima
UGT1A1.
Ao contrario dos outros fármacos, não está associado a dislipidemias,
redistribuição das gorduras ou síndrome metabólica.
22
Existe grande potencial de interações medicamentosas com o atazanavir,
devido a um aumento da toxicidade pelo bloqueio enzimático da glicuronidação
(UGT1A1).
Fosamprenavir
Pró-fármaco do amprenavir que é rapidamente hidrolisado por enzimas no
epitélio intestinal. Os comprimidos podem ser ingeridos com ou sem alimento.
Substituiu o amprenavir para adultos uma vez que possui uma carga diária
significantemente mais baixa de comprimidos.
Indinavir
Deve ser consumido com o estômago vazio para sua absorção máxima. A
biodisponibilidade oral é de cerca de 65% e o fármaco apresenta grande
penetração no LCR. A meia-vida sérica é de 1,5 h a 2 h. A excreção é
sobretudo fecal. Exige redução da dose no contexto de insuficiência hepática.
Os efeitos adversos consistem em hiperbilirrubinemia indireta e nefrolitíase,
devido a cristalização do fármaco. A nefrolitíase pode ocorrer poucos dias após
o início de administração do fármaco, podendo estar associada a insuficiência
renal. É importante o consumo diário de pelo menos 1,5 L de água para manter
uma hidratação adequada e evitar o desenvolvimento da nefrolitíase. Também
foi relatada trombocitopenia, elevação dos níveis sérios de aminotransferases,
náuseas, diarreia e irritabilidade.
Sua associação com o ritonavir (reforço) permite a administração do fármaco 2
vezes/dia em lugar de 3 vezes/dia e elimina a restrição de alimento associada
ao seu uso. Todavia, existe o potencial de aumento da nefrolitíase com tal
associação.
Lopinavir/Ritonavir
O lopinavir 100/ritonavir 400 é uma associação aprovada, em que as doses
subterapêuticas do ritonavir inibem o metabolismo do lopinavir, resultando
assim em maior exposição ao lopinavir. Mantém uma potente supressão viral
além de proporcionar uma barreira farmacológica contra o desenvolvimento de
23
resistência. O ritonavir nessa associação, portanto, atua mais como
intensificador farmacocinético do que como agente anti-retroviral. A associação
melhora a adesão do paciente ao tratamento devido a menor carga de
comprimidos e em geral, é bem tolerada. A absorção do lopinavir aumenta na
presença de alimento.
Os efeitos adversos mais comuns consistem em diarreia, dor abdominal,
náuseas, vômitos e astenia.
Nelfinavir
Sofre maior absorção quando tomado com alimento, é metabolizado pelo
CYP3A e excretado principalmente nas fezes. A meia-vida plasmática é de 3,5
a 5 h. Os efeitos adversos mais comuns consistem em diarreia e flatulência.
Possui um perfil de segurança e farmacocinética favorável para mulheres
grávidas em comparação com os outros IP.
Ritonavir
É inibidor das proteases do HIV-1 e do HIV-2 com alta biodisponibilidade (cerca
de 75%), que aumenta quando o fármaco é ingerido com alimento. Sua
transformação em metabólito ativo ocorre por intermédio das isoformas CYP3A
e CYP2D6 e sua excreção ocorre sobretudo pelas fezes. Deve-se ter cautela
em pacientes com insuficiência hepática.
Os efeitos adversos mais comuns consistem em distúrbios gastrintestinais,
parestesias (circum-orais e periféricas), elevação dos níveis séricos de
aminotransferases, alteração do paladar e hipertrigliceridemia. Tipicamente
ocorrem náuseas, vômitos e dor abdominal nas primeiras semanas de terapia.
Recomenda-se um escalonamento lento da dose no decorrer de um período de
4 a 5 diaspara diminuir os efeitos colaterais que limitam a dose. O ritonavir é
um potente inibidor da CYP3A4, sendo necessário ter cautela quando o
fármaco é administrado concomitantemente com agentes intensamente
metabolizados pela CYP3A, sendo essa característica explorada para elevar a
concentração mínima e prolongar a meia-vida de agentes IP mais potentes e
menos
tóxicos.
Além
disso,
sua
meia-vida
prolongada
permite
sua
24
administração a intervalos menos frequentes em comparação com outros
agentes IP, aumentando a adesão ao tratamento.
Saquinavir
As formulações de saquinavir devem ser tomadas 2 h após uma refeição
gordurosa para aumentar sua absorção. Possui um grande volume de
distribuição, porém a penetração no LCR é insignificante. A meia-vida de
eliminação é de 12 h. A excreção ocorre primariamente nas fezes. Os efeitos
adversos relatados incluem desconforto gastrintestinal (náuseas, diarreia,
vomito, dor abdominal, dispepsia) e rinite.
