MARA SANCHES GUARAGNA
“SÍNDROME NEFRÓTICA EM CRIANÇAS: AVALIAÇÃO
MOLECULAR EM UMA CASUÍSTICA BRASILEIRA”
CAMPINAS
2014
i
ii
iii
iv
v
vi
RESUMO
A Síndrome Nefrótica (SN) é a principal doença renal na infância, sendo caracterizada por
proteinuria, edema, hipoalbuminuria e hiperlipidemia. Usualmente é classificada de acordo com
a idade em que se apresenta em Congênita (SNC), quando se manifesta intraútero ou durante os
primeiros três meses de vida, Infantil durante o primeiro ano de vida, na Infância, entre o
primeiro ano até os 12 anos de idade e Juvenil, entre os 12 e os 18 anos de idade,
aproximadamente. De acordo com a resposta ao tratamento com corticoesteróides os pacientes
podem ser divididos em córtico-resistentes (CR), córtico-sensíveis (CS) ou córtico-sensíveis com
recidiva frequente (CS,RF). Uma disfunção na barreira de filtração glomerular leva às
manifestações clínicas decorrentes da síndrome, tais como proteinúria maciça na urina. Mutações
em diversos genes vêm sendo correlacionadas com a SN em crianças. No entanto, os genes mais
estudados e responsáveis pela maioria dos casos são os genes NPHS1, NPHS2 e WT1. Os
objetivos deste trabalho foram identificar e verificar a distribuição de mutações nestes três genes
em uma casuística brasileira com SN ou proteinúria isolada e avaliar cada alteração identificada
utilizando diversas predições in silico, com o intuito de se esclarecer suas respectivas funções
biológicas. Para isto foi realizada a análise molecular de 150 crianças e adolescentes, sendo três
não aparentados com SNC e os demais 147 com SN Infantil, SN na infância, SN juvenil e
proteinúria isolada, dos quais 134 eram não aparentados. Além destes 150 pacientes, também
foram analisados os materiais de biópsias renais fixadas em bloco de parafina de sete pacientes
que já foram a óbito com SNC. Para verificar a segregação alélica nas famílias, para os casos nos
quais foram identificadas alterações, a análise molecular dos pais foi realizada, quando possível.
Um grupo controle de indivíduos saudáveis foi incluído para se avaliar a frequência de alterações
vii
não depositadas em bancos de dados. No estudo do gene NPHS1 para os casos com SNC
identificamos mutações missense, frameshift e em região de splicing nos três pacientes
encaminhados no período da tese e em um dos materiais proveniente de biópsia renal. Assim, um
total de quatro pacientes com SNC apresentaram mutações que se correlacionam com o grave
quadro clínico apresentado. Para os pacientes com SN infantil, SN infância/juvenil e proteinúria
isolada, foram triados o gene NPHS2 e os éxons 8-9 do gene WT1. No estudo do gene NPHS2
foram identificadas duas alterações em heterozigose nos padrões de herança autossômica
recessiva em 2,7% (4/147), todos CR, enquanto que apenas uma alteração em heterozigose
simples foi identificada em 9,5% (14/147) em casos de SN com apresentação menos grave e
tardia. Em três dos 14 pacientes com uma alteração no gene NPHS2, alterações no gene NPHS1
foram também identificadas, todas já descritas como polimorfismos frequentes na população em
geral. Para o gene WT1, com herança autossômica dominante, foram identificadas mutações em
heterozigose simples em 2,04% (3/147). A avaliação da frequência e distribuição de mutações
nestes genes em crianças com SN é inédita no Brasil e traz um direcionamento para a análise
molecular de grupos específicos das crianças com SN. Além disto, este trabalho contribui para o
estabelecimento das bases moleculares da doença na população brasileira, o que tem uma
repercussão importante na conduta dos pacientes, uma vez que, nos que apresentam mutações,
pode-se considerar o transplante renal a partir de doador vivo, pois para estes considera-se um
risco menor de recidiva de glomérulo esclerose focal e segmentar após transplante do que para
pacientes sem mutações.
viii
ABSTRACT
Nephrotic syndrome (NS) is the main kidney disease in children. It is characterized by
proteinuria, edema, hypoalbuminemia and hyperlipidemia. Acccording to the age of the
diagnosis, it is usually classified as Congenital (CNS) when it manifests in utero or during the
first three months of life, Infantile when the event occurs during the first year of life, in
Childhood when symptoms occur between one year and 12 years and Juvenile, with onset
between 12 and 18 years old. NS is traditionally separated on the basis of the response to
standard steroid treatment as steroid-resistant (SRNS), steroid-sensitive (SSNS) or steroid
sensitive with frequent relapses. A dysfunction in the glomerular filtration barrier leads to the
characteristic clinical manisfestations of the syndrome such as massive loss of essential proteins
in the urine. Mutations in different genes have been associated with NS in children. However, the
most studied genes are NPHS1, NPHS2 and WT1, which are responsible for the great majority of
the cases. The aims of this study were to identify and verify mutation distributions in those three
genes in a Brazilian cohort with NS or isolated proteinuria and to characterize each identified
variation by using different in silico prediction programs in order to understand its biological
functions. For that, we performed molecular analyses of 150 children and adolescents, being
three unrelated children with CNS and the remaining 147 with Infantile, Childhood, Juvenile NS
and isolated proteinuria, being 134 unrelated. Besides those 150 patients, paraffin-embebbed
renal biopsies of seven patients who had died from CNS have been also analysed. To verify
allelic segregation in the family, molecular analyses were also held, whenever possible, for
parents of patients in whom mutations were identified. A healthy control group was included in
the study to evaluate the frequency of alterations that were absent in the databanks. In the
ix
NPHS1 study for CNS cases, we identified missense, frameshift and splicing mutations in the
three patients included during the thesis project and in the paraffin-embedded renal biopsy tissue
of a patient who had died of CNS. Therefore a total of four CNS cases bore mutations that are
associated with the disease. NPHS2 gene and exons 8-9 of WT1 were screened for the 147
patients with infantile NS, childhood/juvenile NS and isolated proteinuria. In the NPHS2 study,
two heterozygous alterations compatible to an autossomal recessive inheritance have been
identified in 2,7% (4/147), all of the cases were SRNS; whereas, only one heterozygous
alteration was identified in 9,5% (14/147), such cases had a less severe and late onset form of
NS. Three out of those 14 patients presented sequence variations also in NPHS1 gene, but they
have been described as neutral polymorphisms. For the WT1 gene whose mutations present a
dominant pattern of inheritance, heterozygous alterations have been identified in 2,04% (3/147).
This is the first study focusing frequency evaluation and mutation distribution in Brazilian
children with NS and provides a guidance to the molecular analysis of specific case groups of
SN. Moreover, this work contributes to the establishment of the molecular bases of this disease
in the Brazilian population, what reflects mainly in the conduction of patients, since those
bearing mutations can be considered for receiving a kidney from a living donor, because for
them it is considered a lower risk of recurrent focal segmental glomerulosclerosis after kidney
transplant than for patients without mutations.
x
SUMÁRIO
RESUMO.................................................................................................................................. vii
ABSTRACT.............................................................................................................................. ix
LISTA DE ABREVIATURAS.............................................................................................. xxiii
INTRODUÇÃO........................................................................................................................
1
1. UM POUCO DE HISTÓRIA..........................................................................................
3
2. SISTEMA RENAL DE FILTRAÇÃO..............................................................................
4
3. A SÍNDROME NEFRÓTICA.........................................................................................
9
3.1 Síndrome Nefrótica Congênita................................................................................. 12
3.2 Síndrome Nefrótica Infantil, na Infância e Juvenil.................................................. 14
4. A ORIGEM GENÉTICA DA SÍNDROME NEFRÓTICA..............................................
15
4.1 O gene NPHS1 e a proteína nefrina........................................................................ 22
4.2 O gene NPHS2 e a proteína podocina..................................................................... 24
4.3 O gene WT1 e o fator de transcrição WT1.............................................................. 29
5. MUTAÇÕES NOS GENES NPHS1, NPHS2 E WT1...................................................... 31
JUSTIFICATIVA...................................................................................................................
41
OBJETIVOS............................................................................................................................. 45
CASUÍSTICA E MÉTODOS.................................................................................................. 47
xi
1. CASUÍSTICA................................................................................................................. 49
2. MÉTODOS..................................................................................................................... 56
2.1 Obtenção das amostras............................................................................................. 57
2.2 Extração de DNA genômico...................................................................................... 57
2.2.1 A partir de sangue total.................................................................................... 57
2.2.2. A partir te tecido fixado em parafina.............................................................. 59
2.3 Quantificação do DNA genômico.............................................................................. 59
2.4. Integridade do DNA genômico................................................................................. 60
2.5 Amplificação dos genes por reação de polimerase em cadeia – PCR...................... 60
2.6 Purificação dos produtos de PCR............................................................................. 64
2.7 Reação de sequenciamento........................................................................................ 65
2.8 Purificação da reação de sequenciamento................................................................ 65
2.9 Amplificação alelo específica por PCR..................................................................... 67
2.10 Estudos in silico........................................................................................................ 70
2.11 Cálculos estatísticos para avaliação dos SNPs e Análise de Desequilíbrio de
ligação.............................................................................................................................. 74
2.12 Classificação das mutações e correlação genótipo-fenótipo................................... 75
RESULTADOS E DISCUSSÃO............................................................................................. 79
1.
SN CONGÊNITA E TRIAGEM DO GENE NPHS1...................................................... 80
1.1 Material fixado em parafina...................................................................................... 80
1.2 Pacientes com SNC.................................................................................................... 86
xii
2. PACIENTES COM SN INFANTIL, NA INFÂNCIA, JUVENIL E COM PROTEINÚRIA
ISOLADA............................................................................................................................. 110
2.1 Gene NPHS2............................................................................................................ 110
2.1.1 Pacientes com duas alterações em heterozigose............................................. 111
2.1.2 Alterações em heterozigose simples................................................................ 132
2.1.2.1 Alterações missense em heterozigose simples......................................... 132
2.1.2.2 Mutações em heterozigose simples na região 5’ reguladora................... 148
2.2 Gene NPHS1........................................................................................................... 165
2.3 Polimorfismos identificados nos genes NPHS2 e NPHS1...................................... 171
2.4 Gene WT1............................................................................................................................ 184
2.4.1 Mutação c.1227+5G>A, paciente 1............................................................... 184
2.4.2 Mutação c.1227+4C>T, paciente 2................................................................ 186
2.4.3 Mutação c.1278C>T – p.Ser393Phe, paciente 3............................................ 187
CONCLUSÕES....................................................................................................................... 197
REFERÊNCIAS...................................................................................................................... 203
ANEXOS................................................................................................................................. 227
Anexo I.......................................................................................................................... 228
Anexo II......................................................................................................................... 229
xiii
xiv
“ Quando uma criatura humana desperta para um grande sonho
e sobre ele lança toda a força de sua alma,
todo o universo conspira a seu favor”.
Johann Wolfgang von Goethe
xv
xvi
A todas as crianças com Síndrome Nefrótica
e às suas famílias,
dedico.
xvii
xviii
AGRADECIMENTOS
Agradeço à querida orientadora e amiga Maricilda. Orientadora de outros tempos, da
minha iniciação científica e que me acolheu de braços abertos 10 anos mais tarde! Nunca vou me
esquecer do dia em que voltei e ela, na mesma sala, virou-se e disse: “o bom filho a casa torna!”
- com tanta felicidade, que eu soube, na hora, que era bem vinda novamente! Maricilda: muito
obrigada por tudo, por todos os conselhos, paciência, correções, ensinamentos. Obrigada por
compreender e aceitar meu novo momento, com filhos!
Ao Dr. Gil Guerra-Jr e à Dra. Vera Belangero (uma mãe...), agradeço imensamente pelo
presente que foi este projeto. Pela disposição sempre imediata em me ajudar.
À Dra. Anna Cristina Lutaif, agradeço pela disposição em tirar TODAS as minhas
dúvidas com relação aos pacientes (e foram TANTAS!). Junto com meu projeto ganhei uma
amiga e companheira.
A toda a equipe do Ambulatório de nefropediatria (APA) pelo carinho com que fui
acolhida.
À Dra. Andréa Trevas Maciel Guerra, tão calma e querida, pelas correções e trocas de
emails nos artigos!
À Dra. Edi Lúcia Sartorado, tão dinâmica e ativa, muito obrigada por nos contagiar com
seu ânimo e seus cursos!
Às secretárias do CBMEG: Sandra, Tania, Gabi: muito obrigada pelo constante e
eficiente suporte com relação à burocracia para viagens a congressos, dúvidas de compras, etc,
etc, etc... e à Lourdes, nossa incansável secretária da pós-graduação! Obrigada por tudo!
Ao bioestatístico Rodrigo Secolin, tão prestativo e atencioso! Obrigada pelas reuniões
valiosas!
Aos amigos do Laboratório de Genética Molecular e Humana do CBMEG, antiquíssimos,
antigos e atuais: Marcela, Ju Assumpção, Lee, Ed, Luzinha, Fer3, Renan, Fran, Denise, Daiane,
Nathy Gavioli, Mari, Zélo, Van, Xuxa, Pri, Creyto, Paulão, Ana Letícia, Pamela, Carol Lincoln,
xix
Rose, Nathy Zocal, Foca, Ana Paula, Bela, Laura, Ju Gabriel, Larissa, Marcel, Aliane, Luana,
Marcela, Juanito, Batatinha, Fábio, Lilian, Alessandra, Nádia, Priscila Jacob. Muito obrigada
pelo convívio, por tantas risadas, limpezas em grupo, cafés da manhã, confraternizações de fim
de ano!
Em particular gostaria de agradecer aos amigos: Débora, por toda a paciência do mundo
nas inúmeras vezes que tirou minhas dúvidas com relação aos cálculos! Taci, por ter me ajudado
com os milhões de PCRs e purificações enlouquecedoras! Cris, por extrair o DNA de inúmeros
pacientes. Você é DEMAIS! Heleninha, obrigada por ser companheira, sempre pronta para
ajudar, você mora no meu coração. Dri, mulher maravilha! Muito obrigada por ser sempre tão
solícita, alegre, eficaz! Por ser a nossa médica de plantão! Suuuuuu!!! Obrigada por me
compreender, por ter vindo para este “lab” para eu ter alguém “como eu” para conversar. Fer:
obrigada por me ensinar tanto! Luli: Com sua curiosidade nos contamina e nos faz querer
aprender sempre mais! Regi: tantas e tantas risadas! Florzinha: tantas lembranças, tantas
conversas, risadas e tropeços! Obrigada por ser minha amiga e sempre me compreender.
Aos meus queridíssimos amigos de vida! Drizinha, Márcião, Lu e Trolha! Tantas
conversas e desabafos! Tantos conselhos e opiniões! Sempre me incentivando e me orientando.
Meus respeitosos agradecimentos pelas valiosas contribuições dos integrantes da prébanca, Dra. Cláuda Vianna Maurer Morelli e Dra. Maria Almerinda Vieira Fernandes Ribeiro
Alves e pela participação dos membros da banca de defesa, Dr. Carlos Eduardo Steiner, Dra.
Maria de Fátima Sonati, Dr. Paulo César Koch Nogueira e Dra. Maria Isabel de Souza Aranha
Melaragno.
Agradeço aos órgãos financiadores FAPESP e CNPq, sem os quais este projeto não seria
viável.
A todos os pacientes e seus familiares, obrigada por literalmente doarem o seu sangue
para esta pesquisa. Sua contribuição é valiosa para continuarmos estudando esta doença.
A todos os que contribuíram direta ou indiretamente para a realização deste trabalho.
A toda a minha família. Muito obrigada pela paciência que tiveram comigo nesta fase!
xx
AGRADECIMENTOS ESPECIAIS
Aos meus amados e queridos Pais e ao meu irmão, meu ídolo!
Sempre prontos para me ouvir e me aconselhar.
Ao meu amor e companheiro de vida Duzinho e aos meus queridos
filhos, meus tesouros, meus amores. Sem vocês não sei existir.
xxi
xxii
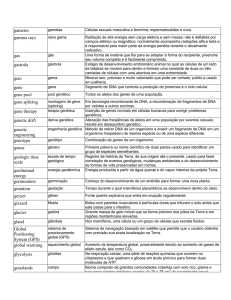
LISTA DE ABREVIATURAS
BFG
Barreira de Filtração Glomerular
BSA
Albumina Sérica Bovina
cM
Centimorgan
DDS
Síndrome de Denys-Drash
DLM
Doença de Lesões Mínimas
DMSO
Dimetilsulfóxido
dNTP
Desoxirribonucleotídeo Trifosfato
EDTA
Ácido Etileno Diamono Trifosfatado
EMD
Esclerose Mesangial Difusa
ESE
Exonic Splicing Enhancer
ESS
Exonic Splicing Silencer
GESF
Glomérulo Esclerose Segmentar e Focal
GNMP
Glomérulo Nefrite Membranoproliferativa
HEK
Human Embryonic Kidney
ISE
Íntronic Splicing Enhancer
ISS
Íntronic Splicing Silencer
KTS
Lisina, Treonina, Serina
LD
Linkage Disequilibrium
LM
Lesão Mínima
MBG
Membrana Basal Glomerular
OMIM
Online Mendelian Inheritance on Man
xxiii
pb
Pares de base
PDB
Protein Data Bank
PHB
Prohibitin
PM
Proliferação Mesangial
RNAi
RNA de interferência
FS
Síndrome de Frasier
shRNA
Short RNA
SN
Síndrome Nefrótica
SNC
Síndrome Nefrótica Congênita
SNCR
Síndrome Nefrótica Córtico Resistente
SNCS
Síndrome Nefrótica Córtico Sensível
SNCS,RF
Síndrome Nefrótica Córtico Sensível, com Rescidiva Frequente
SNF
Síndrome Nefrótica Finlandesa
SNI
Síndrome Nefrótica Infantil
SNP
Single Nucleotide Polymorphism
Ta
Temperatura de annealing (anelamento)
Taq
Thermus aquaticus (enzima polimerase)
TBE
Tris – Borato – EDTA
TE
Tris – EDTA
Tm
Temperatura de melting (fusão)
ZF
Zinc Finger (dedo de zinco)
xxiv
INTRODUÇÃO
1
2
INTRODUÇÃO
1. UM POUCO DE HISTÓRIA
A relação entre proteinúria e doença renal é conhecida há centenas de anos. O registro
mais remoto é do século V, época em que Hipócrates fez a seguinte observação: “quando bolhas
estão presentes na superfície da urina, isto indica doença dos rins e a queixa permanece por
longo tempo” (Chadwick & Mann apud Cattran, 2011). Nos dias de hoje, os nefrologistas
utilizam esta premissa como um dos critérios de avaliação na anamnese dos pacientes com
objetivo de identificar o início da proteinúria. Um longo período de tempo se passou após as
referidas observações de Hipócrates até que Roelans, já no século XV, descrevesse “um inchaço
generalizado” no corpo de crianças com problemas renais por ele avaliadas, característica clínica
que, hoje se sabe, está associada ao edema generalizado (Roelans, 1484 apud Cattran, 2011). No
entanto, foi somente no início do século XVIII que Theador Zwinger III da Basiléia, na Suíça,
documentou toda a evolução clínica da doença até atingir a insuficiência renal crônica, dando as
primeiras descrições do quadro de síndrome nefrótica (Zwinger, 1722 apud Cattran, 2011).
Quase 50 anos mais tarde Cotugno de Nápoles, na Itália, fez as primeiras correlações entre
proteinúria e o quadro de edema, mas foi somente no século XIX que John Blackall estabeleceu
a associação entre proteinúria e doença renal, sendo assim a síndrome nefrótica reconhecida pela
primeira vez como uma entidade patológica (Blackall, 1818, Cotugno, 1974 apud Cattran, 2011).
Contudo, quem descreveu a tríade proteinúria, edema e doença nos rins foi Richard Bright, o que
o consagrou como o “pai da nefrologia” (Bright, 1827 apud Cattran, 2011).
3
O termo “nefrose” foi introduzido por Friedrich Von Müller no início do século XX,
indicando uma lesão degenerativa nos rins (Müller, 1905 apud Cattran, 2011). Contrapunha-se
ao termo “nefrite”, que define as doenças renais de origem inflamatória.
O debate sobre qual seria a estrutura anatômica primária responsável pela perda de
proteínas - glomerular e/ou tubular - perdurou durante a primeira metade do século XX. Após
várias teorias terem sido propostas e discutidas por diversos pesquisadores, Addis, em 1948,
durante uma conferência intitulada “O Mecanismo da Proteinúria”, ressaltou o que na época já
era um senso comum: os pacientes com proteinúria tinham um dano na membrana glomerular
que permitia um aumento da permeabilidade às proteínas (Addis, 1948 apud Cattran, 2011).
2. SISTEMA RENAL DE FILTRAÇÃO
Os rins são órgãos excretores e reguladores que, nos seres humanos, estão situados no
retroperitônio do abdômen. Como parte do sistema urinário, suas funções principais são a
excreção de solutos e água que removem do sangue, a manutenção do equilíbrio ácido base do
soro corporal, bem como a regulação da homeostase dos fluidos e da pressão sanguínea. Para
exercerem suas funções, os rins se dividem em estruturas, cada uma com características
específicas (figura 1). A figura 1A ilustra as características macroanatômicas do rim humano
onde a camada mais externa é o córtex e a região interna, a medula. Os néfrons que são as
unidades funcionais dos rins se situam tanto no córtex como na medula. A figura 1B, por sua
vez, mostra o esquema de um néfron que é formado pelo corpúsculo renal, por um túbulo
4
proximal, pela alça de Henle, por um túbulo distal e pelo sistema de ductos coletores. Já o
corpúsculo renal consiste em capilares glomerulares (glomérulo) e na cápsula de Bowman.
A unidade filtradora renal mais importante é o glomérulo que é composto pelos seguintes
tipos celulares: células epiteliais parietais da cápsula de Bowman, células endoteliais
glomerulares, células epiteliais viscerais (podócitos) e células mesangiais (figura 1C). O sangue,
portanto, entra nos glomérulos por uma arteríola aferente que se ramifica formando uma rede
capilar, é filtrado através da barreira de filtração formando o filtrado glomerular que é coletado
no espaço de Bowman e processado no sistema tubular renal. O sangue filtrado sai do glomérulo
pela arteríola eferente.
A barreira de filtração glomerular (BFG), que separa a corrente sanguínea do espaço
urinário, é responsável pela ultrafiltração do plasma no processo de formação da urina (figura 2).
É composta por três camadas especializadas: a camada de células endoteliais glomerulares, a
membrana basal glomerular (MBG) e a camada do epitélio glomerular, os podócitos. A urina
começa a se formar pela passagem de um ultrafiltrado do plasma dos glomérulos para o espaço
de Bowman.
A primeira camada da BFG é constituída de células achatadas, as chamadas células
endoteliais, que separam o sangue do tecido e regulam o tônus vasomotor e a homeostase. Em
humanos, o endotélio dos glomérulos é fenestrado, contendo numerosos poros com
aproximadamente 70 a 100 nm de diâmetro que correspondem a uma área de 20 a 50% da
superfície capilar total. Originalmente acreditava-se que o endotélio fenestrado contribuía pouco
para a permeabilidade seletiva do glomérulo, já que o tamanho dos poros é suficiente para
permitir a passagem da albumina.