A co-administração com o ritonavir foi adotada devido aos níveis mais elevados
e mais eficazes de saquinavir, permitindo, ao mesmo tempo, uma redução da
dose diária e da frequência de administração do saquinovir.
Tipranavir
É outro IP mais recente. Possui biodisponibilidade precária, porém aumenta
quando o fármaco é administrado com uma refeição rica em gordura. É
metabolizado pelo sistema microssômico hepático. Deve ser administrado em
associação com o ritonavir para obter níveis séricos efetivos.
Está contra-indicado para pacientes com insuficiência hepática e que possuem
alergia conhecida à sulfa.
Os efeitos adversos mais comuns consistem em diarreia, náuseas, vômitos, dor
abdominal e exantema, sendo este último mais comum em mulheres. Foi
observada a ocorrência de hepatotoxicidade, incluindo descompensação
hepática potencialmente fatal, que é mais comum em pacientes com hepatite B
ou C crônica. Outros efeitos incluem depressão, elevação dos níveis de
colesterol total, triglicerídios e amilase e contagem diminuída de leucócitos.
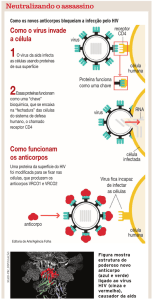
D) Inibidores de fusão
Apesar da evolução das abordagens terapêuticas antiretrovirais, há muitos
25
doentes com intolerância aos medicamentos utilizados ou infectados com vírus
resistentes à ação de tais medicamentos. Além disso, os inibidores da
transcriptase reversa e os inibidores de protease atuam após a instalação da
infecção e a replicação o vírus nos linfócitos. Dessa forma, os inibidores de
fusão constituem uma promissora área de pesquisa na prevenção da entrada
do vírus HIV na célula hospedeira.
O ciclo reprodutivo do HIV depende da sua fusão com o linfócito CD4+, a qual
baseia-se na interação com complexo gp120-gp41 com os receptores CD4 e
CCR5 e/ou CXCR4 do linfócito CD4+. Os inibidores de fusão incluem, portanto:
1) Inibidores da interação da gp120 com o CD4, 2) Inibidores da interação do
gp120 com os co-receptores CXCR4 e CCR5 e 3) Inibidores da interação com
a gp41.
Dentro das classes dos inibidores de fusão há muitos estudos sobre
substâncias passíveis de serem utilizadas como parte da terapia anti-retroviral.
Contudo, os inibidores da interação com a gp41 revelaram-se promissores na
terapêutica. Utilizando-se peptídeos T-20 e T-1249 é possível impedir que a
glicoproteína gp41 entre em contato com a superfície celular. O peptídeo T-20,
também conhecido como Enfuvirtida (evidenciada em 2003), é uma reprodução
sintética exata da cadeia de 36 aminoácidos de sequencia idêntica a da hélice
próxima ao terminal carboxílico e à região transmembranar da gp41. Na fusão
normal do vírus com a célula hospedeira, o domínio HR2 da gp41 liga-se ao
domínio HR1, promovendo a aproximação e a infecção da célula hospedeira.
Ao introduzir uma cópia de T-20 livre no organismo, ela mimetiza o HR2 e se
liga de forma irreversível ao HR1, impedindo a ligação verdadeira entre os
domínios HR2-HR1 da gp41 e a conseqüente fusão entre vírus e linfócito.
Entretanto, pelo alto custo da terapia com o T-20, é mais indicada contra HIV
multi-resistente. O T-1249 representa uma geração mais recente do T-20.
Estudos preliminares indicam que a droga é cerca de 100 vezes mais ativa do
que o T-20, sendo indicado como dose única diária injetável.
26
E) Em fase de investigação
A
FDA
aprovou
recentemente
novos
inibidores
não-nucleosídico
da
transcriptase reversa, o Rilpivirina em 2011 e o Etravirina, em 2088
Em 2007 foi lançado no mercado o Maviroque, o qual é um antagonista dos
receptores CCR5, os quais são essenciais na internalização do HIV-1 com
tropismo para CCR5 nas células T do hospedeiro. Há outros antagonista de
CCR5 em estudos, como o Vicriviroque, que já se encontra em ensaios clínicos
de fase III. Entretanto, a eficácia, toxicidade e efeitos adversos dos
antagonistas de CCR5 de uso crônico ainda não estão estabelecidas.
O Darunavir é um inibidor de protease com atividade anti-HIV-1 recente no
mercado, aprovado pelo FDA em 2006. O Raltegravir é um inibidor de
integrase também recente, aprovado pelo FDA em 2007.