5
Figura 1. Os rins e principais estruturas que formam a rede de filtração do sangue. A. Corte longitudinal de
um
rim
com
a
região
do
córtex
e
medula
ampliada
mostrando
a
localização
dos
néfrons
(http://fau.pearlashes.com/anatomy/Chapter%2041B/Chapter%2041B.htm). B. Esquema de um néfron e suas
principais estruturas, com ampliação do glomérulo. O sangue entra pela arteríola aferente, retorna para a circulação
pela
arteríola
eferente
e
a
urina
formada
sai
pelo
(http://www.medicinapratica.com.br/2010/03/24/saude-medicina-pratica/nefron-o-que-e/).
túbulo
C.
coletor
Ilustração
do
corpúsculo renal, onde são identificados os principais tipos celulares. Adaptado de Leeuwis et al., 2010.
No entanto, recobrindo as células endoteliais há uma malha superficial carregada
negativamente, composta por proteoglicanos sulfatados, o glicocálix e que forma projeções do
tipo escova ao redor dos poros. Deste modo, o endotélio possui um papel ativo na filtração
glomerular, auxiliando na repulsão das proteínas (Ballermann & Stan, 2007).
6
Figura 2. Corpúsculo renal e o sistema de filtração. A. O corpúsculo renal é mostrado no esquema à esquerda; à
direita ilustra-se a ampliação da barreira de filtração glomerular Adaptado de Patrakka, 2001. B. À esquerda, corte
histológico
de
um
corpúsculo
renal
(coloração
de
Shiff)
(http://upload.wikimedia.org/wikipedia/commons/thumb/5/54/Renal_corpuscle.jpg/220px-Renal_corpuscle.jpg),
visualizado ao microscópio óptico; à direita, micrografia eletrônica da barreira de filtração glomerular
(http://www.uni-mainz.de/FB/Medizin/Anatomie/workshop/EM/externes/Wartenberg/Niere12E.html).
A segunda camada da BFG é a membrana basal glomerular (MBG), que é descrita como
uma malha semelhante a um gel, com volume de água entre 90 a 93% e com íons carregados
negativamente. É uma rede heteropolimérica composta por colágeno tipo IV, laminina,
fibronectina, nidogêneo e quantidades abundantes de proteoglicanas de heparan sulfato (1% do
7
peso seco da MBG). A MBG exerce diversas funções, tais como a filtração e a
compartimentalização das moléculas, além de participar na adesão, migração e diferenciação
celulares.
Os podócitos são células altamente especializadas e diferenciadas que recobrem a face
externa (urinária) da MBG e formam a terceira camada da BFG. Possuem um complexo de Golgi
bem desenvolvido, retículos endoplasmáticos (lisos e rugosos) abundantes, numerosas
mitocôndrias e vários lisossomos; estas características indicam que os podócitos exercem uma
atividade metabólica intensa necessária para sustentar a estrutura complexa das fendas do
diafragma e sintetizar a maioria dos componentes da MBG (Pavenstädt et al., 2003). Seu
formato, com longas projeções citoplasmáticas, os pedicelos, é mantido graças a um
citoesqueleto rico em proteínas especializadas, tais como a sinaptopodina e a α-actinina. Mundel
et al. demonstraram que a sinaptopodina tem um papel importante na organização da actina e na
motilidade celular (Mundel et al., 1997). A α-actinina-4 é uma molécula que atua na ligação
cruzada entre actinas e também atua na motilidade celular (Honda et al., 1998).
O corpo celular dos podócitos localiza-se no espaço urinário e os pedicelos, quando
tocam a superfície dos capilares, ramificam-se formando interdigitações (figura 3A). As
interdigitações entre os pedicelos formam poros, as chamadas fendas de diafragma, cuja
ultraestrutura lembra um “zíper” e foi revelada nos estudos de Rodewald e Karnovsky (1974)
(figura 3B). Alguns estudos sugerem que a membrana plasmática das fendas do diafragma seja
constituída por “rafts” de lipídios (Simons et al., 2001; Huber et al., 2003). Os “rafts” de lipídios
são microdomínios especializados da membrana plasmática com uma composição específica
enriquecida de colesterol e glicoesfingolipídios e que atuam no recrutamento e formação de
“clusters” de proteínas de uma maneira seletiva e dinâmica. Os “rafts” têm sido propostos como
8
sendo plataformas para muitos processos celulares importantes, tais como endocitose, exocitose,
adesão celular, arranjo polarizado de proteínas de membrana e transdução de sinal (Simons et al.,
1997; Smart et al., 1999).
Figura 3. Estrutura dos podócitos. A. Microscopia eletrônica de varredura mostrando um podócito observado da
face urinária. Os pedicelos podem ser visualizados ramificando-se cada vez mais até alcançarem a superfície de um
capilar. Os pedicelos se interdigitam com os pedicelos de outros podócitos. O espaço entre os pedicelos são as
fendas de diafragma por onde passa o líquido filtrado proveniente do plasma sanguíneo. Adaptado de Smoyer et al.
(Smoyer & Mundel, 1998). B. No lado esquerdo observa-se a micrografia eletrônica da fenda de diafragma do
glomérulo de um rato. É possível a visualização detalhada das ligações cruzadas formando um padrão do tipo
“zíper” nas fendas de diafragma entre as membranas de pedicelos adjacentes. A região das marcas é ilustrada de
forma ampliada à direita, em um esquema com detalhamento das medidas e que demonstra o padrão do tipo “zíper”
e consequente formação dos poros. Adaptado de Rodewald & Karnovsky, 1974.
3. A SÍNDROME NEFRÓTICA
A Síndrome Nefrótica (SN) é definida como a entidade clínica caracterizada por
proteinúria maciça, edema, hipoalbuminemia e, geralmente, hiperlipidemia. Corresponde a uma
das doenças renais mais comuns na infância, podendo levar à insuficiência renal crônica. Nos
primeiros anos de vida, as crianças com esta condição apresentam inchaço periorbital, com ou
9
sem edema generalizado. A doença ocorre devido à disfunção funcional ou estrutural da barreira
de filtração glomerular (BFG), resultando na perda de proteínas essenciais através da urina.
Fisiologicamente, o fígado tenta compensar esta perda excessiva aumentando a síntese de
proteínas e lipoproteínas sendo que a SN se desenvolve quando a perda de proteínas pela urina
excede à taxa de síntese de albumina pelo fígado, resultando em hipoalbuminemia e edema
(Gbadegesin & Smoyer, 2008).
A SN se classifica de acordo com a idade em que se apresenta em: Congênita (SNC),
quando se manifesta intraútero ou durante os três primeiros meses de vida; Infantil (SNI),
quando ocorre no primeiro ano de vida; SN na Infância, quando se manifesta a partir do primeiro
ano até aproximadamente os 12 anos de idade e Juvenil, com idade de desenvolvimento entre os
12 e os 18 anos de idade (Benoit et al., 2010). No entanto, na prática clínica é identificada
também em relação à resposta ao tratamento com corticoides. Assim, de acordo com este
critério, os pacientes podem ser subdivididos em: córtico-sensíveis (SNCS); córtico-resistentes
(SNCR); córtico-dependentes (SNCD); e, córtico-sensíveis com recidiva frequente (SNCS, RF)
(ISKDC, 1981).
Na tabela 1 estão detalhados alguns termos e características clínicas utilizadas para a
classificação dos vários tipos de SN, principalmente em relação à resposta ao tratamento com
corticoides.
A incidência anual da SN na maioria dos países do hemisfério ocidental é estimada em 2
a 7 casos novos a cada 100.000 crianças (Nash et al., 1992; Srivastava et al., 1999; Hogg et al.,
2000; Niaudet, 2004;). Diversos estudos mostram que a incidência e a gravidade da SN são
influenciadas tanto pela geografia como pela etnia. Em um estudo realizado no Reino Unido, por
10
exemplo, foi demonstrado que a incidência da SN em descendentes asiáticos que vivem no Reino
Unido é seis vezes maior do que entre os seus congêneres europeus (Sharples et al., 1985).
Tabela 1 – Características clínicas usadas na condução de pacientes com SN.
Termo
Síndrome nefrótica
Remissão
Recidiva
SN córtico-sensível
SN córtico-resistente
SN córtico-dependente
SN córtico-sensível com
recidiva frequente
Definição
Edema, proteinúria maciça (>40 mg/m2/h ou taxa de proteína urinária/creatinina >
2,0 mg/mg) e hipoalbuminemia (< 2,5 g/dl)
Redução significativa da proteinúria (<4 mg/ m2/h ou albumina urinária de 0 por três
dias consecutivos), sem edema e albumina sérica de até 3,5 g/dl)
Recorrência da proteinúria maciça (>40 mg/m2/h ou taxa de proteína
urinária/creatinina > 2,0 mg/mg), na maioria das vezes associada a edema
Pacientes que entram em remissão somente com tratamento com corticoides
Pacientes que não entram em remissão após oito semanas de tratamento com
corticoides. Algumas variações podem ser encontradas (ISKDC, 1981; Niaudet et
al., 1998).
Pacientes que necessitam de um tratamento contínuo, com uma dose baixa de
corticoides para prevenir o desenvolvimento ou a recidiva (Schulman et al., 1988).
Pacientes que apresentam quatro ou mais recidivas em um período de 12 meses
Entre os descendentes afro-americanos o risco de desenvolver glomérulo esclerose
segmentar e focal (GESF) é quatro vezes maior do que entre os americanos sem descendência
africana. Atualmente sabe-se que há fatores genéticos associados a esta maior predisposição de
desenvolvimento de GESF entre os africanos e seus descendentes (Kopp et al., 2008; Genovese
et al., 2010). Não existem descrições de dados epidemiológicos sistemáticos realizados no Brasil.
11
Tabela 2: Características histológicas após biópsia renal.
TÉCNICAS
CARACTERÍSTICAS
Microscopia
Óptica
MO
Imunofluorescência
IMUNO
Microscopia
Eletrônica
ME
Lesão Mínima
(LM)
Normal
Negativo
Perda de pedicelos
Aumento do
mesângio
Negativo
Perda de pedicelos
Proliferação mesangial
(PM)
Glomérulo esclerose
focal e segmentar
(GESF)
Normal
Negativo
Perda de pedicelos e
esclerose de alguns
glomérulos +
segmentos de
glomérulos afetados
Fonte: Departamento de anatomia patológica da Unicamp.
Outras características importantes dizem respeito aos achados histológicos após a biópsia
renal e indicam os graus de evolução e de gravidade da doença. No departamento de Anatomia
Patológica do Hospital das Clínicas da Unicamp são realizadas três técnicas para análise do
tecido após biópsia renal: 1) microscopia óptica (MO); 2) imunofluorescência (Imuno); e, 3)
microscopia eletrônica (ME). Na tabela 2 encontram-se os resultados produzidos pelas três
técnicas, que caracterizam histologicamente o tecido renal derivado de biópsia de pacientes com
SN.
3.1. Síndrome Nefrótica Congênita (SNC)
A SNC é uma doença rara, com herança autossômica recessiva e é a principal causa de
proteinúria maciça nos primeiros três meses de vida. O tipo mais comum de SNC é a do tipo
12
Finlandês (SNF), que recebeu este nome por ter sido primeiramente identificada na população
finlandesa na qual apresenta uma incidência aproximada de 1 em cada 8.500 nascimentos (SNF –
OMIM 256300) (Kestilä et al., 1994).
Em 1999, Bolk et al. observaram que numa população da Velha Ordem Menonita
residente na Pensilvânia, a incidência da SNC era ainda maior do que a da Finlândia, ocorrendo
em aproximadamente 1 a cada 500 nascimentos (Bolk et al., 1999). A SNC ocorre em outros
países do mundo, mas com incidências menores (Fuchshuber et al., 1996).
Na SNC a proteinúria é maciça e já se inicia intraútero, sendo acompanhada de edema nas
duas primeiras semanas de vida, além de hipoalbuminemia e hiperlipidemia. O prognóstico deste
tipo de nefrose é de declínio progressivo da função renal, infecções frequentes e, normalmente,
ocorre o óbito nos primeiros anos de vida devido à septicemia ou uremia (Norio, 1966). As
alterações renais histopatológicas características desta síndrome são glomérulos imaturos,
proliferação das células mesangiais e fusão glomerular dos pedicelos, juntamente com dilatação
pseudocística dos túbulos proximais, que podem refletir no desenvolvimento defeituoso dos
podócitos (Haltia et al., 1998; Huttunen et al., 1980). Não existe tratamento definitivo
estabelecido e não ocorre resposta após tratamento com glicocorticoides e drogas
imunossupressoras (Guez et al., 1998; Hamed & Shomaf, 2001). O transplante renal fornece
baixas taxas de sobrevivência no início da doença, pois além da dificuldade com os riscos usuais
de rejeição, a SN pode recidivar nos rins transplantados (Kim et al., 1998).
13
3.2 Síndrome Nefrótica Infantil, na Infância e Juvenil
Estas síndromes são caracterizadas por proteinúria e progressão à insuficiência renal. As
características histológicas reveladas após biópsia renal são: lesões mínimas (se a biópsia for
realizada logo no início do desenvolvimento da síndrome), proliferação mesangial difusa e
glomérulo esclerose focal e segmentar. Entre os casos com desenvolvimento na infância, os
pacientes geralmente são classificados de acordo com a resposta à terapia aos corticóides. Entre
os córtico-sensíveis acredita-se que a origem da doença seja imunológica, já que são responsivos
ao tratamento com imunossupressão e ocorre recidiva da proteinúria após transplante renal. Em
1972, Hoyer et al. sugeriram que um fator de permeabilidade poderia estar presente na circulação
dos pacientes CS e que poderia alterar a BFG sendo responsável pela recidiva da proteinúria
após transplante (Hoyer et al., 1972). Estudos indicam que fatores solúveis circulantes podem se
depositar nos glomérulos prejudicando a permeabilidade da BFG em pacientes que respondem ao
tratamento com agentes imunossupressores e/ou que sofrem recorrência da proteinúria após o
transplante renal (Savin et al., 1996). Wei et al. (2008) demonstraram a indução da expressão do
do receptor solúvel de uroquinase (suPAR) em podócitos durante doença renal e como a
presença deste fator leva à fusão dos pedicelos resultando em proteinúria, com um possível papel
fisiológico na regulação da permeabilidade dos rins. Este fator foi recentemente descrito como
marcador também para pacientes com mutação no gene NPHS2 (Cara-Fuentes et al., 2013).
Entre os pacientes que são resistentes ao tratamento com corticoides, nos últimos anos
vários genes vêm sendo correlacionados a formas herdadas de defeitos estruturais da BFG, o que
justifica o fato de estes pacientes não responderem aos corticoides. Na maioria destes casos não
14
ocorre recidiva após transplante renal, já que a causa da doença é genética, sendo importante esta
informação pois estes pacientes podem ser encaminhados para transplante de doadores vivos.
4. A ORIGEM GENÉTICA DA SÍNDROME NEFRÓTICA
A etiologia da SN varia dependendo da idade em que se apresenta. Na SNC, por
exemplo, a grande maioria dos casos é de origem genética, enquanto que na SNI considera-se
que aproximadamente 65% dos casos sejam de origem genética (Hinkes et al., 2007). A maioria
dos casos da SN na infância é de origem idiopática e, após os primeiros 12 anos de vida aumenta
o número de casos com origem secundária a outras doenças, tais como doenças sistêmicas,
infecções, neoplasias e em decorrência ao uso de medicamentos.
Entre os pacientes infantis e com SN na infância, em especial aqueles que apresentam
resistência ao tratamento com corticoides, vêm sendo identificadas mutações em diversos genes
que codificam proteínas importantes para a manutenção da estrutura e da função da barreira de
filtração glomerular. Estas proteínas são expressas principalmente nos podócitos e estão
envolvidas direta ou indiretamente na organização das fendas de diafragma e do citoesqueleto de
actina. Geralmente as mutações ocorrem em um só gene (herança monogênica), embora alguns
casos de mutações com herança digênica já tenham sido descritos (Löwik et al., 2008). Além
disso, a SN pode seguir um modelo de herança autossômica recessiva ou autossômica dominante
e pode se apresentar de maneira isolada ou como parte de uma doença multissistêmica.
Na tabela 3 estão descritos dez genes que já foram relacionados com diferentes formas de
SNCR não sindrômica. A maioria das mutações identificadas nestes genes se concentra nos
genes NPHS1 e NPHS2 que codificam a nefrina e a podocina, respectivamente (Kestilä et al.,
15
1998; Boute et al., 2000). Estas duas proteínas, juntamente com a CD2AP, correspondem aos
elementos estruturais mais importantes na composição da fenda diafragmática (Gigante et al.,
2009). A nefrina, uma proteína transmembrânica da superfamília das imunoglobulinas, interage
através da sua porção C-terminal com a podocina. A nefrina também interage com a CD2AP,
uma proteína adaptadora encontrada na superfície das células T e das células natural-killer.
Mutações no gene CD2AP (OMIM 604241; 6p12.3) foram identificadas em pacientes com
SNCR tanto com herança autossômica dominante (Kim et al., 2003) como com herança
autossômica recessiva (Löwik et al., 2007).
A PLCε1, codificada pelo gene PLCE1 (Hinkes et al., 2006), é uma fosfolipase que
catalisa a hidrólise dos fosfolípideos da membrana para gerar seg mensageiros, o 1,4,5-trifosfato
inositol (IP3) e o diacilglicerol (DAG), que iniciam a via intracelular de crescimento e
diferenciação. Mutações no gene PLCE1 (OMIM 608414; 10q23) foram identificadas em
crianças com padrão histológico mesangial difuso e apresentam herança autossômica recessiva.
O gene TRPC6 (Reiser et al., 2005) codifica o receptor de potencial transitório dos
canais de cálcio, membro 6 da subfamília C que está localizada no super complexo lipídico de
membrana juntamente com a podocina e regula a sensação mecânica na fenda diafragmática;
enquanto que os genes ACTN4 (Kaplan et al., 2000) e INF2 (Brown et al., 2010) que codificam
α-actinina-4 e a proteína reguladora da actinina da família da formina, respectivamente, estão
envolvidos na dinâmica do citoesqueleto. Mutações nos genes TRPC6 (OMIM 603652; 11q21q22), ACTN4 (OMIM 604638; 19q13) e INF2 (OMIM 613237; 14q32.33) foram identificadas
em famílias com herança autossômica dominante e desenvolvimento tardio de SNCR embora
haja relatos de casos com desenvolvimento precoce (Heeringa et al., 2009). O fato de se
encontrar mutações em vários desses genes nos casos de SN pode ser entendido pelas interações
16
que suas proteínas estabelecem ao formar a BFG, cujo modelo funcional está mostrado na figura
4 (Gigante et al., 2011).
Ilustrando a variabilidade genética relacionada à apresentação da SN, principalmente nos
casos de SNCR, podemos exemplificar a ocorrência de mutações em genes que codificam
proteínas que não estão expressas somente nos podócitos, mas também em outros tecidos e tipos
celulares.
Figura 4. Modelo funcional da barreira de filtração glomerular. A figura ressalta a complexidade das interações
protéicas que se estabelecem na fenda do diafragma da barreira de filtração glomerular (BFG) onde MBG é a
membrana basal glomerular. Adaptado de Gigante et al., 2011.
Embora menos frequentes e se apresentando sob formas sindrômicas da SNCR, podem
ocorrer mutações no gene WT1 (Pritchard-Jones et al., 1990) e LMX1B (Dreyer et al., 1998), que
são fatores transcricionais); LAMB2 (Zenker et al., 2004) e ITGA3 (Kambham et al., 2000), que
são componentes da MBG; SCARB2 (Berkovic et al., 2008), COQ2 (Salviati et al., 2005),
PDSS2 (Mollet et al., 2007), MTTL1 (Kobayashi et al., 1990) e SMARCAL1 (Boerkoel et al.,
17
2002), que são proteínas lisossômicas e mitocondriais ou mediadoras da reestruturação do
nucleossoma DNA (tabela 4).
18
Tabela 3: Genes envolvidos em formas não-sindrômicas de SNCR
Gene
Cromossomo
Número de
mutações*
Expressão
(Seres humanos)
Proteína
Fenda do diafragma
Nefrina
Modo de
Herança
Fenótipo
Histologia Renal
SNC
Dilatação microcística dos túbulos
e esclerose mesangial progressiva
SNCR
SNCR
DLM, GNMP, GESF
HGMD Profissional
NPHS1
(29 éxons)
19q13.1
227
AR
OMIM 602716
NPHS2
(8 éxons)
OMIM 604766
1q25.2
CD2AP
(17 éxons)
OMIM 604241
6p12
6
Vários tecidos e membrana
podocitária
Proteína associada ao CD2
AR/AD
PLCE1
(34 éxons)
OMIM 608414
10q23.33
30
Vários tecidos e citoplasma
dos podócitos
Fosfolipase C epsilon
AR
TRPC6
(13 éxons)
OMIM 603652
11q21-q22
22
Túbulos renais, podócitos,
células mesangiais e
endoteliais
Canal 6 do Receptor de
Potencial Transitório
catiônico
AD
19q13
12
Vários tecidos e pedicelos
Alfa-actinina 4
AD
SNCR
desenvolvimento
tardio
GESF
14q32
**
Células de Schwann e
podócitos
Formina 2
AD
SNCR
GESF
Membrana apical dos
podócitos
Receptor da proteína tirosina
fosfatase
AR
DLM
GESF
Membrana dos podócitos
Miosina não muscular classe
I
AR
SNCR
GESF
Podócitos
Proteína ativadora da RHO
GTPase
-
SNCR
GESF
ACTN4
(21 éxons)
OMIM 604638
INF2
(23 éxons)
OMIM 613237
PTPRO
(27 éxons)
OMIM 614196
MYO1E
(28 éxons)
OMIM 614131
ARHGAP24
(10 éxons)
OMIM 610586
12p12
15q21
4q20
127
**
**
**
Membrana dos podócitos
Podocina
AR
SNC
SNCR
Desenvolvimento
tardio/SNI
SNCR
SNCR
GESF
GESF
EMD
GESF
GESF
AR: autossômica recessiva; AD: autossômica dominante. DLM: doença de lesões mínimas; GNMP: glomérulo nefrite membranoproliferativa.
*Número de mutações depositadas no banco de dados HGMD (The Human Gene Mutation Database) até dezembro de 2013.
** Número de mutações não disponível até dezembro de 2013.
19
Mutações no gene WT1 se associam a SN com quadros de maior ou menor gravidade
como tumor de Wilms (Haber et al., 1991), síndrome de Denys-Drash (Bruening et al., 1992) e
Síndrome de Frasier (Barbaux et al., 1997). No entanto, mutações nos éxons 8 e 9 do gene WT1
podem ser encontradas em aproximadamente 9% dos casos esporádicos de SNCR de
manifestação precoce ou tardia, com padrão histológico de Esclerose Mesangial Difusa (EMD) e
também em casos isolados de glomérulo esclerose segmentar e focal (GESF) (Mucha et al.,
2006).
O LMX1B é um fator de transcrição com homeodomínio LIM que é expresso em
podócitos glomerulares, sugerindo um papel regulatório nesta célula. De fato, 40% dos pacientes
portadores da síndrome ônicopatelar, com anormalidades dermatológicas e músculo-esqueléticas
e apresentando mutações dominantes no gene LMX1B, são afetados com doença renal, com
hematúria, proteinúria e podendo evoluir para doença renal crônica (Harendza et al., 2009;
Lemley, 2009).
Com o advento de técnicas high-throughput de sequenciamento, fatores moleculares de
risco e também modelos de herança complexa de GESF causada por interações gene-gene e
gene-ambiente vêm sendo descritos. Por exemplo, alguns genes tais como o MYH9 (OMIM
612551; 22q12.3) (Kopp et al., 2008) e o APOL1 (OMIM 603743; 22q12.3) (Genovese et al.,
2010) foram descritos recentemente como fatores de risco para GESF em Afro-Americanos.
20
Tabela 4: Genes envolvidos em formas sindrômicas de SNCR
Número de
mutações*
Gene
Cromossomo
WT1
(10 éxons)
HGMD profissional
Expressão
(seres humanos)
Proteína
Modo de
Herança
OMIM 602575
LAMB2
(32 éxons)
OMIM 150325
ITGA3
(26 éxons)
130
Núcleo e citoplasma dos podócitos
Tumor de Wilms 1
AD
SNCR isolado
DDS
FS
GESF, EMD
EMD
GESF
17q11
164
Núcleo e citoplasma dos podócitos
Fator de transcrição 1 beta do
homeobox LIM
AD
Síndrome de Nail Patela
GESF
3p21
58
Membrana basal glomerular
Cadeia B2 da laminina
AR
Síndrome de Pearson
EMD
GESF
OMIM 602257
coQ6
(12 éxons)
OMM 614647
coQ2
(7 éxons)
OMIM 609825
PDSS2
(8 éxons)
OMIM 610564
MT-TL1
(1 éxon)
OMIM 590050
SMARCAL1
(18 éxons)
SNCR
CNS
Doença pulmonar
intersticial
Epidermólise Bolhosa
Síndrome Nefrótica com
depósitos de C1q
Epilepsia mioclônica
progressiva
-
17q21.33
**
Células epiteliais
Integrina alfa-3
-
4q21.1
20
Membranas lisossomais de céls
glomerulares
Proteína integral de membrana
tipo 2 do lisossomo
AR
14q24.3
**
Mitocôndrias
COQ6
AR
SNCR e surdez
neurosensorial
GESF
4q21.22
7
Mitocôndrias
Polipreniltransferase
hidroxibenzoato 4
AR
Deficiência de CoQ10
Glomerulopatia colapsante
6p21
2
Mitocôndrias de células renais
Prenil difosfato sintase,
subunidade 2
-
Deficiência de CoQ10
Síndrome de Leigh
GESF
t-RNA-LEU
**
Mitocôndrias de células renais
RNA-t mitocondrial da
Leucina
materna
MELAS
GESF
2q35
55
Núcleos dos podócitos e túbulos
proximais
Regulador dependende de
actina
AR
Displasia imuno óssea de
Schimmke
GESF
OMIM 605025
SCARB2
(12 éxons)
Histologia Renal
11p13
OMIM 607102
LMX1B
(8 éxons)
Síndrome
-
OMIM 606622
DDS: Síndrome de Denys-Drash; FS: Síndrome de Frasier; AR: autossômica recessiva; AD: autossômica dominante.
*Número de mutações depositadas no banco de dados HGMD (The Human Gene Mutation Database).** Número de mutações não disponível (dezembro/2013).
21
4.1 O gene NPHS1 e a proteína nefrina
Em 1994, após triagem de 17 famílias afetadas com SNF, uma região gênica candidata de
150 kb foi identificada no cromossomo 19 (Kestilä et al., 1994). Em 1998, o gene NPHS1
(OMIM 602716) foi identificado em 19q13.1, ocupando 26 kb de extensão com 29 éxons (figura
5A) (Kestilä et al., 1998). O gene NPHS1 codifica uma proteína de membrana de 180 kDa
denominada nefrina. Esta proteína é formada por um peptídeo sinal na região N-terminal, oito
domínios C2 do tipo imunoglobulina (Ig1 – Ig8), um motivo de fibronectina tipo III, uma região
transmembrânica e uma cauda citosólica na região C-terminal (Figura 5B) (Schoeb et al., 2010).
A nefrina é o componente principal das fendas de diafragma e conecta os pedicelos com um
padrão do tipo “zíper” (Figura 5C), conforme foi citado anteriormente (Figura 3) (Rodewald &
Karnovsky, 1974). O primeiro éxon do gene NPHS1 codifica o peptídeo sinal, os éxons 2 ao 20
codificam a região dos oito domínios do tipo imunoglobulina, os éxons 21 ao 23, o motivo de
fibronectina tipo III, o éxon 24, o domínio transmembrânico e a cauda citosólica é codificada
pelos éxons 25 a 29 (Schoeb et al., 2010).
A nefrina sofre N-glicosilação, uma modificação pós-tradução importante para seu
enovelamento e para sua correta localização na membrana dos podócitos (Yan et al., 2002).
Vários trabalhos sugerem que uma função importante da nefrina seja a manutenção tanto do
contato célula a célula quanto do citoesqueleto dos podócitos e demonstram que a nefrina se liga
à actina, provavelmente via CD2AP, sendo, portanto, um elo entre o citoesequeleto e as fendas
do diafragma (Lehtonen et al., 2002; Shih et al., 2001). Além da função estrutural, a nefrina
possui mecanismos de ação, tais como o reconhecimento de superfície, envolvimento na resposta
imune e sinalização celular (Benzing, 2004).
22
Figura 5. Esquemas do gene NPHS1 e da nefrina. A. Distribuição genômica dos 29 éxons do gene NPHS1. B.
Correspondência dos éxons aos domínios da nefrina: o éxon 1 codifica o peptídeo sinal, éxons 2 ao 20 codificam os
oito domínios do tipo imunoglobulina, éxons 22 e 23 codificam o domínio de fibronectina tipo 3, éxon 24 o domínio
transmembrana e os éxons 25 ao 29 codificam o domínio intracelular. C. Padrão do tipo “zíper” formado pelas
proteínas nefrinas inseridas nas membranas dos pedicelos. Adaptado de Tryggvason, 2006.
O domínio intracelular da nefrina possui vários resíduos de tirosina que são fosforilados
por proteínas pertencentes à família das quinases Src (Lahdenperä et al., 2003; Verma et al.,
2003; Qin et al., 2009; New et al., 2013). Assim, acredita-se que oligômeros de nefrina se
associem aos “rafts” de lipídios nas membranas das fendas do diafragma (Simons et al., 2001),
atuando como moléculas receptoras e transdutoras de sinal. A nefrina possui um papel
fundamental na manutenção da integridade das fendas de diafragma. Algumas evidências que
corroboram o papel crítico da nefrina na manutenção da permeabilidade seletiva dos glomérulos
são, por exemplo, a expressão máxima que pode ser observada nos pedicelos na região das
fendas do diafragma (Ruotsalainen et al., 1999); ou, o fato de que experimentos com
23
camundongos knockout de nefrina revelam o desenvolvimento de proteinúria intraútero e morte
nas primeiras 24 horas de vida (Putaala et al., 2001); outro estudo recente compara a expressão
normal da proteína nefrina e outras proteínas nas paredes dos capilares glomerulares de tecido
renal sem alterações, com uma redistribuição ou perda de expressão destas proteínas em
pacientes com proteinúria devido à síndrome nefrótica (figura 6) (Arias et al., 2009).
Figura 6. Microscopias dos capilares glomerulares mostrando a expressão da nefrina. À esquerda vê-se a
expressão nas paredes de um tecido normal; à direita, em pacientes com proteinúria nefrótica. Imunofluorescência
indireta para a cauda C-terminal da nefrina (Arias et al., 2009).
4.2 O gene NPHS2 e a proteína podocina
Em 1995 Fuchshuber et al. descreveram um grupo de pacientes com um tipo de síndrome
nefrótica familiar córtico-resistente (SNCR), e a descreveram primeiramente como SRN (OMIM
60099). Este tipo de SN se caracterizava por um modo de herança autossômica recessiva, idade
de desenvolvimento entre os 3 meses e os 5 anos de idade, resistência ao tratamento com
corticoesteroides, rápida progressão para insuficiência renal, ausência de recorrência após
transplante renal e ausência de outras doenças que não fossem renais. Histologicamente, lesões
24
mínimas eram observadas nos glomérulos em amostras de biópsias realizadas no início da
doença e em estágios mais avançados da doença havia o desenvolvimento de GESF. Esse grupo
mapeou um gene e o denominou SRN1 já que localizava-se no cromossomo 1, dentro de uma
região de 9 cM (Fuchshuber et al., 1995). O mesmo grupo, em 2000, mapeou, por clonagem
posicional, o gene NPHS2 (OMIM 604766) em 1q25-1q31, que ocupa 25 kb do DNA genômico
e é organizado em 8 éxons (figura 7A) (Boute et al., 2000). O gene NPHS2 codifica a proteína
podocina, uma proteína transmembrânica de aproximadamente 42 kDa, 383 aminoácidos e com
homologia à família de estomatina banda-7 (figura 7B). O domínio transmembrânico é único e
curto sendo que os domínios N e C-terminais estão localizados no citosol, formando uma
estrutura do tipo “hairpin”, uma característica das proteínas da família das estomatinas e
caveolinas (Boute et al., 2000). De maneira análoga ao gene NPHS1 da nefrina, a expressão do
gene NPHS2 nos rins localiza-se nos podócitos.
A podocina pertence a uma família com mais de 1300 proteínas conservadas ao longo da
evolução. Esta família de proteínas de membrana possui um domínio com aproximadamente 150
aminoácidos que apresentam similaridade às proteínas mitocondriais proibitinas (PHB) (figura
7B). Já foi demonstrado que a podocina recruta o colesterol nas fendas do diafragma através do
domínio PHB e de dois domínios palmitoil hidrofóbicos adjacentes ao domínio PHB. Desta
maneira, a podocina forma multímeros de proteínas com alto peso molecular que recrutam o
colesterol contribuindo para a criação de um supercomplexo de proteínas e lipídios (Huber et al.,
2007). Este supercomplexo é essencial para as interações que a podocina exerce. Huber et al.
demonstraram que a podocina interage com a cauda citoplasmática da nefrina e que esta
interação induz e potencializa a ação de sinalização celular exercida pela nefrina (Huber et al.,
2001; Huber et al., 2006).
25
Figura 7: Esquema do gene NPHS2 e da proteína podocina. (A) Distribuição genômica dos oito éxons do gene
NPHS2. (B). Proteína podocina inserida na bicamada lipídica. Os éxons 1 e 2 codificam o domínio N-terminal, o
éxon 3, a região transmembrana, éxons 4, 5, 6 e 7, o domínio PHB e o éxon 8, o domínio C-terminal. A seta cinza
mostra a posição em “hairpin” que a podocina é inserida na membrana, com os domínios N e C terminal
direcionados para o citoplasma. Adaptado de Relle et al., 2011.
Quack et al. propuseram um modelo funcional da dinâmica dos “rafts” de lipídios, no
qual resíduos de tirosina presentes na cauda citoplasmática da nefrina, como citado
anteriormente, são fosforilados por proteínas tirosinas quinases da família Src, sendo a
fosforilação importante para a regulação de algumas interações realizadas pela nefrina. Uma
destas interações é a interação da nefrina com a podocina (Quack et al., 2006). Neste modelo, a
fosforilação de um resíduo de tirosina na cauda citoplasmática da nefrina atua na regulação da
26
interação da podocina com a nefrina nas fendas de diafragma. Se a estrutura estiver intacta, com
as nefrinas inseridas corretamente na membrana plasmática formando interações homofílicas nas
fendas, a tirosina quinase Yes fosforila o sítio de tirosina citoplasmático e a podocina interage e
ancora corretamente a nefrina na membrana. Se a nefrina não estiver corretamente inserida na
membrana ou se houver alguma mutação nos sítios de fosforilação, outra proteína citoplasmática,
a β-arrestina, reconhece este sítio não fosforilado e internaliza a nefrina, não havendo a interação
da podocina com a nefrina (figura 8).
Figura 8: A interação podocina-nefrina dependente do status da fosforilação de resíduos de tirosina
citoplasmáticos da nefrina.
A. Estrutura intacta da interação nefrina-podocina nas fendas de diafragma. A
homodimerização de moléculas de nefrina nas fendas de diafragma leva à fosforilação da cauda C-terminal da
nefrina por proteínas quinases da família Src (por exemplo, a Yes). Consequentemente, a proteína podocina liga-se
ao sítio fosforilado, ancora a nefrina à membrana plasmática e aumenta a sinalização celular realizada pela nefrina.
B. Se por algum problema, por exemplo, a ausência de um sinal extracelular, não houver homodimerização, ocorre a
desfosforilação do sítio citoplasmático e a podocina não consegue mais interagir e ancorar a nefrina.
Consequentemente, a proteína citoplasmática β-arrestina liga-se ao sítio não fosforilado e internaliza a nefrina,
interrompendo a sinalização celular. Adaptado de Quack et al. 2006.
27
Ainda com relação à interação da podocina com a nefrina, Roselli et al. em 2004 fizeram
experimentos de colocalização da podocina normal, compararam com variantes mutantes em
células humanas embrionárias de rim (HEK293) e demonstraram que muitas das variantes
mutantes ficam retidas no retículo endoplasmático, não conseguindo atingir a membrana
plasmática das células, o que resulta na consequente alteração do tráfego de nefrina (Roselli et
al., 2004).
Outra interação estabelecida pela podocina e para a qual o supercomplexo lipídico é
importante, é com o canal iônico TRPC6. Já foi demonstrado que o TRPC6 é regulado pela
podocina nas fendas de diafragma, mais especificamente nos “rafts” de lipídios (Huber et al.,
2006). Experimentos recentes sugerem que a podocina possa influenciar na preferência pelo
mecanismo de ativação do TRPC6 (Anderson et al., 2013). Outra possível função para a
podocina é na interação entre o TRPC6 com a subunidade catalítica da NADPH oxidase, a
NOX2. Após a expressão de um shRNA (um tipo de RNAi) tendo a podocina como alvo a
interação de TRPC6 com NOX2 deixou de acontecer, apesar dos níveis destas duas proteínas
continuarem normais; o grupo que fez esse estudo sugere que a podocina seja necessária para a
interação entre TRPC6-NOX2 nos “rafts” de lipídios (Kim et al., 2013).
Portanto, a função da podocina vai além das funções de uma proteína estrutural; ela é
uma proteína estrutural que fornece um microambiente especial, rico em proteínas e lipídios,
necessário para o funcionamento e sinalização do complexo proteico das fendas de diafragma.
Recentemente uma isoforma menor da podocina foi caracterizada. Esta isoforma possui
315 aminoácidos, não apresenta o éxon 5 contendo uma parte do domínio PHB, é traduzida em
proteína, porém fica retida no retículo endoplasmático assim como muitas podocinas resultantes
de mutações. Esta diferença na localização celular sugere um papel fisiológico diferente para esta
28
isoforma, talvez no sequestro de componentes que possam interagir com lipídios e proteínas no
retículo endoplasmático (Völker et al., 2013).
4.3. O gene WT1 e o fator de transcrição WT1
O gene WT1 humano, localizado em 11p13, codifica um fator de transcrição do tipo dedo
de zinco que regula vários genes importantes para o desenvolvimento do sistema genitourinário
normal. A falta da sua função leva principalmente ao desenvolvimento do tumor de Wilms sendo,
portanto, considerado como gene supressor deste tipo de tumor (Call et al., 1990; Gessler et al.,
1990). O WT1 possui 10 éxons, com 50 kb aproximadamente. Os éxons 1 a 6 codificam uma
região rica em prolina e glutamina, que está envolvida na repressão ou ativação da transcrição
(Wang et al., 1993) e também para um domínio envolvido na homodimerização da proteína
(Moffett et al., 1995). Os éxons 7 a 10 codificam quatro dedos de zinco correspondentes ao
domínio de ligação ao DNA (figura 9A e B). Cada dedo de zinco é formado por 28 a 30
aminoácidos, incluindo um par de cisteínas e histidinas que são ligados a um átomo de zinco.
Um aminoácido básico, geralmente uma arginina, está localizado no topo do dedo de zinco.
A proteína WT1 pode variar de tamanho entre 52 a 54 kDa. Esta diferença é devido às
mais de 24 isoformas que podem ser formadas como resultado de diferentes combinações de
início de tradução, sítios de splicings alternativos e edição de RNA distintos. Quatro isoformas
principais podem ser formadas a partir de dois splicings alterativos: um deles pode incluir ou
excluir 17 aminoácidos codificados pelo éxon 5 (+ ou – 17 aa); o outro envolve dois sítios
doadores de splicing localizados no íntron 9, levando à presença ou ausência de uma trinca de
aminoácidos (lisina, treonina e serina, KTS) entre o terceiro e quarto dedos de zinco, designadas
como KTS (+) e KTS (-), respectivamente (Haber et al., 1991) (figura 9C). É necessária uma
29
correta proporção entre estas quatro isoformas para que o WT1 exerça sua função tanto durante a
nefrogênese quanto na vida adulta (Morrison, 2008).
Mutações no gene WT1 que levam ao desenvolvimento de proteinúria isolada na ausência
de anormalidades genitais e de tumor de Wilms podem ser explicados devido à função exercida
pela proteína WT1 e que é diferente da regulação tradicional da proliferação celular e da
regulação do desenvolvimento do trato genitourinário. O WT1 é expresso nos podócitos adultos
e aí influencia na arquitetura do citoesqueleto destas células.
Figura 9. Estrutura do gene WT1 e de sua proteína. A. Esquema dos 10 éxons do gene WT1. Os principais sítios
de splicing alternativos estão identificados. B. Fator de transcrição WT1, mostrando a região N-terminal com os
domínios ricos em prolina e glutamina e os quatro dedos de zinco, além dos sítios de splicing alternativo. C. Quatro
isoformas principais formadas a partir dos splicings identificados em A e B: um deles pode incluir ou excluir 17
aminoácidos codificados pelo éxon 5 (+ ou – 17 aa); o outro envolve dois sítios doadores de splicing localizados no
íntron 9, levando à presença ou ausência de uma trinca de aminoácidos (lisina, treonina e serina, KTS) entre o
terceiro e quarto dedos de zinco, designadas como KTS (+) e KTS (-), respectivamente.
30
A manutenção eficaz da estrutura dinâmica e complexa da BFG requer uma integração
entre múltiplas vias de sinalização entre as células endoteliais, mesangiais e os podócitos. Vários
estudos têm demonstrado que o WT1 interage potencialmente com genes alvo presentes nestas
células, que incluem genes que codificam fatores de transcrição (PAX2), fatores de crescimento
(TGF-β1 e PDGF-α) (Morrison et al., 2008), além de proteínas dos podócitos tais como a nefrina
e a podocalixina (Palmer et al., 2001; Guo et al., 2004; Morrison et al., 2008). Ainda, um estudo
proteômico realizado com podócitos apresentando o fenótipo de DDS mostraram que estes não
expressam proteínas associadas à arquitetura do citoesqueleto (cofilina, calpolina, hsp27 e
vinculina) e que os níveis totais dos filamentos de actina ficam reduzidos (Viney et al., 2007).
Em outro estudo foi demonstrada a regulação do filamento intermediário nestina pelo WT1
(Wagner et al., 2006). Estas observações confirmam a importância do estudo molecular do gene
WT1 em pacientes com proteinúria isolada ou associada a GESF, especialmente os que
apresentam histórico familiar.
5. MUTAÇÕES NOS GENES NPHS1, NPHS2 E WT1
Mutações no gene NPHS1 constituem a principal causa da SNC. Duas mutações
principais
são
responsáveis
pela
síndrome
na
população
finlandesa:
Finmajor
(p.Leu41Valfx*91), uma deleção de CT no éxon 2 que causa uma mutação frameshift e Finminor
(p.Arg1109*) uma mutação nonsense (CGA>TGA) que cria um stop codon prematuro no éxon
26. Estas mutações correspondem a 78% e 16% entre os alelos mutantes nos pacientes
finlandeses, respectivamente, o que deve ser causado por um provável efeito fundador (Lenkkeri
31
et
al.,
1999).
Em
outros
países,
mais
de
225
mutações
foram
identificadas
(http://www.hgmd.cf.ac.uk/ac/all.php), correspondendo a aproximadamente 40% das mutações
no gene NPHS1 como causa de SNC em crianças (Schoeb et al., 2010). Estas compreendem
mutações nonsense, missense, frameshifts causadas por inserções/deleções e mutações em região
de splicing (Lenkkeri et al., 1999; Beltcheva et al., 2001; Frishberg et al., 2007; Schoeb et al.,
2010) Ainda, são encontradas sempre em homozigose ou heterozigose composta e estão
presentes em diferentes éxons, em íntrons nas regiões de splicing e na região reguladora do gene
NPHS1 indicando que não há neste gene um hot-spot preferencial para ocorrência de mutações
(Figura 10) (Beltcheva et al., 2001; Koziell et al., 2002).
Figura 10. Distribuição de algumas mutações em todos éxons do gene NPHS1. As mutações ocorrem em todos
os éxons, o que resulta em alterações nos diversos domínios da proteína nefrina, mostrando que não há hot spot
preferencial para mutações nesta proteína. Adaptado de Schoeb et al., 2010.
Mutações em heterozigose composta no gene NPHS1 foram descritas em crianças com
desenvolvimento dos sintomas de SN após os três meses de idade (Philippe et al., 2008),
mudando a ideia inicial de que mutações no gene NPHS1 estariam relacionadas somente com
32
síndrome nefrótica congênita. O grupo de Philippe identificou estas mutações em um caso
familiar e em nove casos esporádicos e demonstrou que a maioria das variantes não atrapalhou o
tráfico da nefrina e a correta inserção na membrana plasmática, sendo sua função parcialmente
mantida. Estas mutações foram classificadas como “atenuadas” (do inglês, mild). O grupo de
Santín, uma pesquisadora da Espanha, também identificou mutações em heterozigose composta
no gene NPHS1 em pacientes com SNCR e GESF, com idade de início variando entre os oito
meses e os sete anos de idade (Santín et al., 2009). Além disto, esse grupo descreveu um
paciente com mutações no gene NPHS1 que desenvolveu GESF com 27 anos de idade e
respondeu parcialmente ao tratamento com imunossupressores.
A presença de pelo menos uma mutação “atenuada” pode explicar o início tardio e o
desenvolvimento de uma forma menos grave da doença entre estes casos. Desta maneira,
mutações “atenuadas” podem ser assim designadas quando são mutações missense que permitem
que a função de tráfego da nefrina na célula continue normalmente, ou quando são mutações que
criam sítios de splicing alternativos permitindo eventualmente que em uma parcela dos
transcritos haja splicing correto, resultando em uma proporção de nefrina que mantenha sua
função normal; ainda, quando mutações que levam ao truncamento da proteína como as de
frameshifts acontecerem somente bem na extremidade C-terminal. Assim, estas mutações ditas
“atenuadas” ainda permitem que uma função parcial da proteína nefrina seja mantida.