Tratamento
Os protocolos sobre terapia antiretroviral foram primeiramente publicados em
2002, resumidos e simplificados em 2003 e atualizados em 2006. Agora, as
novas recomendações são baseadas nos dados disponíveis mais atuais.
As principais modificações nas recomendações da OMS para a terapia
antiretroviral (TAR) são:
A última orientação da OMS, feita em 2006, sugeria que os portadores do vírus
iniciassem o tratamento assim que seus níveis de células T CD4 (célula de
defesa) fossem iguais ou inferiores a 200 células por milímetro cúbico de
sangue. De acordo com a nova recomendação, o tratamento para adultos e
adolescentes HIV positivos deve começar mais precocemente, quando a
contagem for igual ou inferior a 350 células por milímetro cúbico de sangue. A
nova diretriz serve para todos os pacientes com HIV, inclusive grávidas,
independente da presença ou ausência de sintomas da doença. Esta nova
diretriz tem como base que o início precoce da TAR estimula o sistema imune,
diminui o risco de mortalidade e morbidade associados ao HIV e também reduz
o risco de transmissão de HIV e tuberculose.
27
Efeitos colaterais irreversíveis e de longo prazo, como a neuropatia periférica e
a lipodistrofia (perda de gordura corporal), estão presentes com o uso da
Estavudina (d4T), droga usada amplamente em países em desenvolvimento
por sua disponibilidade e baixo custo. A OMS recomenda que a estavudina
seja, na medida do possível, substituída pela Zidovudina (AZT) ou pelo
Tenofovir (TDF).
Para prevenir a transmissão de mães HIV positivas para seus filhos, a OMS
recomenda que a TAR deva ser iniciada mais cedo na gravidez (com 14
semanas de gestação) e continuar até que a amamentação tenha se
encerrado. É uma mudança em relação a 2006, que dizia para iniciar a TAR no
terceiro trimestre da gestação, começando na 28ª semana da gravidez.
A OMS recomenda que a amamentação siga até 12 meses de vida, desde que
mães HIV positivas ou crianças recebam a TAR durante este período. Duas
alternativas para mulheres HIV positivas que estão amamentando e não estão
recebendo TAR são:
Para mulheres que receberam AZT durante a gravidez, Nevirapina está
recomendada diariamente para a criança, do nascimento até o final da
amamentação.
Para as mulheres que receberam esquema de 3 drogas durante a gravidez, o
regime deve ser mantido até que a amamentação se complete.
Existem dois desafios principais. O primeiro é disponibilizar os medicamentos
para alguns países com recursos limitados. O segundo é fazer com que mais
pessoas
façam
testes
de
HIV
de
maneira
voluntária
e
recebam
aconselhamento antes de desenvolverem os sintomas da doença.
Caso as novas recomendações sejam seguidas, mais pessoas vão necessitar
de tratamento, aumentando os custos. Mas os gastos com hospitalizações vão
diminuir, aumentando a produtividade, já que os infectados poderão trabalhar
por mais tempo sem apresentar os sintomas da doença. Menos crianças serão
abandonadas com o vírus e, com a divulgação dos benefícios do início precoce
28
da terapia, mais pessoas ficarão motivadas a fazer testes de HIV e a receber
aconselhamento sobre a doença.
Conclusão
Nos últimos anos, o grande avanço verificado no campo da farmacologia dos
anti-retrovirais (talvez a área de maior crescimento e desenvolvimento) mudou
a vida das pessoas vivendo com AIDS, possibilitando um aumento da
sobrevida e melhora da qualidade de vida. Apesar deste avanço, a facilidade
com que os vírus adquirem resistência, torna imprescindível o desenvolvimento
de novos medicamentos. Neste sentido, a Farmacêutica tem um papel
fundamental seja no estudo direto de novos fármacos ou na atenção
farmacêutica trabalhando a questão da adesão ao medicamento. A adesão ao
medicamento é essencial para a manutenção da baixa carga viral e menor
desenvolvimento de resistência. Através da genotipagem, hoje adotada apenas
para pacientes com falha terapêutica, é possível uma terapia de resgate mais
personalizada de acordo com o perfil de mutações apresentadas. A ampliação
do arsenal terapêutico aliado a politerapia baseada na genotipagem para todos
os pacientes com indicação de uso poderá quem sabe no futuro garantir o
controle farmacológico desta infecção por períodos maiores, auxiliando no
controle da epidemia.
29
Bibliografia
Farmacologia Básica & Clínica – Katzung – 10ª. Edição, 2008.
http://drauziovarella.com.br/doencas-e-sintomas/aids/
http://www.roche.pt/sida/virus/life.cfm
30