Com relação ao gene NPHS2, mutações foram identificadas como causa de SNC entre
crianças da Europa Central o que indica certa variabilidade na origem genética da doença e
reforça a necessidade da análise não só do gene NPHS1, mas também do gene NPHS2 para se
realizar o correto diagnóstico molecular nestes casos (Weber et al., 2004).
33
As causas genéticas nos casos de SN infantil, na Infância e Juvenil têm sido atribuídas na
última década a mutações em diversos genes, o que veio a explicar muitos casos idiopáticos,
principalmente os córtico-resistentes. Os principais genes envolvidos na forma SNCR de origem
genética são os genes NPHS2 e o WT1, seguidos pelo gene NPHS1.
A maioria dos casos de SNCR que têm origem genética apresentam mutações no gene
NPHS2, sendo a taxa de mutações neste gene de 40% em casos familiares e de 6 a 17% em casos
esporádicos (Karle et al., 2002; Caridi et al., 2003; Hinkes et al., 2008; Benoit et al., 2010; Ruf
et al., 2004; Weber et al., 2004). Até o momento existem mais de 120 mutações deletérias já
descritas (http://www.hgmd.cf.ac.uk/ac/all.php), incluindo mutações nonsense e frameshift,
variantes em região de splicing e alterações missense, distribuídas em todos os éxons do gene.
Ainda é difícil se estabelecer uma correlação direta entre o genótipo e o fenótipo, porém há
estudos que mostram que pacientes com mutações frameshift, mutações nonsense e a mutação
missense em homozigose p.R138Q manifestam os sintomas da doença mais precocemente
(Caridi et al., 2003; Weber et al., 2004). Acredita-se que a mutação p.R138Q seja de origem
fundadora no gene NPHS2. Localizada no éxon 3, foi originalmente descrita por Boute et al.
(Boute et al., 2000) em pacientes com SNCR familiar. A proteína resultante da mutação
p.R138Q é retida no retículo endoplasmático, o que faz com que não recrute a nefrina nos “rafts”
de lipídios. Este fato é considerado como uma possível justificativa para a gravidade do fenótipo
associado à homozigose desta mutação (Huber et al., 2003; Roselli et al., 2004). A figura 11
mostra a distribuição e a correlação entre algumas mutações e seu efeito funcional na proteína
podocina.
É importante ressaltar também uma variante muito frequente, a p.R229Q, que, quando
associada a outra mutação no gene NPHS2, leva ao desenvolvimento tardio de Síndrome
34
Nefrótica (Tsukaguchi et al., 2002; Machuca et al., 2009). A análise de alelos do gene NPHS2
em 91 indivíduos com GESF indicou que 6% dos alelos carregavam a variante Gln229 da
proteína (Tsukaguchi et al., 2002). Estudos in vitro indicaram que a proteína mutante resultante
da alteração p.R229Q apresentou uma diminuição na ligação da podocina à nefrina, o que pode
justificar seu papel deletério. No entanto, frequências que variam entre 1,6 e 3,6% da variante
entre indivíduos normais fazem com que o valor aumentado nos indivíduos com GESF não tenha
significância estatística (Tsukaguchi et al., 2002).
Figura 11. Estrutura da podocina e distribuição das mutações. Estrutura da proteína podocina mostrando sua
forma em “hairpin”, domínio transmembrana, domínios N-terminal e C-terminal citosólicos. Mutações em
vermelho: já foram relacionadas a um fenótipo mais grave, sendo que a podocina fica retida no retículo
endoplasmático. Mutações em azul: correspondem a um fenótipo menos grave e a podocina é inserida na membrana
plasmática. Em verde ressalta-se a variante p.R229Q, que associada a outra mutação está envolvida com
desenvolvimento tardio de SN. Em cinza: mutações com função biológica desconhecida. Adaptado de Weber et al.,
2004.
35
Outros estudos, entre eles destacam-se um brasileiro e um europeu (Pereira et al., 2004;
Di Duca et al., 2006), mostraram que diferentes alterações no gene NPHS2 estão associadas a
graus variados de proteinúria e a diferentes doenças renais. No estudo brasileiro foi relatado que
indivíduos com microalbuminúria apresentavam uma maior frequência do alelo p.R229Q, em
heterozigose (Pereira et al., 2004). No outro estudo, Di Duca et al. (2006) mostraram que
variações funcionais na região reguladora do gene NPHS2 estão associadas com proteinúria em
dois grupos distintos com doença renal na Europa: um grupo apresenta Síndrome Nefrótica e o
outro, Nefropatia IgA (IgAN). Em um estudo realizado no norte da China sugere-se que
determinados polimorfismos e haplótipos do gene NPHS2 estejam relacionados com uma maior
susceptibilidade genética e também com o grau de proteinúria em doenças renais tais como a
Doença de Lesão Mínima (DLM) e diferentes subgrupos de IgAN (Zhu et al., 2009).
Mutações constitutivas em heterozigose no gene WT1 podem dar origem a diferentes
síndromes, tais como Síndrome de Denys-Drash (DDS, OMIM 194080) (Pelletier et al., 1991) e
Síndrome de Frasier (FS, OMIM 136680) (Barbaux et al., 1997; Demmer et al., 1999; Denamur
et al., 1999). Na grande maioria dos casos as mutações estão localizadas no hot-spot preferencial
de mutações que compreende os éxons 8 e 9 e o íntron 9. Quase 80% das mutações que causam
DDS são mutações missense localizadas nos éxons 8 e 9 que codificam os dedos de zinco 3 e 4,
respectivamente (Bruening et al., 1992). Estas mutações parecem atuar de modo dominante
negativo, impedindo a homodimerização e interferindo na atividade do WT1 nas células (Reddy
et al., 1995). A DDS é caracterizada pela tríade de genitália ambígua, glomerulopatia progressiva
com histologia de esclerose mesangial difusa (DMS) e alto risco de desenvolver tumor de Wilms
(WT) (Denys, 1967; Drash, 1970). No caso da FS, geralmente os pacientes apresentam mutações
que afetam o seg sítio doador de splicing do íntron 9 (Barbaux et al., 1997). O resultado é um
36
decréscimo da isoforma KTS (+) e uma reversão da proporção normal que é de 2 KTS (+): 1
KTS (-), o que leva ao desenvolvimento da doença (Klamt et al., 1998). A definição clássica
inclui pacientes com cariótipo 46,XY e genitália externa normal, glomérulo esclerose focal e
segmentar e alto risco de desenvolver gonadoblastoma (Frasier, 1964). No entanto, há diversos
relatos na literatura de mulheres com cariótipo 46,XX, com desenvolvimento genital e puberdade
normais e com glomerulopatia isolada, que podem apresentar mutações (missense e de splicing)
no gene WT1 (Demmer et al., 1999; Tsuda et al., 1999; Loirat et al., 2003; Ruf, et al., 2004;
Mucha, et al., 2006).
Jeanpierre et al. (1998) descreveram quatro pacientes com fenótipo de esclerose
mesangial difusa, cariótipo normal e nenhum sinal de DDS, Tumor de Wilms ou anormalidades
extrarrenais e nos quais foram identificadas mutações no gene WT1. Características clínicas
similares também foram descritas por outro grupo (Schumacher et al., 1998) em duas meninas
com cariótipo normal e SNC, que também apresentaram mutações no gene WT1. Assim, esses
estudos sugerem que a pesquisa de mutações no gene WT1 deve ser realizada em crianças com
desenvolvimento precoce de SN, principalmente em casos com padrão histológico de esclerose
mesangial difusa (EMD), pois estas crianças têm um alto risco de desenvolver Tumor de Wilms.
Estima-se que mutações dominantes de novo no gene WT1 sejam encontradas nos éxons 8
e 9 (hot spot para mutações) em aproximadamente 9% dos casos esporádicos de SNCR com
manifestação precoce e tardia (Mucha et al., 2006). Em jovens do sexo feminino menores de 18
anos a prevalência aumenta, sendo de 12% (Aucella et al., 2006). Atenção especial deve ser dada
a este grupo de pacientes, com screening do WT1, juntamente com o gene NPHS2.
37
38
JUSTIFICATIVA
39
40
JUSTIFICATIVA
Desde o final da década de 90 mutações em diversos genes que codificam estruturas da
barreira de filtração glomerular vêm sendo identificadas em pacientes com Síndrome Nefrótica.
No entanto, estes estudos abrangem principalmente casuísticas provenientes dos Estados Unidos,
Europa Ocidental, alguns países da Europa Oriental e da Ásia, não havendo descrições de
frequência e distribuição de mutações em crianças brasileiras. Assim, a proposta inicial deste
trabalho foi a triagem de mutações nos genes NPHS1, NPHS2 e WT1 em pacientes pediátricos,
para uma primeira avaliação das bases genéticas da doença no Brasil. No entanto, o escopo desta
tese abrange, além da identificação e registro de mutações, a caracterização de cada alteração
identificada com estudos de predição in silico, visando formular hipóteses para correlação das
mesmas com suas respectivas funções biológicas e procurando estabelecer uma correspondência
com os quadros clínicos apresentados.
Este estudo traz benefícios tanto para os pacientes como para seus familiares, pois a
partir da identificação de mutações em pacientes córtico-resistentes torna-se possível a
programação da retirada do tratamento com esteroides e com outros imunossupressores que não
esteja sendo efetivo; outro benefício é a possibilidade de um transplante com baixo ou nenhum
risco de rejeição que pode ser, inclusive, a partir de doador vivo; ainda, se necessário, pode-se
programar o diagnóstico pré natal para a família do paciente. Além da contribuição direta no
diagnóstico, este estudo traz avanços científicos para a comunidade de nefropediatras e
pesquisadores da área, pois pela primeira vez temos uma abordagem genética ampla em um
grupo de pacientes pediátricos brasileiros com SN e proteinúria isolada.
41
42
OBJETIVOS
43
44
OBJETIVOS
GERAL
O objetivo deste estudo foi investigar alterações moleculares nos genes NPHS1, NPHS2 e
WT1, em casos de SNC, SN infantil, na infância e juvenil e proteinúria isolada no sentido de
contribuir para uma análise crítica tanto da correlação das alterações moleculares com suas
respectivas funções biológicas quanto da associação dos achados moleculares com as
manifestações clínicas da SN em uma casuística brasileira.
ESPECÍFICOS
Realizar a análise molecular do gene NPHS1 nos casos de SNC. Para casos com mutação
em heterozigose, realizar a análise dos genes NPHS2 e WT1.
Realizar a análise molecular dos genes NPHS2 e WT1 em pacientes com SN infantil, na
infância e juvenil e proteinúria isolada. Para casos com mutação em heterozigose, realizar
a análise do gene NPHS1.
Realizar análise em grupo controle saudável para as alterações que forem identificadas e
ainda não estiverem depositadas nos bancos de dados.
Realizar estudos de predição in silico para as alterações identificadas, com o objetivo de
formular hipóteses da correlação das mesmas com as respectivas funções biológicas.
Estimar a frequência das mutações nestes genes em cada uma das formas da doença em
nossa casuística, fazendo-se uma correlação genótipo-fenótipo.
45
46
CASUÍSTICA E MÉTODOS
47
48
CASUÍSTICA E MÉTODOS
1.CASUÍSTICA
A casuística deste trabalho foi composta por 150 crianças e adolescentes, com idades de
início de desenvolvimento da Síndrome Nefrótica entre 0 e 18 anos e que foram encaminhados
durante o período da tese. Também compuseram a casuística sete materiais provenientes de
biópsias renais fixadas em bloco de parafina de pacientes que já foram a óbito com SNC. Três
dos 150 pacientes são casos não aparentados com SN Congênita. Os outros 147 pacientes, sendo
134 não aparentados, apresentam SN infantil, na infância, juvenil e proteinúria isolada.
A tabela 5 mostra a procedência dos pacientes que compuseram a casuística deste
trabalho,
o diagnóstico clínico e quais genes foram triados para cada grupo. Na tabela 6
encontram-se alguns dados clínicos dos 150 pacientes.
Também foi realizada, quando possível,a análise molecular dos pais de alguns pacientes
nos quais foram identificadas alterações, para verificar a segregação alélica na família. Assim,
foram analisados os éxons 8-9 do gene WT1 da mãe da paciente 2 e do pai da paciente 3; éxons
2 e 4 do gene NPHS1 dos pais do paciente 150; éxons 10 e 24 do gene NPHS1 e éxon 5 do gene
NPHS2 dos pais da paciente 6; éxons 5 e 7 do gene NPHS2 dos pais da paciente 10; éxons 5 e 8
do gene NPHS2 da mãe da paciente 81 e éxon 5 do gene NPHS2 do pai do paciente 87. Para
alterações não depositadas no banco de dados foi realizado o estudo em grupo controle saudável.
Para as substituições missense que foram identificadas, seja no gene NPHS2 ou no gene NPHS1,
foram analisados de 90 a 110 controles saudáveis e para as alterações na região promotora do
gene NPHS2 foram analisados 278 controles.
49
Tabela 5: Composição da casuística e genes triados para cada grupo clínico.
TOTAL DE PACIENTES: 150
Procedência
Unidade de Nefrologia Pediátrica do
Departamento de Pediatria do Hospital de
Clínicas da UNICAMP
Diagnóstico Clínico
n° Pacientes
SN infantil
1
SN infância e juvenil
123
Proteinúria Isolada
9
Gene triado
133
Setor de Nefrologia do Departamento de
Pediatria da Universidade Federal de São
Paulo
SN infância
4
Hospital Municipal José de Carvalho
Florence São José dos Campos
SN infância
6
Hospital Estadual da Criança, Feira de
Santana, BA
SN infância
1
Hospital Regional de Mato Grosso do Sul,
Campo Grande, MS
SN infância
1
Rio de Janeiro, Clínica Particular
SN infância
2
Hospital Universitário de Brasília, Brasília,
DF
SNC
1
Santa Casa de Belo Horizonte, Belo
Horizonte, MG
SNC
1
Hospital Universitário Pedro Ernesto,
Universidade Estaual do Rio de Janeiro, RJ
SNC
1
NPHS2 e
éxons 8-9
WT1
NPHS1 em
caso de
heterozigose
simples no
NPHS2
NPHS1
MATERIAL FIXADO EM BLOCO DE PARAFINA: 7
Procedência
Diagnóstico Clínico
n° Pacientes
Gene triado
Departamento. Anatomia Patológica da FCM
- UNICAMP
SNC
7
NPHS1
50
Tabela 6: Dados clínicos dos 150 pacientes.
N° paciente
Gêneroa
Raçab
Familiar /
esporádicoc
Idade
iníciod
Resposta terapia
corticóidee
Biópsiaf
Evolução para DRCg
classe V / idade
Transplante
renal
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
F
F
F
F
F
F
M
F
F
F
M
F
M
M
F
M
M
F
M
M
F
M
F
M
M
F
M
M
M
M
M
M
F
M
M
B
B
B
Pa
B
B
B
B
B
B
P
B
B
B
P
B
B
B
B
B
B
B
B
B
B
B
B
B
B
Pa
B
B
B
B
B
esp
fam
fam
fam
esp
esp
fam
esp
fam
fam
esp
esp
esp
esp
fam
esp
esp
esp
esp
fam
esp
esp
esp
fam
esp
esp
esp
fam
esp
esp
fam
esp
esp
fam
esp
4a5m
2a
16 a
5a
4a
9a
7a
3a9m
1a
12 a
1a6m
7m
5a8m
2a
3a
6a9m
2a
3a
2a4m
2a3m
4 a 10 m
5a7m
12 a
2 a 10 m
2a
1a
1a9m
4a
5 a 10 m
2a
1a8m
2a
4a3m
1a3m
nd
CR
CR
Prot. nefr.
Prot. nefr.
CS,RF
CS,RF
CS,RF
CS,RF
CS
CR
CS,RF
CS
CS,RF
CS,RF
CS,RF
CS
CS,RF
CS
CS
CS,RF
CS,RF
CS,RF
CR
CS,RF
CS,RF
CS,RF
CS
Prot.não nefr.
CS,RF
CS
CS,RF
CS,RF
CS,RF
CS,RF
nd
PM
GESF
PM
N
N
N
N
N
PM
GESF
N
PM
CO/LM/PM /GESF
N
N
N
N
N
CO/LM/PM /GESF
N
N
N
N
N
N
LM
N
N
N
N
N
N
N
PM
nd
S /10 a
S /10 a
N
N
N
N
N
N
N
S /15 a
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
nd
S
S
N
N
N
N
N
N
N
S
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
N
nd
51
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
F
F
M
M
M
F
F
M
M
M
F
M
M
F
F
F
M
M
M
M
F
F
F
F
M
F
F
M
F
F
M
M
F
M
M
F
F
M
M
F
B
B
B
B
B
Pa
B
B
B
B
B
B
B
B
B
B
B
B
Pa
B
B
B
A
B
B
B
B
B
B
B
B
nd
P
P
B
B
B
B
B
P
esp
esp
esp
esp
esp
fam
esp
fam
esp
esp
esp
esp
esp
esp
esp
esp
esp
esp
esp
esp
esp
esp
fam
esp
esp
esp
fam
fam
esp
esp
esp
esp
fam
fam
esp
fam
fam
fam
fam
esp
4 a 11 m
5a
2a5m
7a
1 a 10 m
1a8m
1 a 11 m
7a7m
4a
1a4m
7a
7m
1a
2a1m
3a3m
2a5m
11 a
2 anos
3a5m
2a8m
3 a 11 m
9a
13 a
1a9m
13 a
2a8m
4a
9a
2a
2 anos
2a3m
nd
5a
nd
3a
2a
2a2m
12 a
5a
3 anos
CS
CS
CS
CS,RF
CS,RF
CR
CS
CS
CS,RF
CS,RF
CS,RF
CS,RF
CS,RF
CS,RF
CS,RF
CS,RF
CS
CS
CS,RF
CS
CS
CS
CS,RF
CS
CS
CS,RF
CS,RF
nd
CS,RF
CS
CS,RF
CS,RF
CS
nd
CS,RF
CS,RF
CS,RF
Prot. Nefr.
CR
CS,RF
N
GESF
N
GESF
N
GESF
N
GESF
CO/ LM/PM/GESF
N
CO/LM/PM/GESF
nd
nd
N
N
N
N
N
N
N
N
N
CO/ LM/PM/GESF
N
CO/ LM/PM/GESF
GESF
N
N
N
N
N
nd
N
nd
GESF
N
N
N
GESF
GESF
N
N
N
N
N
S / 10 a
N
N
N
N
N
N
S/2a
N
N
N
N
N
N
N
N
N
N
N
N
S/ 13 a
N
N
N
N
N
nd
N
nd
S/N
N
N
S/S/ 4 a
N
N
N
N
N
S
N
N
N
N
N
N
S
N
N
N
N
N
N
N
N
N
N
N
N
S
N
N
N
N
N
nd
N
nd
S
N
N
N
S
S
52
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
M
M
F
M
M
F
M
F
F
M
M
M
M
M
F
F
M
F
F
M
F
M
M
M
M
F
M
M
M
M
M
F
M
M
M
M
F
M
M
F
B
nd
nd
B
B
B
nd
B
B
B
B
B
B
P
Pa
Pa
Pa
B
B
nd
B
B
B
B
B
B
B
nd
nd
nd
B
B
B
B
B
B
B
B
B
nd
fam
nd
esp
fam
esp
esp
esp
esp
esp
esp
esp
fam
esp
esp
fam
fam
fam
esp
esp
nd
esp
esp
esp
esp
esp
esp
fam
nd
nd
nd
esp
esp
esp
esp
esp
esp
esp
esp
esp
esp
2 anos
2a
nd
4a8m
2a6m
2a2m
4a
6a
3a6m
6 a 11 m
5a6m
16 a
2a4m
2a
4a
11 a
7a
5a
10 m
nd
1a2m
13 a
2a
1a9m
4a
8a
3a
nd
nd
nd
1a7m
2a
2a
4a
1a7m
2a
2a6m
2a6m
2a3m
1 a 11 m
CS,RF
CR
CS
CS
CS,RF
CR
CR
nd
CS,RF
CR
CS,RF
CR
CS,RF
CS
CR
Prot. nefr.
Prot. não nefr.
CS,RF
nd
CR
CS
Prot.nefr.
CS
CS,RF
Prot. nefr.
CS,RF
CS,RF
nd
nd
nd
CS,RF
nd
CS,RF
CS,RF
CS,RF
CS,RF
CS,RF
CS,RF
CS,RF
CS
N
nd
nd
N
LM
PMD
CO/LM/PM/GESF
nd
N
GESF
CO/LM/PM/GESF
GESF
N
N
GESF
N
N
N
finlandesa
GESF
N
PM
N
N
GESF
N
N
nd
nd
nd
N
nd
N
N
N
N
N
GESF
N
N
N
nd
nd
N
N
N
N
nd
N
S/6a
N
S / 18 a
N
N
S
N
N
N
nd
nd
N
N
N
S/7a
N
N
nd
nd
nd
N
nd
N
N
N
N
N
N
N
N
nd
nd
N
N
N
N
nd
N
N
N
S
N
N
S
N
N
N
nd
nd
N
N
N
N
N
N
nd
nd
nd
N
nd
N
N
N
N
N
N
N
53
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
M
F
F
M
M
M
M
F
F
F
M
F
M
F
M
F
F
F
M
F
F
F
M
M
F
F
M
F
M
F
M
M
F
M
M
B
nd
B
B
B
B
B
B
B
Pa
P
B
nd
nd
nd
nd
nd
B
B
B
P
B
B
P
B
B
nd
nd
nd
nd
nd
nd
B
nd
nd
esp
nd
esp
esp
esp
esp
esp
esp
esp
esp
esp
esp
nd
nd
nd
fam
fam
esp
esp
esp
esp
nd
fam
esp
esp
esp
nd
nd
nd
nd
fam
fam
fam
nd
nd
10 a 2 m
3a
2a6m
3a4m
3a
4a5m
2a
4a
10 a
1a
11 a
3m
nd
nd
3m
1a2m
2a
3a
2a
8 a5 m
9a
12 a
4a
3a
2a
1a
5d
nd
7m
nd
nd
14 a
4a
15 d
CS,RF
CS,RF
CS,RF
CS,RF
CR
CS
CS
CS
CS
CS
CR
CS,RF
nd
nd
nd
CR
CR
CS,RF
CS,RF
CR
CR
CR
CR
CR
CR
CR
nd
nd
nd
nd
CR
CR
Prot. nefr.
CS,RF
nd
N
N
N
CO/LM/PM/GESF
CO/LM/PM/GESF
N
N
N
N
N
PMg
N
GESF
nd
nd
nd
GESF
LM
N
GESF
GESF
nd
GESF
LM
GESF
nd
finlandesa
nd
GESF
GESF
GESF
N
GESF
N
N
N
N
N
N
N
N
N
N
N
nd
nd
nd
nd
nd
N
N
S/ 2 anos
S / 13 a 4 m
Nh
Nh
Nh
nd
Nh
nd
nd
nd
nd
nd
nd
N
nd
nd
N
N
N
N
N
N
N
N
N
N
N
nd
nd
nd
nd
nd
N
N
N
S
nd
nd
nd
nd
nd
nd
nd
nd
nd
nd
N
nd
nd
a
M: masculino; F: feminino. bB: branca; Pa: parda; A: amarelo; P: preta. cFam: familiar; Esp: esporádico; da: anos; m: meses d: dias; eCR: córtico resistente;
CS,RF: córtico sensível com recidiva frequente; CS: córtico sensível; Prot. Nefr: proteinúria nefrótica; fLM: lesão mínima; PM: proliferação mesangial; GESF:
glomérulo esclerose focal e segmentar; CO: complexo; N: não; S: sim; gesclerose focal em 1 glomérulo; hem uso de ciclosporina.
54
O gráfico 1 mostra a classificação quanto à resposta ao tratamento com corticoesteroides
dos 138 pacientes (137 com SN infância e juvenil e um com SNI). Todos tiveram idade de início
da doença entre os três meses e os 18 anos de idade.
Gráfico 1: Distribuição dos pacientes de acordo com a resposta ao tratamento.
55
2. MÉTODOS
Os critérios de inclusão dos pacientes deste trabalho foram o diagnóstico da
Síndrome Nefrótica ou da proteinúria isolada e a idade de início da doença, que deveria
ser entre zero e 18 anos, não havendo uma seleção preferencial pelo quadro clínico de
resposta ao tratamento com corticoides.
Na figura 12 estão ilustrados os principais passos da metodologia.
Figura 12: Fluxograma dos métodos utilizados neste trabalho.
56
2.1 Obtenção das amostras
As amostras de DNA genômico dos pacientes foram obtidas a partir de 10 a 20 mL de
sangue total periférico, que foram coletados em tubos cônicos contendo 10 gotas de
anticoagulante ácido etilenodiaminotetracético (EDTA) 0,5 M pH 8,0.
2.2 Extração de DNA genômico
2.2.1 A partir de sangue total
Para a extração de DNA foi empregada metodologia de lise com Proteinase K
(Boehringer Mannhein, Germany) padronizada no laboratório de Genética Molecular Humana –
CBMEG (De Araújo, 1996).
As soluções empregadas no protocolo foram:
Solução A:
[ ] final
Solução B (estoque 2x) [ ] final
5 mM
EDTA
20 mM
Solução B
Sacarose
0,32 M
NaCl
20 mM
SDS
Tris HCl pH8,0
10 mM
Tris HCl pH8,0
20 mM
TritonX-100
1%
MgCl2
Solução C:
Proteinase K
[ ] final
0,5x
5%
1 mg/ml
As etapas da extração estão listadas abaixo:
-
Para lise das hemácias foi adicionada solução A ao sangue coletado até completar o
volume de 50 mL permanecendo o homogeneizado em gelo por 30 min;
-
As amostras foram centrifugadas a 2500 rpm por 15 min a 4°C (Centrífuga Eppendorf®
5804R);
57
-
O sobrenadante foi descartado e o precipitado (pellet) ressuspendido em 35 mL de
solução A. Esta etapa foi repetida até o pellet ficar livre de hemácias lisadas;
-
A seguir, o pellet foi ressuspendido em solução B diluída para 1x;
-
Acrescentou-se 250 μL de solução C recém preparada, contendo proteinase K;
-
O pellet com as soluções permaneceu em incubação a 37°C overnight (aproximadamente
18 horas).
-
No dia seguinte, para a extração de DNA dos leucócitos foram adicionados 1 mL de TE
1x (Tris-HCl 10 mM pH 8,0; EDTA 1 mM pH 8,0) e 1,25 mL de fenol saturado com Tris-HCl
10 mM pH 8,0;
-
A mistura foi homogeneizada por inversão lenta do tubo durante 5 min;
-
Os tubos foram centrifugados a 2500 rpm por 15 min à temperatura ambiente;
-
Após centrifugação, procedeu-se com a recuperação da fase superior, a fase aquosa;
-
A esta fase aquosa que foi retirada e colocada em outro tubo, acrescentou-se igual
volume de fenol:clorofórmio:álcool isoamílico na proporção de 25:24:1, v:v:v;
-
Os tubos foram homogeneizados por mais 5 min e levados para centrifugação por mais
15 min a 2500 rpm para separação e recuperação da fase aquosa;
-
O procedimento foi repetido com clorofórmio:álcool isoamílico (24:1,v:v);
-
Para precipitação do DNA, foi acrescentado 0,1 volume de acetato de sódio 3 M pH 5,5 e
2,5 volumes de etanol absoluto gelado;
-
O DNA precipitado foi recuperado com auxílio de uma haste plástica esterilizada e
lavado com etanol 70% para retirada do excesso de sal, antes de ser ressuspendido em TE 1x
(200 a 500 μL).
58
2.2.2 A partir de tecido fixado em parafina
O método para a obtenção de DNA de tecido fixado com parafina foi realizado seguindose o protocolo já padronizado e previamente descrito (Maciel-Guerra et al., 2008).
- Inicialmente 25 mg de cada amostra foi tratada com 1200 µl de xileno (Synth Chemical Co.,
São Paulo, Brasil).
- As amostras foram vigorosamente homogeneizadas no vórtex e, em seguida, centrifugadas
por 5 min a 3000 rpm, à temperatura ambiente (Centrífuga Eppendorf® 5804R).
- Para remover o resíduo de xileno, foram efetuadas duas lavagens com 1200 µl de etanol
100% (Merck Chemical, Darmstadt, Alemanha).
- Entre uma lavagem e a outra, as amostras foram centrifugadas por 5 min a 3000 rpm, à
temperatura ambiente, descartando-se o sobrenadante a cada lavagem.
- Para eliminar o etanol residual as amostras foram incubadas por 10 min a 37°C.
- Para a extração do DNA foi então utilizado o DNeasy Blood and Tissue Kit (QIAGEN,
Hilden, Alemanha), de acordo com instruções do fabricante.
2.3 Quantificação do DNA genômico
A concentração do DNA foi obtida através de leitura de absorbância óptica a 260 ηm, em
espectrofotômetro Nanodrop 8000 - Multi-Sample Micro-Volume UV-Vis Spectrophotometer
(Thermo Scientific, USA). Para calibrar o aparelho (branco) foi utilizado TE 1x, pH 8,0. A
leitura da absorbância foi realizada com 1 μL de cada amostra. A pureza do DNA foi verificada
através da proporção de leitura 260/280. Para o DNA ser considerado puro, livre de
contaminação de proteínas, fenol ou qualquer outro contaminante que possa absorver luz UV a
59
280 ηm, a relação entre a abosrbância de 260 ηm e a de 280 ηm deve ser de aproximadamente
1,8.
2.4 Integridade do DNA genômico
A verificação da integridade do DNA genômico extraído foi realizada por meio de
eletroforese em gel de agarose 0,8% em TBE 1x (TBE 10x: ácido bórico 55 g; Tris 108 g; EDTA
0,2 M pH 8,0 100 mL; H20 destilada q.s.p. 1000 mL) preparado seg protocolo descrito por
Sambrook et al. (Sambrook et al., 1989). A amostra foi aplicada no gel juntamente com tampão
de eletroforese (0,25% de azul de bromofenol; 50% glicose) na razão de 6:1. As condições de
eletroforese variaram entre 90 a 110 V. O marcador de peso molecular utilizado foi DNA ladder
de 1 kb, 1 kb plus (Invitrogen Corporation, Estados Unidos), em concentração de 0,15 μg/μL. O
gel foi imerso em solução diluída de brometo de etídio (0,5 µL/mL de água destilada) durante 15
min, sendo visualizado em transluminador de luz ultravioleta e fotografado utilizando uma
câmera digital acoplada a um computador (Kodak Electrophoresis Documentation and Analysis
System-EDAS, Kodak Digital Science, Estados Unidos) ou pelo sistema MiniBis Pro®
(BioSystematica, Reino Unido).
2.5 Amplificação dos genes por reação de polimerase em cadeia – PCR
A amplificação foi realizada com primers específicos para cada um dos genes. Estes primers
foram desenhados não só para amplificação dos éxons, mas também se estendendo por pelo
menos 50 pb da região intrônica nos limites dos éxons, para verificação da região de splicing.
60
Também foram desenhados primers para as regiões não traduzidas 5` e 3` (5`UTR e 3`UTR). Os
pares de primers utilizados para os genes NPHS2, NPHS1 e WT1 estão especificados nas tabelas
7, 8 e 9, respectivamente. Para o gene NPHS1, como o DNA a ser amplificado (material de
biópsia antigo fixado em parafina) apresentava-se muito degradado, houve a preocupação de
desenhar primers que amplificassem regiões curtas. Portanto, apesar do gene possuir 29 éxons,
foram elaborados 32 pares de primers.
Tabela 7 – Primers para amplificação e sequenciamento do gene NPHS2.
Fragmento
Primer sense
5`
3`
Primer antissense
5`
3`
Taa
o
Tamanho
( C)
(pb)
5`s-1.2as
GTAGGGAGGAGAGAAAGGCATC
TGCCCTCTTGTTCTCCTTGTG
54
750
1s-1as
GGGCGCAGTCCACAGCTC
GGCCCCGAGACCAGTATATAGTG
59
717
2s-2as
GCCCTGTGAACTCTGACTACTCTG
GGAGCACCAGGAAGGGAATG
58,4
488
3s-3as
ATCAAAATTCTGTCTATGGGTTC
TGGCATGTGGGCTCTCTG
54
528
4s-4as
CCATGACTAAAAGGACCACACAG
CATTCCCTAGATTGCCTTTGC
54
515
5s-5as
TTAAATAAAGGGTAGGCCAACTCC
CCTAAGGGATGGAACTGGCC
56
441
6s-6as
CCTCTTGGGGTAACATTCACAG
TGCGCCTGGCCTAAAATG
55,5
615
7s-7as
CAAAACCTGCTGTGCTGATAATG
GAGGGATTGATGTGTGTGGAGG
56
636
8s-8.1as
TTCTATGCTTAACCGTGCTTGC
GCTGTTTCCCATAATTGCTCTGb
54
1126
8.1s-8as
ATGTTATAGGAAGGATGGGGC b
CTCCCTCAGATTTTAAGCCAC
a
A Ta é a temperatura média entre as Tm de cada primer nos pares de PCR.
O éxon 8 é muito longo (tamanho total 1126pb) sendo necessários primers internos para sequenciamento.
b
61
Tabela 8 – Primers para amplificação e sequenciamento do gene NPHS1.
Fragmento
Primer sense
5`
3`
Primer antissense
5`
3`
Taa
Tamanho
(°C)
(pb)
58,5
292
5`s – 1.1as
CCTGGCCTGCTGGACTCTG
GCCAGGTTGATCTCAGACTCTTT
1.1s – 1.2as
TTAGACAAGGAGAGAAAGATGG
GTTACTCTCCTCCCTTTCTCG
53
304
ACAGGGAAGAGGGGAAGAGG
GGGAAGGTAAGTGGGAAATGG
57
285
2s-2as
GATCCCAGCCTTGTACCCAG
GCTTCCGCTGGTGGCTGA
59
357
3s-3as
GACCCTCAGCCACCAGCG
GCACTGAGAAGGACTTGAAGATTG
58,5
316
4s-4as
CCAGCCTCTCCTCTCCCAG
ATCTTTTCTGGGGCCCTTAG
54
324
5s-5as
CAGGCAGTCCAGAAAGTCGG
ATGAAGAATTGGGTCCCAGATG
56,5
372
6s-6as
GACTCCCCAAATTTCAGATG
GCCCATCCACTCTTTCCAG
53
315
7s-7as
TCAGGCACTCAGAGAAACATGG
GTCCCCCCATTCCCCATG
57,5
263
8s-8as
AATGGGGGGACAGTGGGG
TCACAGACCAGCCCAGACAG
59,5
374
9s-9as
TCCTGTTCTGTCTGGGCTGG
AAGGAGAAAGCCCCCCAG
58
332
10s-10as
CATGACTCGTGGATAGGGAAGG
TGAGGCTTGGGGGCATTG
57,5
375
11s-11as
AATGCCCCCAAGCCTCAG
CTCATAGCATTTGTGTCTTTCCTG
61,4
357
12s-12as
TTCCACTCCCCACTGCTTTG
CTGGCTCTGTCCCTCCCG
59
414
13s-13as
GGAGGGACAGAGCCAGGTG
GAGGCTGGAGAGGCACTAGG
59,5
390
14s-14as
CCTCTCTGGTCTGGCCTCG
TTAGGGTCAAGAAGGCATCG
60
326
15s-15as
CTTAGCTCTGAACTTGTTACCTTG
AGAATAAGGGACCTGGCAGG
55
350
16s16as
AGGGAAGTGGTCATGGGGAG
GGAGACTCCACAATGGGCAAG
62,8
276
17s-17as
CTAAGACATCCCTCCCACCTG
CATGCCCTGGCGAGTATATAG
60
336
18s-18as
GTAAATGGATAGATGGATGACAGG
GGGTTCAGTGGCAGGTCTTG
60,5
376
19s-19as
TTACCCAAACACCTGACTTCAAG
TCCTCCACCCATTCGTCTTC
60,9
361
20s-20as
GTGGATGAAAAGATAAATGGATG
ACTCCATCCTCACACATACACAG
58
408
21s-21as
GCCAGAGCAGTGTTCACCATG
GGACAGGGGGATAGTAAATTCAG
61,9
341
22s-22as
GAAGGGAATGGGCTAGGG
CTTTTACTAGTTGTGTGACCTTGG
58
383
23s-23as
TTAGCGTTACCATTAGAATTGC
ATCACATGCCTTGGCCTC
57
420
24s-24as
GGCAGAAAGGGTGGGCAC
GGTCTCCACCCTGGCAGG
61,5
305
25s-25as
CCTGTGGTTGCTGCATACTGAAG
GGCTCTCCTCATATTCGTTCCTG
64
260
26s-26as
GTAAAGAAGGCTCTGAGGGAGG
CCCCACACGCAAAACAAAC
61
304
27s-27as
CACAATCAGGGCACCGACG
AGGCACCCAGTCCAGGCG
64
282
28s-28as
GCCTGGACTGGGTGCCTTG
GTTGGGATTACAGGCATGGACC
64,5
331
29.2s-29.2as
GGAGGCGGAGGTTGCAGTG
GGAAACCTCTGCTGTGCTCTCTG
64,9
468
29.3s-29.3as
GAGGAAAAGAATATGTACTGTGTG
GATCCTCTTACTGGGCTGTTA
55,5
325
1.2s-1as
a
A Ta é a temperatura média entre as Tm de cada primer nos pares de PCR.
62
Tabela 9: Primers para amplificação e sequenciamento do gene WT1.
Primer sense
Fragmento
5`
Primer antissense
3`
5`
3`
Taa
Tamanho
(oC)
(pb)
8-9
b
TACCCTAACAAGCTCCAGCG
b
55,1
1037
-
c
TGAGGCAGATGCAGACATTG
c
-
-
TCTCTCAACTGAGTCTAAACCTTAG
GAGAATCATGAAATCAACCCTAG
a
A Ta é a temperatura média entre as Tm de cada primer nos pares de PCR.
Como os éxons 8 e 9 do gene WT1 são curtos e ficam próximos, é possível realizar a sua amplificação com o
primer 8 sense e 9 antisense, gerando o fragmento 8-9, de 1037 pb.
c
Primers internos utilizados somente para reação de sequenciamento.
b
O ciclo geral de PCR foi:
Desnaturaçao inicial: 95°C ---------------------------- 5 min
Desnaturação:
95°C ---------------------------- 1 min
Anelamento:
53,5°C a 64°C ----------------- 1 min
Extensão:
72°C ---------------------------- 1 a 6 min
Extensão final:
72°C --------------------------- 10 min
Espera:
15°C --------------------------
30 ciclos
∞
Todas as reações tiveram volume final de 50 l e foram realizadas com Taq DNA
polymerase recombinant (Invitrogen Corporation, Estados Unidos). A reação geral de PCR está
ilustrada na tabela 10.
Para as amostras de DNA extraídas de material contido em parafina foram efetuadas duas
etapas de PCR para se evitar que a não amplificação numa primeira etapa fosse subestimada. Na
primeira etapa, uma reação de PCR com volume final de 25 µl foi realizada, utilizando-se o PCR
Master Mix Kit (Promega, Madison, WI), seguindo-se o protocolo do fornecedor. Para a segunda
etapa de PCR utilizou-se como molde 4 µl do produto da primeira PCR.
63
Tabela 10: Reagentes utilizados para a reação de PCR
REAGENTES
[ ] FINAL
Tampão Invitrogen (10x)
1x
MgCl2 Invitrogen (50 mM)
1,5 mM
dNTP Invitrogen (2 mM)
0,1 mM
Primer sense (20 pmoles)
20 pmoles
Primer antisense (20 pmoles)
20 pmoles
Enzima Taq DNA polymerase recombinante Invitrogen (5 U/µL)
2U
DNA genômico
1 a 2 µg
DMSO 5% ou BSA 1%
Se necessário
H20 deionizada q.s.p.
50 µL
2.6 Purificação dos produtos de PCR
Os fragmentos, após amplificação por PCR, foram purificados com o Kit Wizard SV Gel
and PCR Clean-UP System (Promega) seguindo o protocolo fornecido pelo fabricante. Após
purificação, as amostras foram quantificadas no equipamento Nanodrop 8000 - Multi-Sample
Micro-Volume UV-Vis Spectrophotometer (Thermo Scientific, USA).
64
2.7 Reação de Sequenciamento
As reações de sequenciamento foram realizadas utilizando-se o sistema BigDyeTM
Terminator Cycle Sequencing Kit V3.1 Ready Reaction (ABI PRISM/Life Technologies, USA)
em placa de 96 tubos, aonde foram adicionados os reagentes descritos na tabela 11.
Tabela 11: reagentes utilizados na reação de sequenciamento
REAGENTES
Volume utilizado
Tampão Big Dye 5x (fornecido pelo fabricante)
2 µL
Primer sense ou antisense 5 pmoles
1 µL
Big Dye (fornecido pelo fabricante)
0,6 µL
1 µL
DNA purificado (40 a 80 ng/µL)
q.s.p. 10 µL
H20 deionizada
O ciclo geral de sequenciamento foi:
Desnaturação inicial: 95°C ----------- 1 min
Desnaturação:
95°C ----------- 20 seg
Anelamento:
50°C ----------- 20 seg
Extensão:
60°C ------------ 4 min
Espera:
4°C ------------- ∞
26 ciclos
2.8 Purificação da reação de sequenciamento
Após reação de sequenciamento, as seguintes etapas de purificação da reação foram
seguidas:
65
- Adicionou-se 2,5 μL de EDTA (125 mM) em cada tubo, seguidos de 25 μL de etanol
100% (Merck);
- A mistura foi deixada à temperatura ambiente e na ausência de luz por 15 min;
- Centrifugou-se à 13.000 rpm e 4 ºC por 35 min;
- A placa foi invertida e o sobrenadante foi descartado, a fim de remover o etanol;
- Adicionou-se 30 μL de etanol 70% (Merck), e realizou-se uma nova centrifugação por
10 min a 13.000 rpm e 4 ºC;
- A placa foi invertida e o sobrenadante foi descartado;
- A placa foi deixada à temperatura ambiente por cerca de 40 min e depois colocada no
termociclador por 3 min a 80 ºC para completar a secagem, em seguida, foi estocada a -20 ºC.
No momento de levar as placas para o sequenciador, os produtos da reação de
sequenciamento purificados foram vigorosamente ressuspendidos em 10 μL de Formamida HiDi (Applied Biosystems-Applera Corporation, Estados Unidos) e a 94 ºC por 5 min para
desnaturação.
As sequências de bases nitrogenadas dos fragmentos foram obtidas em um sequenciador
automático ABI3500 genetic analyzer (Applied Biosystems®). As sequências obtidas foram
analisadas e comparadas com as sequências de referência dos genes NPHS2 (ID:
ENSG00000116218), NPHS1 (ID: ENSG00000161270) e WT1 (ID:ENSG00000184937),
contidas no site www.esembl.org, com o auxílio dos programas Chromas Lite e CLC Sequence
Viewer 6.3 que são de distribuição gratuita.
66
2.9 Amplificação alelo-específica por PCR
A técnica de amplificação por PCR alelo específico foi utilizada para a triagem, em 100
controles saudáveis, de algumas mutações que foram identificadas em heterozigose na nossa
casuística. Foram desenhados primers sense ou antisense denominados de âncora e primers
específicos que se acoplam na posição do alelo mutante e na respectiva posição do alelo normal.
Duas reações foram realizadas, uma com primer âncora e com o primer normal (reação normal)
e a outra com o primer âncora e com o primer mutante (reação mutante) (figura 13). Através da
visualização no gel de agarose 1% foi possível verificar a aplificação positiva ou negativa para
estes 100 controles.
Figura 13: Esquema de PCR alelo específico. Na reação normal é utilizado um par de primers: o primer âncora + o
primer contendo a sequência com a base normal, no exemplo da figura, com uma adenina (A). Na reação mutante é
utilizado o mesmo primer âncora, porém com o primer contendo a substituição, no exemplo acima, uma guanina (G).
No momento da visualização no gel de agarose após corar com brometo de etídeo, é possível visualizar se o indivíduo
é homozigoto normal, heterozigoto ou homozigoto mutante, como mostrado na figura, nos poços 1, 2 e 3,
respectivamente.
67
Os primers específicos para as mutações estão ilustrados na tabela 12.
Tabela 12: Primers utilizados para PCR alelo específico.
Primer Normal
5`
3`
Primer mutante
5`
3`
Primer âncora
5`
3`
Ex1s (P20L) N
Ex1s (P20L) M
Ex1as
GGCGAGGCGGCAGGACTCC
GGCGAGGCGGCAGGACTCT
GGCCCCGAGACCAGTATATAGTGATA
Ex5.1as (R229Q) N
Ex5.1as (R229Q) M
Ex5s
TAGAAGAATTTCAGTGAGGGATC
TAGAAGAATTTCAGTGAGGGATT
TTAAATAAAGGGTAGGCCAACTCC
Ex5as (A242V) N
Ex5as (A242V) M
Ex5s
TATCTAAGTACCTTTGCATCTTGGG
TATCTAAGTACCTTTGCATCTTGGA
TTAAATAAAGGGTAGGCCAACTCC
Ex6as (E264Q) N
Ex6as (E264Q) M
Ex6s
GCTAGTTAATTTCCTACCCACATTTC
GCTAGTTAATTTCCTACCCACATTTG
CCTCTTGGGGTAACATTCACAG
Ex 7as (A284V) N
Ex7as (A284V) M
Ex7s
CACTTTGGCTTGTCTTTGCG
CACTTTGGCTTGTCTTTGCA
CAAAACCTGCTGTGCTGATAATG
Ex8as (E310K) N
Ex8as (E310K) M
Ex8s
AGGGGTGCCTGACAGAATCTC
AGGGGTGCCTGACAGAATCTT
TCTCTATGTTGGCAAAATCCTAATC
Tamanho
Fragmento (pb)
500
310
340
360
330
365
68
Na tabela 13 podem ser observados os reagentes utilizados.
Tabela 13: reagentes utilizados na reação de PCR alelo específico.
REAGENTES
Volume
Tampão Invitrogen (10x)
3 µL
MgCl2 Invitrogen (50 mM)
0,6 a 0,9 µL
dNTP Invitrogen (2 mM)
2,5 µL
Primer sense (20 pmoles)
1 µL
Primer antisense (20 pmoles)
1 µL
Enzima Taq DNA polymerase recombinant Invitrogen (5U/µL)
0,3 µL
DNA genômico
1 µL
BSA 1%
0,3 µL
H20 deionizada q.s.p.
30 µL
O ciclo geral das reações foi:
Desnaturação inicial: 95°C ----------- 5 min
Desnaturação:
95°C ------------ 1 min
Anelamento:
Ta°C*----------- 1 min
Extensão:
72°C ------------ 1 min
Extensão final:
72°C ------------- 5 min
Espera:
15°C ------------- ∞
30 ciclos
* A Ta é a temperatura média entre as Tm de cada primer nos pares de PCR.
69
2.10 Estudos in silico
Estudos in silico são utilizados como ferramentas de predição que avaliam o potencial
efeito funcional que as alterações identificadas podem causar no transcrito ou na proteína.
Assim, diversos programas foram utilizados nesta tese, dependendo da região aonde as alterações
foram identificadas.
Para alterações identificadas na região 5’reguladora dos genes foi utilizado o programa
AliBaba2 Gene Regultation (http://www.gene-regulation.com/pub/programs.html), que avalia a
existência de motivos reguladores. O programa faz uma comparação entre a sequência normal e
a mutante (incluídas no site em formato “fasta”, pelo usuário) e verifica se a alteração está
localizada em uma região que altera o reconhecimento por fatores de transcrição e, se for o caso,
identifica quais os fatores de transcrição que se ligam a este sítio.
Para alterações identificadas em sítios de splicing, foram utilizados os programas de
predição
“Splice
site
prediction
(http://www.fruitfly.org/seq_tools/splice.html),
by
o
neural
network
“HumanSplicingFinder
homepage”
2.4.1”
(http://www.umd.be/HSF/) e o “SpliceAid2” (http://www.íntroni.it/splicing.html). O primeiro
verifica os sítios aceptores e doadores de splicing, atribuindo scores para cada sítio encontrado,
tanto para a sequência normal como para a sequência mutante, ambas inseridas pelo usuário no
formato fasta no website. O website “HumanSplicingFinder (HSF)” (Desmet et al., 2009) é uma
ferramenta que não só prediz o efeito de mutações nos scores de splicing mas também identifica
as sequências em cis exônicas e intrônicas que são reconhecidas pelas proteínas da maquinaria de
splicing. O SpliceAid2 (Piva et al., 2009) compara o seu transcrito primário com um extenso
banco de dados de RNAs primários e verifica quais as proteínas regulatórias de splicing se ligam
70
na sua região de interesse e se o reconhecimento por estas proteínas é prejudicado após a
mutação.
Para as mutações missense foram utilizados três métodos computacionais que avaliam o
impacto da troca de aminoácido na proteína, indicando se esta alteração pode ser prejudicial para
sua função e se pode contribuir ou não para o fenótipo do indivíduo afetado. Os programas
foram: SIFT (Sorting Intolerant From Tolerant) (http://sift.jcvi.org/); PolyPhen2 (Polymorphism
Phenotyping) (http://genetics.bwh.harvard.edu/pph2/); Align-GVGD (http://agvgd.iarc.fr/).
A ferramenta SIFT tem como função determinar o efeito funcional causado por
substituições de resíduos de aminoácidos nas proteínas, baseando-se no pressuposto que a
evolução proteica está correlacionada com a função proteica, mostrando a conservação de
resíduos de aminoácidos localizados em posições importantes para a função proteica. O
programa realiza diversos passos, a partir de uma sequência “query” que possui a mutação: 1)
procura sequências similares, 2) escolhe sequências intimamente relacionadas que possam ter
função similar, 3) alinha estas sequências e 4) calcula probabilidades normalizadas para todas as
substituições possíveis para cada posição do alinhamento. As substituições que apresentam um
valor de tolerância menor do que 0,05 são consideradas intolerantes ou deletérias, enquanto que
as que apresentam um valor maior do que 0,05 são consideradas tolerantes. A precisão da
ferramenta é de 60%, sendo que as taxas de “falso-negativos” e “falso-positivos” registradas
foram iguais a 31% e 20%, respectivamente (Ng & Henikoff, 2001; Ng & Henikoff, 2006).
O PolyPhen classifica as variantes tomando-se como base a anotação e o alinhamento de
sequências, além de parâmentros estruturais. Dois tipos de banco de dados são utilizados pelo
programa para a realização destas análises. O primeiro é o HumDiv, que compara a sequência
contendo a substituição do aminoácido “query” com 3.155 alelos (anotados no Uniprot)
71
causadores de doenças mendelianas humanas e que afetam a estabilidade e a função das
proteínas, juntamente com 6.321 diferenças consideradas não prejudiciais entre proteínas
humanas e suas homólogas de mamíferos próximos. Portanto, o HumDiv é indicado para a
avaliação de doenças mendelianas, nas quais é necessária a distinção de mutações de efeito
drástico entre todas as variações humanas, mesmo as com efeito menos grave. O seg banco de
dados é o HumVar, que consiste em 13.032 mutações causadoras de doenças humanas (Uniprot),
além de 8.946 polimorfismos não sinônimos humanos frequentes (nsSNPs, MAF>1%), tratados
como não prejudiciais. O HumVar deve ser utilizado para avaliar loci de alelos raros com
potencial envolvimento com fenótipos complexos e regiões densas de mapas identificados por
estudos GWAS (genome wide association studies). Os valores calculados vão de 0 a 1,0, sendo
que quanto mais alto for este valor, mais raramente (ou nunca) a substituição estudada é
observada na família proteica, sendo considerada mais deletéria. A precisão é de
aproximadamente 81%, com uma taxa de “falso-negativos” e “falso-positivos” de 31% e 9%,
respectivamente (Ramensky et al., 2002).
O programa Align-GVGD foi baseado no método da Diferença de Grantham (Grantham,
1974), utilizado para calcular as distâncias bioquímicas entre pares de aminoácidos e que leva
em consideração a composição (C), a polaridade (P) e o volume (V) molecular dos aminoácidos
mutante e selvagem. Cada aminoácido é plotado em um gráfico 3D, tendo como eixos os valores
de C, P e V. Todos os aminoácidos resultantes do alinhamento de múltiplas sequências são
plotados no gráfico e recebem valores GV (Grantham Variant) - quantidade de variação
bioquímica observada para uma determinada posição no alinhamento - e GD (Grantham
Difference) - medida da diferença bioquímica entre a variação mutante e a observada naquela
posição do alinhamento de múltiplas sequências (Mathe et al., 2006; Tavtigian et al., 2006).
72
Estes valores predizem se a mutação de interesse está dentro de um espectro deletério ou neutro.
Assim, a partir dos cálculos desses valores (GV e GD), a troca é classificada dentro de classes
(C0 – C65), sendo que quanto mais próximo de C65, mais deletéria a mutação é considerada.
Para verificação da conservação de uma posição proteica onde houve a troca de
aminoácidos, as sequências normal e mutante das proteínas podocina, nefrina e WT1 humanas
foram comparadas a sequências correspondentes de várias espécies de vertebrados (UniProt,
http://www.uniprot.org/, acesso livre) (Bairoch et al., 2005), utilizando-se o programa de
alinhamento múltiplo de sequências ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/,
acesso livre) (Thompson et al., 1994).
Para a avaliação estrutural de uma alteração nova no gene WT1, foi realizado o download
do banco de dados Protein Data Bank do modelo resolvido (PDB-ID 2PJA: domínio de dedo de
zinco do WT1 humano complexado com o DNA), obtido por espectrometria de ressonância
magnética nuclear (espectometria NMR) (Stoll et al., 2007). Foi solicitado o modelamento da
estrutura
molecular
(normal
e
mutante)
pelo
software
SWISSMODEL
(http://www.swissmodel.expasy.org) (Arnold et al., 2006; Kiefer et al., 2009). A visualização e a
manipulação das estruturas foram realizadas através da interface gráfica gerada pelo programa de
acesso livre STING Millennium (suíte BlueStar STING, www.cnptia.embrapa.br) (Neshich et al.,
2003) e pelo programa PyMol Viewer (http://www.pymol.org/).
A figura 14 apresenta um fluxograma dos programas de predição in silico utilizados e a
região gênica correspondente à predição.
73
Figura 14: Fluxograma dos programas de predição in silico utilizados.
2.11 Cálculos estatísticos para avaliação dos SNPs e Análise de Desequilíbrio de
ligação
Os cálculos das frequências esperadas e do qui-quadrado (x2) foram realizados através do site
OEGE (http://www.oege.org/software/hwe-mr-calc.shtml) (Rodriguez et al., 2009). O cálculo do
p-value foi realizado com a função qui-quadrado da planilha Excel. O software HaploView4.2
(Barrett et al., 2005) foi utilizado para a construção dos mapas de desequilíbrio de ligação (LD).
74
2.12 Classificação das mutações e correlação genótipo-fenótipo
À medida que as mutações missense, alterações em sítio de splicing e na região
promotora foram sendo identificadas, algumas etapas foram seguidas como estratégia de
classificação destas variantes e tentativa de correlacioná-las com o fenótipo apresentado pelos
pacientes. Assim, a classificação destas variantes baseou-se na (1) análise de cromossomos
controle, (2) observação da cosegregação do alelo com a doença na família (quando possível),
(3) avaliação de dados biofísicos e bioquímicos entre os aminoácidos normal e mutante, (4)
análise de conservação do resíduo entre os ortólogos ao longo da evolução e (5) verificação do
potencial efeito patogênico através de programas de predição in silico, seja com programas de
avaliação da região promotora, da região de splicing ou de avaliação do efeito deletério de
substituições de aminoácidos não sinônimos.
Com relação ao grupo controle utilizado neste trabalho, este foi constituído por parentes
de casos já triados no nosso laboratório e também por alunos de medicina da Unicamp
selecionados para um estudo caso:controle. Não foi realizada uma triagem inicial para saber se
os indivíduos controle são portadores de doença renal.
75
76
RESULTADOS E DISCUSSÃO
77
78
RESULTADOS E DISCUSSÃO
O fluxograma da figura 15 apresenta um resumo dos resultados identificados após a
análise dos genes NPHS1, NPHS2, e WT1 entre os 150 pacientes.
Figura 15: Fluxograma apresentando um resumo dos resultados dos genes NPHS1, NPHS2 e WT1 nos pacientes
com SNC e SN infantil, SN na infância, SN juvenil e proteinúria isolada.
79
1. SN CONGÊNITA E TRIAGEM DO GENE NPHS1
1.1 Material fixado em parafina
Para as amostras de DNA extraídas de material contido em parafina a amplificação foi
mais difícil, considerando a qualidade do DNA, que, devido aos processos de fixação e extração,
encontra-se muito degradado. Várias tentativas de amplificação dos 29 éxons do gene NPHS1
foram realizadas, todas sem sucesso. Quando se conseguia a amplificação de algum éxon, a
concentração de DNA após a etapa de purificação era muito pequena e o resultado do
sequenciamento era de baixa qualidade. Tentou-se o sequenciamento direto do produto de PCR,
sem prévia purificação, mas também sem êxito. Após tentativas de amplificação, realizadas em
duas etapas de PCR para não subestimar a amplificação em uma primeira etapa, obteve-se um
resultado satisfatório para alguns fragmentos (região 5’reguladora e éxons 2, 3, 4, 5, 6, 7, 10 e 12)
de um dos materiais (M7, anexo II) que estava em maior concentração. A partir do
sequenciamento destes éxons foi identificada a mutação c.59-5G>C, em heterozigose, no íntron 1
(figura 16). Esta mutação já foi identificada por Caridi et al. (Caridi et al., 2009) em heterozigose
em um paciente com desenvolvimento tardio de SN e boa resposta aos corticoides. No caso do
paciente aqui descrito, que já foi a óbito por SNC, como a SNC possui um padrão de herança
autossômica recessiva (Kestilä et al., 1998), é possível que exista outra mutação em algum dos
éxons não investigados.
80
Figura 16: Eletroferograma da mutação c.59-5G>C. A heterozigose G/C encontra-se ressaltada em amarelo na
sequência acima dos picos do eletroferograma e, abaixo, está denotada em vermelho onde se indica sua posição no
gene NPHS1. A seta indica o sítio aceptor de splicing localizado na junção do íntron 1 com o éxon 2. Esta mutação
foi identificada no material M7 (anexo II) proveniente de autópsia de paciente com SNC.
A mutação c.59-5G>C localiza-se na região aceptora de splicing do íntron 1. Esta região
corresponde a um dos elementos de ação cis altamente conservados geralmente localizados nos
limites íntron-éxon que auxiliam na correta remoção dos íntrons a partir do RNAm primário em
vertebrados (Berget, 1995; Black, 1995). Em mamíferos, íntrons que passam por splicing são
quase em sua totalidade flanqueados por 5`-GT ... AG-3`. Outros elementos já bem descritos são a
sequência de pirimidinas na posição 3` do íntron (trato polipirimidínico), além do ponto de
ramificação (“branch site”), que consiste em uma adenina reconhecida pela maquinaria
responsável pelo splicing (figura 17). No entanto, estas sequências consenso nem sempre são
suficientes para que os íntrons sejam removidos corretamente. Outros elementos de ação cis, com
ação positiva ou negativa, são necessários para que o mecanismo de splicing seja eficiente. As
sequências de ação positiva melhor estudadas são os elementos denominados “exonic splicing
enhancers” (ESEs) (Blencowe, 2000), que são sequências nucleotídicas curtas encontradas
primariamente em éxons com splicing que é regulado. Estes éxons geralmente estão localizados
81
adjacentes a íntrons com sinal fraco para splicing e precisam destas sequências exônicas (ESEs)
para serem reconhecidos. Deleções de ESEs podem causar éxon skipping e, no caso de éxons
finais, não permitem a remoção do último íntron. As ESEs são reconhecidas essencialmente por
proteínas ricas em serina e arginina (proteínas SR), proteínas estas indispensáveis para o
mecanismo de splicing, já que promovem o reconhecimento dos éxons, atuando tanto no splicing
constitutivo quanto no splicing alternativo. A SC35 é uma proteína humana rica em serina (SR)
cuja sequência consenso ESE já foi bem caracterizada (Liu et al., 2000).
Figura 17: Sequências importantes para o splicing presentes no pré-RNAm. São mostradas as regiões
conservadas que são essenciais para que o processo de splicing ocorra, a saber: 5`SS: sequência consenso no sítio
doador de splicing de mamíferos (GU); PR: ponto de ramificação; TPP: trato polipirimidinico; 3`SS: sequência
consenso no sítio aceptor de splicing de mamíferos (AG); ESE: Exonic splicing enhancer; ESS: Exonic splicing
silencer. R: purina; Y: pirimidina. Adaptado de Blencowe, 2000.
Realizamos algumas predições in silico para verificarmos se a mutação c.59-5G>C
identificada no material M7 prejudica o sítio aceptor de splicing entre o íntron 1 e o éxon 2.
A primeira predição de sítio de splicing realizada foi através do programa “Splice site
prediction by neural network homepage” (http://www.fruitfly.org/seq_tools/splice.html) e foi
verificada uma significativa redução no score do sítio aceptor de splicing após esta alteração
82
(figura 18A). Isto significa que este sítio com a mutação pode não ser mais reconhecido como um
sítio aceptor preferencial pelas proteínas envolvidas na maquinaria de splicing.
Figura 18: Resultados obtidos nas análises in silico para a mutação c.59-5G>C. A. Análise realizada com o
programa fruitfly (http://www.fruitfly.org/seq_tools/splice.html); circulado em vermelho é possível ver a redução
significativa do sítio de reconhecimento de splicing após a alteração. B. Histograma gerado na predição in silico
realizada com o programa SpliceAid2 (http://www.íntroni.it/splicing.html). As barras acima da sequência
correspondem a proteínas que reconhecem sequências facilitadoras ou inibidoras de definição do éxon, ou seja,
sequências exonic splicing enhancer (ESEs) ou íntronic splicing silencer (ISSs), respectivamente. As barras abaixo
correspondem a sequências que facilitam ou inibem a definição dos íntrons, isto é, sequências exonic splicing silencer
(ESSs) ou íntronic splicing enhancer (ISEs), respectivamente. As barras possuem largura e altura variáveis, o que
está relacionado ao tamanho da sequência e ao grau de afinidade de ligação, respectivamente. Normal: Quando a
sequência normal sem a mutação foi inserida no programa, a proteína SR SC35 (barra amarela) reconheceu a ESE
“GUCCUCAG” (C ressaltado na sequência). Mutante: Após inserir a sequência mutante, com a troca de um C por
um G (c.59-5C>G), a proteína regulatória SC35 não identifica mais a sequência “GUCGUCAG” alterada.
83
Outro programa de predição utilizado foi o programa SpliceAid2 que avalia se existem
proteínas regulatórias que se ligam à região aonde se encontra a mutação e se esta ligação é
prejudicada. Este programa disponibiliza a análise tecido específica das proteínas regulatórias,
que são coletadas a partir dos bancos de dados Human Protein Atlas, HPRD, Human
Proteinpedia, Transcriptome Map e CGAP. Deste modo, realizamos uma análise restrita para
proteínas regulatórias que são expressas nas células glomerulares do tecido renal. O resultado
desta predição pode ser observado na figura 18 B. Nota-se que no gene normal a sequência
“GUCCUCAG” (ESE) é reconhecida pela proteína SR SC35 e que, no mutante, o sítio de
reconhecimento para a SC35 ficou alterado e esta proteína não mais constou do resultado.
O
terceiro
programa
de
predição
utilizado
foi
o
Human
Splicing
Finder
(http://www.umd.be/HSF/) (Desmet et al., 2009), que avalia a região de splicing baseando-se em
diversas matrizes disponíveis para análise de elementos de ação cis exônicas e intrônicas. Após
inserir a sequência normal e a mutante, o programa compara a região ao redor da troca
nucleotídica em busca de sítios de splicing e motivos de reconhecimento de proteínas
regulatórias. Esta análise apontou resultados semelhantes, pois identificou que a mutação c.595C>G está contida em alguns elementos de ação cis reconhecidos por proteínas regulatórias,
como, por exemplo, a sequência: “GTCCTCAG”, reconhecida pela proteína SC35, o que valida o
resultado observado através da predição anterior, com o programa SpliceAid (figura 19).
Portanto, a partir destas predições pode-se inferir que esta mutação afete o splicing desta
região, porém somente estudos funcionais in vitro usando a técnica de minigene indicarão o real
impacto desta mutação no RNAm transcrito.
Nenhuma outra alteração foi identificada nos outros fragmentos analisados (região
5’reguladora e éxons 2, 3, 4, 5, 6, 7, 10 e 12) para este material.
84
A proposta inicial era a de avaliarmos a distribuição e frequência de mutações em
pacientes com síndrome nefrótica congênita a partir de sete amostras provenientes de autópsias
e/ou biópsias fixadas em blocos de parafina cedidos pelo departamento de anatomia patológica da
FCM da Unicamp. Porém, dadas as dificuldades mencionadas anteriormente nos processos de
extração e amplificação do DNA e frente ao recebimento de amostras de três pacientes vivos com
SNC, optamos por não mais investir tempo e recursos em tentativas de amplificação de material
tão degradado.
Figura 19: Resultados obtidos na análise in silico realizada com o programa Human Splicing Finder
(http://www.umd.be/HSF/). As sequências normal e mutante estão identificadas acima com as regiões de interesse
ressaltadas: a citosina normal está representada em verde e a guanina alterada, em vermelho. Abaixo está ilustrado o
resultado da predição: Quatro sequências Exonic Splicing Enhancer (ESE) foram identificadas como potencial sítio
de reconhecimento para as proteínas SRp40, SC35, SF2/ASF e SRp55 avaliadas pelas matrizes do ESE finder. A
região para a SC35 encontra-se entre as ESEs abolidas após a mutação, como já identificado anteriormente com o
programa SpliceAid.
85
1.2 Pacientes com SNC
Três pacientes com hipótese diagnóstica de SNC foram encaminhados no decorrer do
período da tese. Para estes três pacientes foram analisados todos os éxons, limites íntron-éxon e
região promotora do gene NPHS1 e foram identificadas alterações em todos eles 3/3 (100%)
(tabela 14). Na figura 20 encontra-se a sequência de aminoácidos da proteína nefrina, da maneira
como proposta por Kestilä et al. em 1998 (Kestilä et al., 1998) e as posições das alterações
identificadas nos pacientes com SNC deste trabalho estão ressaltadas nos respectivos domínios
da proteína.
Tabela 14: Alterações identificadas nos pacientes com SNC
Pacientes
Mutações
128
p.Thr172del (éxon 4)
c.526+1G>T (íntron 4)
143
p.Pro264Arg (éxon 7)
p.Val736Met (éxon 16)
150
p.Ala47Profs*127 (éxon 2)
p.Thr172del (éxon 4)
86
Figura 20: Esquema das alterações identificadas nos pacientes com SNC distribuidas ao longo da sequência
de aminoácidos da proteína nefrina. Os 22 resíduos que codificam para o peptideo sinal N-terminal localizam-se
logo no início da sequência e o sítio de clivagem está indicado com uma seta. Os resíduos 1059 a 1086, que
correspondem ao domínio transmembrana, estão sublinhados e em negrito. A porção extracelular, contendo os oito
domínios do tipo imunoglobulina estão em “caixas” e o domínio de fibronectina tipo 3 está ressaltado em amarelo,
adjacente ao domínio transmembrana. Os resíduos de cisteína são indicados por pontos em negrito e os 10 possíveis
sítios de N-glicosilação na porção extracelular da proteína estão sublinhados. Adaptado de Kestilä et al., 1998. *
Esta alteração foi identificada em dois pacientes com SN na infância descritos na seção 2 dos Resultados e
Discussão. **Alteração em sítio de splicing.
O paciente 128 foi atendido no Hospital Universitário Pedro Ernesto (UERJ), Rio de
Janeiro e encaminhado com hipótese diagnóstica de SNC devido à idade em que desenvolveu a
87
doença, menos de três meses. O resultado da biópsia foi sugestivo de GESF. Pela história de
início precoce e grave evolução, não foi realizado tratamento com corticoides e, após tentativas
de tratamento sem sucesso, foi realizada nefrectomia unilateral. Após triagem do gene NPHS1,
identificamos uma mutação em heterozigose no éxon 4, a deleção de uma trinca de nucleotídeos
c.514-6del3nt que causa a deleção do aminoácido treonina na posição 172 da proteína nefrina
(p.Thr172del) (figura 21). Lenkkeri et al. identificaram esta mutação em um paciente com
origem holandesa e diagnóstico de SNC (Lenkkeri et al., 1999); o paciente estudado por
Lenkkeri também era portador de outra mutação em heterozigose composta, já que a SNC possui
herança autossômica recessiva: uma alteração missense no éxon 18. No caso do paciente 128,
além desta deleção, foi identificada outra alteração, na mesma região, porém, no mesmo alelo:
consiste na troca de uma guanina para uma timina, o G do sítio 5’ “GT” consenso doador de
splicing do íntron 4 (figura 21).
88
Figura 21: Eletroferograma e sequência do fragmento do éxon/íntron 4 do gene NPHS1 do paciente 128.
Acima, no eletroferograma, é possível a visualização dos três nucleotídeos que foram “deletados” na sequência
mutante (c.514-6del3nt) e que estão ressaltados em vermelho e o consequente “truncamento” da análise da
sequência (letras ressaltadas em amarelo), já que a alteração foi identificada em heterozigose. No mesmo alelo e na
mesma região foi identificada outra mutação: a substituição da primeira guanina do sítio doador de splicing do
íntron 4, por uma timina (c.526+1G>T). A guanina que foi trocada por uma timina está ilustrada em laranja. A seta
indica o sítio doador de splicing localizado na junção do éxon 4 com o íntron 4. Abaixo, a região dos limites do éxon
4 e íntron 4 retirada do www.ensembl.org , com o G que foi alterado ressaltado (sítio doador de splicing).
A
predição
in
silico
realizada
através
do
programa
http://www.fruitfly.org/seq_tools/splice.html, que verifica os scores dos sítios doadores e
aceptores de splicing, retornou que a alteração c.526+1G>T suprime o sítio doador do íntron 4
(figura 22 A e B). Este resultado já era previsto, já que alterações que afetam a posição +1 do
89
sítio 5’doador de splicing são as mais comuns (Krawczak et al., 1992) e com consequências
drásticas para o transcrito. Esta posição e a posição +5 são responsáveis pelo pareamento do sítio
doador de splicing com a região complementar da ribonucleoproteína U1snRNP, um dos
primeiros passos do processo de splicing do pré RNAm; portanto, quando estas posições sofrem
mutação, ocasiona um prejuízo significativo para este pareamento (Wittop et al., 1994). Algumas
consequências do não pareamento podem ser o exon skipping (“pular” o éxon), quando a
maquinaria de splicing procura o próximo sítio de splicing disponível, o que pode resultar na
completa remoção do éxon em questão; a retenção do íntron, ou inserções e deleções que
ocorrem devido à utilização de sítios de splicing crípticos, que são sítios de splicing que estão
presentes no genoma dos eucariotos mas que geralmente estão inativos ou são pouco utilizados, a
não ser que mutações os ativem.
Com a predição pelo programa SpliceAid2 pudemos avaliar se há alguma proteína
regulatória que se liga a esta região. Analisando a figura 22 C e D,
observa-se que a
ribonucleoproteína heterogênea A1 (hnRNP A1), que interage com o a sequência “CUGAG”
normal, não reconhece a sequência “CUGAU” mutante. As hnRNPs estão entre as proteínas
nucleares mais abundantes e mais importantes na maturação do pré RNAm dos eucariotos
(Dreyfuss et al., 1993) e possuem um papel fundamental na regulação do splicing. Enquanto as
ESEs interagem com proteínas SR, acredita-se que as sequências Exonic Splicing Silencers
(ESS) interajam com proteínas hnRNP (Jean-Philippe et al., 2013). No caso da mutação
identificada no paciente 128 o G é a primeira base do íntron e foi reconhecido pelo programa
SpliceAid2 como sendo a última base de uma sequência exônica silenciadora de splicing que é
reconhecida pela proteína hnRNP A1 (CUGAG). Quando ocorre a mutação, a proteína não
reconhece a nova sequência, CUGAU.
90
Figura 22: Predição in silico para a mutação c.526+1G>T. A. Predição realizada no site fruitfly
(http://www.fruitfly.org/seq_tools/splice.html) com a sequência normal, mostrando que o sítio doador de splicing
“GT” entre o éxon 4 e o íntron 4 está intacto. B. Sítio de splicing abolido após a mutação. C. Histograma gerado na
predição in silico realizada com o programa SpliceAid2 (http://www.íntroni.it/splicing.html). As barras acima da
sequência correspondem a proteínas que reconhecem sequências facilitadoras ou inibidoras de definição do éxon, ou
seja, sequências exonic splicing enhancer (ESEs) ou íntronic splicing silencer (ISSs), respectivamente. As barras
abaixo correspondem a sequências que facilitam ou inibem a definição dos íntrons, isto é, sequências exonic splicing
silencer (ESSs) ou íntronic splicing enhancer (ISEs), respectivamente. As barras possuem largura e altura variáveis,
o que está relacionado ao tamanho da sequência e ao grau de afinidade de ligação, respectivamente. A
ribonucleoproteína heterogênea A1 (hnRNP A1)(indicada com a seta lilás e barra azul) se liga à sequência CUGAG
normal (sublinhada em vermelho), sendo que a última guanina é o G do sítio doador de splicing GT no íntron 4 e
está ressaltada na figura. D. Mutante: após a troca da guanina para uma timina (uracila na sequência do pré-RNAm)
a sequência CUGAU não é mais reconhecida pela hnRNP A1.
91
As duas mutações do paciente 128 foram identificadas no mesmo alelo. Como o padrão
de herança da SNC é autossômica recessiva seria de se esperar que outra mutação estivesse
presente no outro alelo. Não existem descrições de padrão de herança dominante ou dominante
negativo para a SNC, embora já foram descritas alterações em heterozigose em pacientes com
quadro clínico de SNC (Heeringa et al., 2008). Devido à gravidade do quadro clínico do paciente
128 provavelmente estas mutações estão atuando no desenvolvimento da doença. Ensaios
funcionais in vitro que avaliem o efeito das duas mutações podem auxiliar na avaliação do
potencial efeito destas mutações na produção do transcrito.
A segunda paciente com SNC para a qual realizamos a análise molecular é a paciente
143. Esta paciente foi acompanhada pela Santa Casa de Belo Horizonte, MG, apresentando
proteinúria maciça, hipoalbuminemia e edema desde os 5 dias de vida e resultado da biópsia de
SNF.
Após análise do gene NPHS1, foram identificadas três alterações (figuras 23 e 28). No
éxon 7 foi identificada a troca de uma citosina por uma guanina na posição 791 do cDNA, que
leva à substituição missense de uma prolina por uma arginina no resíduo 264 da proteína nefrina
(figura 24). O resíduo 264 é adjacente a um resíduo de cisteína (figura 20), que estabelece pontes
dissulfeto, ligações extremamente importantes para o correto enovelamento das proteínas de
membrana. Esta mutação já foi previamente identificada por Koziell et al. em um paciente com
origens inglesa e indiana (Koziell et al., 2002). A outra alteração, identificada no éxon 16, ainda
não está anotada no banco de dados: é a troca de uma guanina por uma adenina na posição 2206
do cDNA e que leva à substituição do aminoácido valina pela metionina na posição 736 da
nefrina (c.2206G>A, p.Val736Met) (figura 23A). Por ser nova, fizemos análise de 100
indivíduos controle e não identificamos a variante A em nenhum deles (figura 23B).
92
Figura 23: Eletroferogramas ilustrando as alterações p.Pro264Arg e p.Val736Met identificadas no gene
NPHS1 da paciente 143 e análise de controles para a p.Val736Met. A. As heterozigoses identificadas tanto a
G/C da alteração c.791C>G quanto a G/A da alteração c.2206G>A, estão ressaltadas em amarelo na sequência
acima dos picos dos eletroferogramas e abaixo, estão denotadas em vermelho, onde se indica a sua posição no gene
NPHS1. Na alteração c.2206G>A a seta indica o sítio doador de splicing localizado na junção do éxon 16 e íntron
16. B. Alinhamento realizado com o auxílio do programa CLC Sequence Viewer 6.3 após triagem de 100 indivíduos
controle saudáveis, mostrando que a alteração p.Val736Met não foi identificada em nenhum dos indivíduos controle
analisados. O alinhamento de 20 controles está ilustrado. A letra R indica a substiuição de um G para um A,
identificada somente na paciente.
Como a alteração p.Val736Met localiza-se no éxon 16, mas próxima da região de
splicing, a seis nucleotídeos do íntron 16, foi realizado também o estudo in silico com o
programa SpliceAid2 para verificar se esta alteração pode afetar algum elemento cis de
93
reconhecimento de proteínas regulatórias. O programa não identificou nenhuma proteína
regulatória interagindo com esta região (figura 24).
Figura 24. Predição in silico da alteração c.2206G>A no éxon 16 (p.Val736Met). realizada através do
programa SpliceAid2 (http://www.íntroni.it/splicing.html). As barras acima da sequência correspondem a
proteínas que reconhecem sequências facilitadoras ou inibidoras de definição do éxon, ou seja, sequências exonic
splicing enhancer (ESEs) ou íntronic splicing silencer (ISSs), respectivamente. As barras abaixo correspondem a
sequências que facilitam ou inibem a definição dos íntrons, isto é, sequências exonic splicing silencer (ESSs) ou
íntronic splicing enhancer (ISEs), respectivamente. As barras possuem largura e altura variáveis, o que está
relacionado ao tamanho da sequência e ao grau de afinidade de ligação, respectivamente. A região da alteração está
ressaltada em cinza e a substituição da guanina pela adenina está sublinhada em vermelho. O programa não
identificou sítios de ligação para o reconhecimento por proteínas regulatórias nesta região, nem na sequência
normal, nem na mutante.
94
Não foi possível a análise molecular dos pais, portanto não há como confirmar se estas
mutações estão em heterozigose composta.
Diferentemente do que acontece com mutações frameshift e nonsense, para as quais o
efeito funcional é mais fácil de se prever, com mutações missense isto torna-se mais difícil pois a
consequência que uma única substituição de aminoácidos terá sobre a proteína não é tão
evidente. No caso da proteína nefrina, Koziell et al. (Koziell et al., 2002) sugeriram que
algumas mutações missense que ocorrem em regiões altamente conservadas, removem resíduos
de cisteína ou alteram a carga de certos domínios, atrapalhando o correto enovelamento e função
da nefrina. Outro grupo (Liu et al., 2001) demonstrou que muitas substituições de aminoácidos
afetam o tráfego da nefrina dentro da célula.
Atualmente existem programas de bioinformática estrutural que, a partir das sequências
de proteínas depositadas em bancos de domínio público tais como o Protein Data Bank, PDB
(http://www.rcsb.org/pdb/home/home.do), possibilitam a avaliação e a previsão do impacto
funcional que as substituições de aminoácidos possam exercer sobre proteínas. Através destas
plataformas on line é realizada a comparação entre modelos estruturais da proteína normal e
mutante. No entanto, para a análise estrutural, é necessário que a proteína já tenha sido
cristalizada, que sua estrutura já tenha sido “resolvida” e depositada no banco de dados. As
proteínas de membrana, como é o caso da nefrina, estão complexadas à bicamada lipídica, o que
dificulta o processo de cristalização. A proteína nefrina não possui sua estrutura “resolvida”,
portanto não foi possível este tipo de estudo para a mutação p.Val736Met.
A partir do conhecimento prévio das propriedades dos aminoácidos (Betts et al., 2003)
(figura 25), foi possível fazer algumas inferências com relação aos aminoácidos da mutação em
questão. A valina é um aminoácido hidrofóbico; a metionina pode ser incluída na mesma
95
categoria que a valina, apesar de possuir um átomo de enxofre: ambas contêm cadeias laterais
não reativas e flexíveis, ideais para o empacotamento no interior das proteínas e que
provavelmente não participam de sítios ativos ou de ligação, a não ser uma possível interação
com ligantes hidrofóbicos, tais como lipídios. Portanto, com base nas características de cada
aminoácido, esta troca por si só não teria um impacto funcional importante para a nefrina.
Figura 25: Aminoácidos valina e metionina envolvidos na mutação p.Val736Met. Acima está representada a
estrutura tridimensional e abaixo a fórmula estrutural Em vermelho: átomo de oxigênio; em azul: átomo de
nitrogênio; em preto: átomo de carbono; em branco: átomo de hidrogênio; em amarelo: átomo de enxofre. Adaptado
de http://biochemunclassified.wordpress.com/ e http://www.explicatorium.com/quimica/Aminoacido.
Outra questão importante a ser considerada é com relação à posição que o resíduo 736
afetado ocupa na proteína. Este resíduo localiza-se no éxon 16 que codifica a região entre os
domínios do tipo imunoglobulina 6 e 7 e que fica na face extracelular dos podócitos, nas fendas
de diafragma. A nefrina é uma molécula de adesão celular (CAM) e sua a região extracelular,
96
além de formar homodímeros, forma também heterodímeros com outras proteínas localizadas
nas fendas de diafragma, tais como a Neph1 (Barletta et al., 2003). Portanto, mutações que
afetam esta região podem interferir na capacidade de interação da nefrina, causando danos para a
formação do filtro glomerular.
Também foi realizada a análise de alinhamentos múltiplos pelo programa ClustalW
(http://www.genome.jp/tools/clustalw/) para verificar a conservação do resíduo Val736 ao longo
da evolução. Para isto a sequência da nefrina humana foi comparada com sequências ortólogas
de alguns vertebrados disponíveis no banco de dados Uniprot (http://www.uniprot.org/) (figura
26). Pode-se observar que a valina na posição 736 é conservada entre as espécies de vertebrados
analisados.
HUMANO
RATO
CAMUNDONGO
SAPO
PEIXE ZEBRA
DDGLYQLHCQNSEGTAEARLRLDVHYAPTIRALQDPTEVNVGGSVDIVCTVDANP
DDGFYQLHCQNSEGTAEALLKLDVHYAPTIRALRDPTEVNVGGSVDIVCTVDANP
DDGFYQLHCQNSEGTAEALLKLDVHYAPTIRALKDPTEVNVGGSVDIVCTVDANP
DSGIYNVTCVNKEGKNSVVIRLDVQYSPTIRSLGD-TEVDLGADAEIVCTADANP
DGGDYIIECSNAEGSNRTKLRLDVQYSPTVRMKSDPVFVNIGDTADLLCVADANP
*.* * : * * **. . ::***:*:**:*
* . *::* .:::*..****
Figura 26: Alinhamento das sequências contendo a região vizinha do resíduo 736 (posições 713 a 767) da
proteína nefrina humana com as de quatro espécies de vertebrados. A análise foi realizada pelo programa
Clustal W. O aminoácido valina na posição 736 está ressaltado em amarelo. Este resíduo é , conservado entre os
animais analisados ao longo da evolução, apesar de a região não ser tão conservada. O número de acesso Uniprot
das sequências é: homem (O60500), rato (Q9R044), camundongo (Q9QZS7), peixe zebra (Q1KMS5) e sapo
(Q58QC3). Asteriscos (*) indicam posições que possuem um resíduo conservado e único; Dois pontos (:) indicam
conservação entre grupos de aminoácidos com propriedades similares; Um ponto (.) indica conservação entre grupos
de aminoácidos com propriedades diferentes.
Outras ferramentas de predição in silico que foram utilizadas para avaliar o impacto
potencial da substituição dos aminoácidos na mutação p.Val736Met foram o PolyPhen2
(http://genetics.bwh.harvard.edu/pph2/),
SIFT
(http://sift.jcvi.org/)
e
Align-GVGD
97
(http://agvgd.iarc.fr/), como descrito na seção de Métodos. Os resultados são apresentados na
tabela15.
É importante a validação das predições através de vários programas já que existe uma
taxa de falsos positivos e falsos negativos para todos os métodos. Observando-se a tabela 15,
pode-se perceber que houve diferença entre os resultados gerados pelos três programas: enquanto
a previsão dos programas SIFT e PolyPhen2 foi de prejuízo para a função da proteína após a
mutação p.Val736Met, o Align-GVGD avaliou a substituição como benigna. Considerando-se
que o SIFT e o Polyphen2 avaliam a estrutura e a função biológicas através da comparação
evolutiva entre alinhamentos múltiplos, o resultado de sua análise corrobora o fato de que a
valina é um aminoácido comprovadamente conservado ao longo da evolução (figura 26, análise
ClustalW); já o programa Align-GVGD avalia a composição, polaridade e o volume do
aminoácido e, como já foi visto, estes dois aminoácidos (valina e metionina), são muito
parecidos com relação a estas propriedades, daí o fato de este programa ter avaliado esta troca
como benigna. Como dois sites (SIFT e PolyPhen) classificaram esta substituição como deletéria
e já que este resíduo localiza-se na região extracelular da nefrina, região importante para que a
proteína exerça sua função, considerou-se esta mutação como deletéria.
98
Tabela 15: Predição in silico para a substituição missense p.Val736Met.
Substituição de
aminoácido
p.Val736Met
SIFTa
PolyPhen2b
Align-GVGDc
Deletéria
Provavelmente
deletéria
Classe C15
(0,00)
(1,00)
GV = 0,00; GD = 20,52
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
b
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
c
Align-GVGD: GD (Grantham distance) = pontuação da diferença química entre os residuos normal e
mutante. Quanto maior a pontuação, maior a diferença. GV: Grantham variation = pontuação de acordo com a
diferença química entre 11 ortólogos (abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação
completa entre as ortólogas. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Classe 35 =
probabilidade média de ser deletéria; Classe 25 = não classificada; Classe 15 e C0 = benigna.
A outra variante identificada na paciente 143 foi a p.Pro264Arg (figura 23). Neste caso,
os aminoácidos possuem propriedades bem diferentes. Enquanto a prolina é um aminoácido
pequeno e alifático, a arginina é bem maior, sendo anfipático, ou seja, possui uma cadeia
alifática, hidrofóbica e outra polar, carregada positivamente. Ambos preferem a superfície das
proteínas, no entanto a prolina possui uma estrutura bem peculiar, pelo fato de se ligar duas
vezes ao esqueleto proteico, o que a torna um aminoácido que força voltas abruptas nas
proteínas,
principalmente
em
regiões
com
muitas
prolinas
seguidas
(poliprolinas).
Frequentemente as argininas estão envolvidas em pontes salinas, aonde pareiam com
aminoácidos negativos tais como o aspartato ou o glutamato para criar ligações de hidrogênio
estáveis que podem ser muito importantes para a estabilidade da proteína (Betts et al., 2003)
(figura 27).
99
Figura 27: Aminoácidos prolina e arginina envolvidos na mutação p.Pro264Arg. Acima está representada a
estrutura tridimensional e abaixo a fórmula estrutural. Estes aminácidos possuem propriedades bem diferentes. A
região circulada em laranja na prolina corresponde às cadeias laterais ligadas ao esqueleto de proteína tanto pelo
carbono quanto pelo nitrogênio. Na arginina, o grupamento amino positivo está circulado em vermelho. Vermelho:
átomo de oxigênio; azul: átomo de nitrogênio; preto: átomo de carbono; branco: átomo de hidrogênio. Adaptado de
http://biochemunclassified.wordpress.com/ e http://www.explicatorium.com/quimica/Aminoacido.
Na análise pelo ClustalW, verificou-se que a prolina na posição 264 é conservada
somente entre os três mamíferos e não entre todos os vertebrados avaliados (figura 28).
HOMEM
RATO
CAMUNDONGO
SAPO
PEIXE ZEBRA
VLFPPGPPVIEWPGLDEGHVRAGQSLELPCVARGGNPLATLQWLKNGQPV
ILFPPGPPVIDWPGLNEGHVRAGENLELPCTARGGNPPATLQWLKNGKPV
ILFPPGPPVIDWPGLNEGHVRAGENLELPCIARGGNPPATLQWLKNGKPV
VLFPPNAPIIEG--YTGPSVKAGETLKLICISLSGNPLATLQWSKNDDVV
VYFPPQAPMIMG--LENEEVKAGSFLKVVCMSYGGNPLATLHWTKNGEVL
: *** .*:*
*:**. *:: * : .*** ***:* **.. :
Figura 28: Alinhamento das sequências contendo a região vizinha do resíduo 264 (posições 236 a 285) da
proteína nefrina humana com as de quatro espécies de vertebrados. Análise realizada pelo programa Clustal W.
A prolina na posição 264 está ressaltada em amarelo. Este resíduo não é conservado entre os animais analisados ao
longo da evolução. O número de acesso Uniprot das sequências é: homem (O60500), rato (Q9R044), camundongo
(Q9QZS7), peixe zebra (Q1KMS5) e sapo (Q58QC3). Asteriscos (*) indicam posições que possuem um resíduo
conservado e único; Dois pontos (:) indicam conservação entre grupos de aminoácidos com propriedades similares;
Um ponto (.) indica conservação entre grupos de aminoácidos com propriedades diferentes.
100
A partir da análise com os três programas de predição in silico, confirma-se o que já
havia sido averiguado para a mutação anterior (tabela 16). O site Align-GVGD, que avalia a
composição, polaridade e o volume dos aminoácidos, após análise, retornou um resultado de alto
risco, já que os aminoácidos possuem propriedades bem diferentes. Através da análise do
PolyPhen, que avalia tanto a função biológica quanto a estrutura dos aminoácidos em função da
evolução, esta troca foi classificada em possivelmente prejudicial. Já a classificação através do
programa SIFT, que avalia somente a função protéica ao longo da evolução, o resultado foi
tolerável, considerando-se que este resíduo não é conservado entre todos os vertebrados
analisados.
Tabela 16: Predição in silico para a substituição missense p.Pro264Arg
Substituição de
SIFTa
PolyPhen2b
Align-GVGDc
aminoácido
p.Pro264Arg
Tolerável
(0,1)
Possivelmente
deletéria
(0,675)
Classe C65
GV = 0.00; GD = 102.71
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
b
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas. GD
(Grantham distance) = pontuação da diferença química entre os residues normal e mutante. Quanto maior a
pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class 35 =
probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
Ainda na paciente 143 foi identificada a troca de uma asparagina por uma serina na
posição 1077 da proteína, a p.Asn1077Ser (rs4806213), descrita pela primeira vez por Lenkkeri
et al. (Lenkkeri et al., 1999) (figura 29).
101
Figura 29: Eletroferograma e análise em grupo controle da alteração p.Asn1077Ser identificada na paciente
143. A. Heterozigose A/G ressaltada em amarelo na sequência acima dos pico do eletroferograma e abaixo denotada
em vermelho, onde se indica sua posição na sequência (c.3230A>G) do gene NPHS1.B. Alinhamento realizado com
o auxílio do programa CLC Sequence Viewer 6.3 após triagem de 93 indivíduos controle saudáveis, mostrando
sequências de 17 indivíduos dentre os quais dois apresentaram a heterozigose da substituição de um A por um G (R)
que leva a alteração p.Asn1077Ser.
Outros grupos também já a reportaram como uma variação frequente cuja associação com
a SN não ficou clara. Lahdenkari et al. (Lahdenkari et al., 2004) a identificaram em 3/50 alelos
(6%) de pacientes adultos finlandeses que haviam sido tratados para doença renal de lesões
mínimas na infância e em 9/50 alelos (18%) de indivíduos controle. Caridi et al. (Caridi et al.,
102
2009) a identificaram entre outras variantes em heterozigose, numa triagem onde estudaram
tanto o gene NPHS1 como o NPHS2, e fizeram uma correlação da p.Asn1077Ser com SN tardia
já que não a identificaram em 200 alelos controle analisados.
No presente trabalho identificamos a variante p.Asn1077Ser em heterozigose na paciente
143 e também em outros dois pacientes com SN na infância portadores de somente uma mutação
em heterozigose no gene NPHS2 e para os quais também analisamos o gene NPHS1 como será
descrito mais adiante (pacientes 6 e 12 – tabela 31). Para verificar a frequência desta alteração na
população brasileira, analisamos por sequenciamento 93 indivíduos controle (186 alelos) e
comparamos com as frequências genotípicas e alélicas descritas para algumas populações do
projeto 1000 genomes (Abecasis et al., 2012) (tabela 17). A população do projeto 1000 genomes
é constituída por 1092 indivíduos provenientes das seguintes populações: AFRICA (AFR),
composta por três subpopulações – ASW, americanos descendentes da África; LWK, habitantes
do Quênia e YRI, Yaruba, Nigéria. AMERÍNDIOS (AMR), composta por três subpopulações –
CLM, colombianos; MXL, moradores de Los Angeles com descendência mexicana e PUR,
habitantes de Porto Rico. ASIÁTICOS (ASN), composta por três subpopulações asiáticas –
CHB, Pequim, China; CHS, sul da China e JPT, Tóquio, Japão. EUROPEUS (EUR), composta
por cinco subpopulações europeias – CEU, descendentes de UTAH com descendência do norte e
oeste europeus; FIN, finlandeses; GBR, britânicos na Inglaterra e Escócia; IBS, população
ibérica da Espanha e TSI, região da Toscana, Itália. Nossos controles apresentaram as
frequências semelhantes às das populações AMR e EUR (tabela 17), o que está dentro do
esperado devido à nossa colonização.
103
Tabela 17: Comparação das frequências da variante p.Asn1077Ser (rs4806213) na
população controle brasileira com a de outras populaçõesa.
Populações
Genótipo
Frequência alélica
78 AA
13 AG
2 GG
A = 90,86%
G = 9,14%
AFR
64 AA
28 AG
1 GG
A = 84%
G = 16%
AMRd
78 AA
15 AG
0 GG
A = 92%
G = 8%
91 AA
2 AG
0 GG
A = 99%
G = 1%
(n = 93)b
Controles brasileiros
c
ASN
e
78 AA
A = 92%
15 AG
EUR
G = 8%
0 GG
a
Fonte: site www.ensembl.org, projeto 1000 genomes (Abecasis et al., 2012).
b
Para efeito de comparação, os números das populações foram corrigidos para o mesmo total de controles do atual
trabalho (n = 93).
c
AFR: três subpopulações: ASW: americanos descendentes da África; LWK: habitantes do Quênia e YRI: Yaruba,
Nigéria.
d
AMR: três subpopulações: CLM: colombianos; MXL: moradores de Los Angeles com descendência mexicana e
PUR: habitantes de Porto Rico.
e
ASN: três subpopulações asiáticas: CHB: Beijing, China; CHS: sul da China e JPT: Tóquio, Japão.
f
EUR: cinco subpopulações europeias: CEU: descendentes de UTAH com descendência do norte e oeste europeus;
f
FIN: finlandeses; GBR: britânicos na Inglaterra e Escócia; IBS: população ibérica da Espanha e TSI: região da
Toscana, Itália.
Os aminoácidos asparagina e serina possuem propriedades físico-químicas semelhantes,
ambos são polares e neutros. Observando-se a conservação deste resíduo, este é conservado ao
longo da evolução entre os vertebrados analisados (figura 30). Com relação aos resultados da
104
predição in silico para verificar o potencial efeito da substituição dos aminoácidos na proteína,
dois programas retornaram um resultado prejudicial para a proteína (tabela 18).
Com base nos dados apresentados, não é possível afirmar que esta variante esteja
agravando o quadro clínico da paciente 143.
No banco de dados de mutações humanas, o Human Gene Mutation Database (HGMD)
de acesso gratuito, das 97 mutações missense descritas, somente duas encontram-se entre os
resíduos 1059 e 1086, domínio transmembrana da nefrina: a p.Asn1077Ser e a p.Ala1078Asp. A
grande maioria das mutações (88/97, ~90%) encontra-se na região extracelular compreendida
pelos domínios do tipo imunoglobulina (Ig-1 a Ig-8), como se pode observar na figura 9. Assim,
uma provável justificativa para esta troca de aminoácidos ser menos grave pode residir no fato de
o resíduo Asn1077 estar localizado na porção transmembrana da nefrina, região até o momento
quase não afetada por mutações.
HOMEM
CAMUNDONGO
RATO
SAPO
PEIXE ZEBRA
P-LLPVLFALGGLLLLSNASCVGGVLWQRRLRRLAEGIS
P-LLPVLFAVGGLLLLSNASCVGGLLWRRRLRRLAEEIS
P-LLPVLFAVGGLLLLSNASCVGGLLWRRRLRRLAEEIS
PYILAAVCAGGALLLISNAAFIGCLVKRSRDKSQGGEGE
PVYVVVILVVGGVLFLLNPFICFLLARWKRRRSLKGNTS
* : .: . *.:*:: *.
:
* :
.
Figura 30: Alinhamento das sequências contendo a região vizinha do resíduo 1077 (posições 1060 a 1098) da
proteína nefrina humana com as de quatro espécies de vertebrados. A análise foi realizada através do programa
Clustal W. O aminoácido asparagina na posição 1077está ressaltado em amarelo. Este resíduo é conservado entre os
animais analisados ao longo da evolução, apesar de a região no qual ele se encontra inserido não ser conservada. O
número de acesso Uniprot das sequências é: homem (O60500), rato (Q9R044), camundongo (Q9QZS7), peixe zebra
(Q1KMS5) e sapo (Q58QC3). Asteriscos (*) indicam posições que possuem um resíduo conservado e único; Dois
pontos (:) indicam conservação entre grupos de aminoácidos com propriedades similares. Um ponto (.) indica
conservação entre grupos de aminoácidos com propriedades diferentes.
105
Tabela 18: Predição in silico para a variante p.Asn1077Ser.
Substituição de
SIFTa
PolyPhen2b
Align-GVGDc
aminoácido
p.Asn1077Ser
Deletéria
Benigna
Class C45
(0,03)
(0,01)
GV = 0.00; GD = 46.24
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
b
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas. GD
(Grantham distance) = pontuação da diferença química entre os residues normal e mutante. Quanto maior a
pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class 35 =
probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
Com relação às alterações missense p.Val736Met e p.Pro264Gln identificadas na região
extracelular da nefrina , somente após a realização de estudos de expressão em células HEK293,
avaliação das variantes quanto a sua localização celular e função, quanto ao comprometimento
da interação nefrina-nefrina e/ou nefrina-neph1 é que se poderá afirmar se estas são ou não
causadoras de doença ou se possuem um efeito grave ou menos grave na função da nefrina.
O terceiro indivíduo com SNC, o paciente 150, para o qual realizamos análise molecular,
foi acompanhado pelo hospital Universitário de Brasília, apresentando edema, proteinúria, GESF
e com hipótese diagnóstica de SNC. Após o sequenciamento das regiões codificantes do gene
NPHS1, foram identificadas duas mutações. Uma das mutações localiza-se no éxon 2 e consiste
na deleção de uma guanina na posição 139 do éxon 2, c.139delG. Esta deleção acarreta em um
frameshift de leitura a partir do ácido glutâmico (E) na posição 46 da proteína nefrina (ID do
transcrito: ENST00000378910) gerando um códon de parada de leitura prematura na posição 127
da proteína, p.Ala47Pro4fs*127 (Heeringa et al., 2008). A outra mutação identificada foi a
106
mesma do paciente 128, a c.514-6del3nt que causa a deleção do aminoácido treonina na posição
172 da proteína (p.Thr172del) (Lenkkeri et al., 1999). Após estudo molecular do DNA dos pais
identificou-se que a mutação frameshift p.Ala47Pro4fs*127 foi herdada da mãe e a p.Thr172del,
do pai, confirmando-se assim a heterozigose composta (figura 31).
A mutação c.139delG no éxon 2 já foi identificada em heterozigose em um paciente com
descendência afro americana, com SNCR e biópsia com histologia de lesões mínimas, por
Heeringa et al. (Heeringa et al., 2008). O grupo justificou o fenótipo menos grave do paciente
em questão, que respondeu aos corticoides, devido ao fato de a mutação estar em heterozigose. O
fato dos pais serem portadores destas alterações em heterozigose e não possuírem SN confirma a
herança autossômica recessiva neste caso sendo o genótipo compatível com o quadro clínico de
SNC deste paciente.
Ainda no estudo do gene NPHS1 foram identificadas heterozigoses ou homozigoses de
variantes mais raras de trocas nucleotídicas tanto nos pacientes com SNC como nos 13 pacientes
(em um dos 14 pacientes com apenas uma alteração no gene NPHS2 não foi realizada a análise
de polimorfismos no gene NPHS1) com SN na infância/juvenil para os quais o gene NPHS1
também foi analisado. Para estas variações foi feita uma análise comparativa dos dados das
frequências alélicas entre os pacientes com SNC e uma população controle. Estes dados serão
discutidos mais adiante quando serão apresentados (tabelas 35, 36 e 37).
107
Figura 31: Heredograma da família do paciente 150 e eletroferogramas das mutações identificadas no gene
NPHS1. Do lado esquerdo visualiza-se o heredograma da família do paciente 150, verificando-se a segregação
alélica das alterações que encontram-se em heterozigose composta no paciente. À direita, estão ilustrados os
eletroferogramas das mutações identificadas: a deleção de um G denotado em vermelho na sequência normal
(c.139delG) em heterozigose, que causa o desalinhamento na análise da sequência, como pode ser visto pelas letras
ressaltadas em amarelo após a deleção e que leva à mutação frameshift p.Ala47Profs*127. Deleção c.514-6del3nt,
em heterozigose, correspondente aos 3 nucleotídeos CCA representados em vermelho na sequência normal, que
também causa o desalinhamento da sequência e leva à deleção do aminoácido treonina, p.Thr172del na proteína.
108
A figura 32 ilustra a posição das mutações identificadas na nefrina neste trabalho.
Figura 32: Ilustração da proteína nefrina identificando os domínios aonde foram encontradas as mutações
do gene NPHS1 nos pacientes com SNC. Setas pontilhadas indicam mutações em região de splicing. Observar a
concentração de mutações na região extracelular da nefrina.
109
2. PACIENTES COM SN INFANTIL, NA INFÂNCIA, SN
JUVENIL E COM PROTEINÚRIA ISOLADA
2.1 Gene NPHS2
A figura 33 mostra o resultado geral para os pacientes triados para o gene NPHS2.
Figura 33: Fluxograma geral dos resultados após triagem do gene NPHS2 dos pacientes com SN infantil, SN na
infância, SN juvenil e proteinúria isolada.
110
Os oito éxons, regiões limítrofes íntron/éxon e região promotora do gene NPHS2 foram
analisados em 147 pacientes (características clínicas nas tabelas do anexo II).
2.1.1 Pacientes com duas alterações em heterozigose
Em quatro pacientes foram identificadas duas alterações em heterozigose no gene NPHS2
(4/147) correspondendo a 2,7 % (figura 33 e tabela 19).
Tabela 19: Pacientes com 2 mutações no gene NPHS2.
Pacientes
Mutação
Éxon
Mutação
Éxon
Idade
Início
Resposta ao
tratamento
10
p.Arg229Gln
5
p.Ala284Val
7
12a
CR
81
p.Arg229Gln
5
p.Glu310Lis
8
2a2m
CR
p.Lis239Argfx13*
5
p.Val260Glu
6
131*
132*
3m
CR
1a
*As pacientes 131 e 132 são irmãs e provenientes da UNIFESP, SP; a = anos; m= meses.
A paciente 10, encaminhada pelo departamento de Nefrologia Pediátrica do HC da
Unicamp, desenvolveu SN com 12 anos de idade e teve diagnóstico histológico de GESF após
biópsia renal. No estudo molecular do gene NPHS2 duas variantes foram identificadas, uma no
éxon 5 e outra no éxon 7. Os éxons 5 e 7 dos pais também foram analisados: a variante
111
p.Ala284Val foi identificada na mãe e a p.Arg229Gln, no pai (figura 34), confirmando-se a
heterozigose composta no caso índice.
Figura 34: Heredograma da família da paciente 10 e eletroferogramas das alterações identificadas no gene
NPHS2. Do lado esquerdo visualiza-se o heredograma da família da paciente 10 aonde é possível verificar a
segregação alélica das alterações que encontram-se em heterozigose composta na paciente. À direita, estão
ilustrados os eletroferogramas das alterações identificadas: as heterozigoses G/A e C/T encontram-se ressaltadas em
amarelo nas sequências acima dos picos dos eletroferogramas e abaixo estão denotadas em vermelho, onde se
indicam as alterações c.686G>A (p.Arg229Gln) e c.851C>T (p.Ala284Val), respectivamente.
A variante identificada em heterozigose no éxon 5 foi gerada pela transição de uma
guanina para uma adenina na posição 686 e que leva à troca de uma arginina por uma glutamina
na posição 229 da proteína, a alteração p.Arg229Gln (p.R229Q).
A análise do alinhamento múltiplo de sequências no ClustalW de algumas espécies de
vertebrados para as quais as sequências ortólogas da podocina estão disponíveis no Uniprot
112
(http://www.uniprot.org/) mostra que a arginina na posição 229 é um aminoácido conservado;
somente no Danio rerio (paulistinha, peixe zebra) que a histidina ocupa esta mesma posição, no
entanto este aminoácido possui propriedades similares às da arginina (ambos possuem carga
positiva) (figura 35).
HOMEM
CAMUNDONGO
RATO
SAPO
PEIXE ZEBRA
SLAHVSKAVQFLVQTTMKRLLAHRSLTEILLERKSIAQDAKVALDSVTCIWG
SLAHVSKAIQFLVQTTMKRLLAHRSLTEILLERKSIAQDVKVALDAVTCIWG
SLAHVSKAIQFLVQTTMKRLLAHRSLTEILLERKSIAQDVKVALDSVTCVWG
SVSNISSAFQLLVQTTTKRLLAHRAFLDILLERKSIGEEVKVALDAATCHWG
SFASIPDVMQALTQVSVREILAHHAFNDILLDRKRIAQEIQVTLDSGTCRWG
*.: :....* *.*.: :.:***::: :***:** *.:: :*:**: ** **
Figura 35: Alinhamento das sequências contendo a região vizinha do resíduo 229 (posições 205 a 257) da
proteína podocina humana com as de quatro espécies de vertebrados. Análise realizada pelo programa Clustal
W. O aminoácido arginina na posição 229 está ressaltado. O aminoácido histidina, com propriedades similares e que
ocupa a mesma posição no peixe zebra, está representado em verde. O número de acesso Uniprot das sequências é:
homem (Q9NP85), rato (Q8K4G9), camundongo (Q91X05), peixe zebra (Q52TF2) e sapo (C8C505). Asteriscos (*)
indicam posições que possuem um resíduo conservado e único; Dois pontos (:) indicam conservação entre grupos de
aminoácidos com propriedades similares. Um ponto (.) indica conservação entre grupos de aminoácidos com
propriedades diferentes.
Com relação às propriedades físico-químicas destes aminoácidos, a arginina, como já
mencionado anteriormente, é um aminoácido polar e com carga positiva. Já a glutamina também
é polar, porém não tem carga, o que pode influenciar nas interações que estes aminoácidos
estabelecem dentro da proteína (figura 36).
Assim como a nefrina, a podocina também é uma proteína de membrana e ainda não foi
cristalizada. Não foram encontradas proteínas homólogas depositadas no banco de dados PDB
que pudessem ser utlilzadas para modelamento em estudos estruturais.
De acordo com os programas de predição in silico analisados, esta troca foi classificada
de maneira diferente pelos três programas utilizados (tabela 20). Com relação à função e
113
estrutura dos aminoácidos substituídos, análise realizada pelos programas Polyphen e
AlignGVGD, a substituição foi considerada deletéria e com probabilidade média de ser deletéria,
respectivamente. Já o programa SIFT classificou a variante como tolerável, já que a ortóloga do
peixe zebra apresenta outro resíduo na mesma posição.
Figura 36: Aminoácidos arginina e glutamina envolvidos na alteração p.Arg229Gln. Acima está representada a
estrutura tridimensional e abaixo a fórmula estrutural. Estes aminácidos possuem propriedades bem diferentes. Na
arginina, o grupamento amino positivo está circulado em vermelho. Vermelho: átomo de oxigênio; azul: átomo de
nitrogênio;
preto:
átomo
de
carbono;
branco:
átomo
de
hidrogênio.
Adaptado
de
http://biochemunclassified.wordpress.com/ e http://www.explicatorium.com/quimica/Aminoacido.
114
Tabela 20: Predição in silico para a substituição missense p.Arg229Gln
SUBSTITUIÇÕES
SIFT
PolyPhen
Align-GVGD
Tolerável
(0,11)
Provavelmente
deletéria
(0,903)
Classe C35
GV = 0.00; GD = 42.81
DE AMINOÁCIDOS
p.Arg229Gln
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
b
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas de
podocina. GD (Grantham distance) = pontuação da diferença química entre os resíduos normal e mutante. Quanto
maior a pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class
35 = probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
Sob o ponto de vista de frequência alélica, esta variante pode ser considerada um
polimorfismo, já que a frequência do alelo raro na população em geral é 2% (www.ensembl.org)
(considerando MAF < 0,01 como a frequência limítrofe para uma alteração ser considerada uma
mutação). Karle et al. (Karle et al., 2002), ao estudarem famílias e pacientes esporádicos com
SNCR, descreveram a variante p.Arg229Gln pela primeira vez como um polimorfismo, pois a
identificaram em 3% dos 200 alelos controle estudados. No entanto, ressaltaram o fato de que
nas quatro famílias em cujos casos índice foi identificada a variante p.Arg229Gln, também havia
sido encontrada outra mutação no outro alelo, descrita como deletéria, o que deixava dúvidas
com relação ao efeito que este “polimorfismo” podia ter como causa da doença.
Tsukaguchi et al. (Tsukaguchi et al., 2002), ao estudarem pacientes com início tardio de
GESF em 30 famílias, identificaram a variante p.Arg229Gln nos indivíduos afetados de duas
famílias em heterozigose composta com uma mutação já descrita como patogênica, a
115
p.Arg291Trp. Os membros não afetados das referidas famílias apresentavam somente uma das
duas mutações, comportamento de segregação alélica consistente com um padrão de herança
autossômica recessiva. Em seu trabalho, Tsukaguchi et al. também analisaram 91 pacientes
esporádicos com GESF e identificaram a variante p.Arg229Gln em heterozigose em 11 pacientes
(6%), sendo que em dois desses havia associação com a mutação p.Ala284Val no outro alelo.
Após análise de grupo controle proveniente de três populações distintas, identificaram a variante
p.Arg229Gln em 1,6% (1/64 alelos) de indivíduos com descendência africana, 3,1% (3/98 alelos)
em indivíduos com descendência brasileira e 3,6% (9/248 alelos) em indivíduos com
descendência ocidental (americana e europeia). Intrigados, realizaram estudos funcionais com a
intenção de demonstrarem o efeito biológico da variante p.Arg229Gln. Como já havia sido
demonstrada a colocalização e a interação direta da podocina com a nefrina nas fendas de
diafragma (Huber et al., 2001; Schwarz et al., 2001), Tsukaguchi et al. realizaram ensaios de
ligação à nefrina para verificar se havia diferença biológica entre os peptídios com e sem a
mutação. Experimentos de “pull down” com anticorpos antipodocina demonstraram uma
diminuição da interação da variante p.Arg229Gln com a nefrina se comparada com a variante
normal. Desde modo, esse grupo concluiu que a variante p.Arg229Gln tem sua função biológica
de ligação com a nefrina diminuída e que, associada com outra mutação deletéria no gene
NPHS2, causa desenvolvimento tardio de GESF.
Outros grupos também avaliaram a frequência da variante p.Arg229Gln, tanto em
pacientes, como em controles saudáveis (McKenzie et al., 2007; Ruf et al., 2004; Weber et al.,
2004). Geralmente a frequência encontrada para a variante p.Arg229Gln é maior entre os
pacientes do que entre os controles, assim como no trabalho de Tsukaguchi et al., porém os
trabalhos descrevem a diferença como não significativa estatisticamente para que esta variante
116
seja associada com a doença. Nenhum dos trabalhos encontrou a variante em homozigose no
grupo controle. Baseando-se nesses achados, os autores desses trabalhos concordam que a
variante p.Arg229Gln, quando em heterozigose simples, não é um alelo de risco para
desenvolvimento de GESF, porém, quando associado a uma mutação deletéria em outro alelo,
predispõe ao desenvolvimento tardio de GESF.
Para verificar a frequência do alelo p.Arg229Gln em população controle, analisamos 109
(218 alelos) indivíduos saudáveis com a técnica de PCR alelo-específico (Figura 37). A variante
p.Arg229Gln foi identificada em heterozigose em apenas um indivíduo (0,45%; 1/218).
Figura 37: Análise de indivíduos controle para alteração p.Arg229Gln. Foto de gel de agarose corado com
brometo de etídio e visualizado com luz UV após PCR alelo específico de 109 indivíduos (218 alelos) para a
alteração p.Arg229Gln. Somente parte da análise está ilustrada. M: marcador de peso molecular 1 kb plus
(Invitrogen®). Poço 29: controle negativo. Poço 30: controle heterozigoto. O indivíduo controle no poço 3 é
positivo para a p.Arg229Gln.
117
Um trabalho apresentado recentemente no congresso europeu de genética humana
“European Human Genetics Conference 2013” (Tory et al., 2013) traz novas descobertas para a
questão da variante p.Arg229Gln. Esse grupo observou que seis dos 129 parentes saudáveis de
crianças afetadas com SNCR apresentavam a variante p.Arg229Gln associada com outra
mutação, sendo que esta outra mutação localizava-se sempre entre os éxons 1 e 6. Por outro lado,
curiosamente, as mutações associadas com a p.Arg229Gln nos pacientes afetados, eram
mutações missense localizadas entre os éxons 7-8. Com o intuito de estudar este comportamento
deletério das mutações ao nível celular, esse grupo estudou a localização subcelular da variante
[Arg229Gln] em função da mutação associada, em podócitos cultivados in vitro. Os resultados
foram conclusivos: a podocina com a variante [Arg229Gln] não foi corretamente co-localizada
nos espaços subcelulares quando as mutações associadas se localizavam nos éxons 7 e 8. No
entanto, quando a variante [Arg229Gln] encontrava-se associada a mutações entre os éxons 1-6,
as isoformas possuíam comportamento normal dentro dos podócitos. Esse trabalho traz um
conceito de herança autossômica recessiva dependente da mutação, mecanismo que vai além das
leis de Mendel, pois a patogênese de um alelo depende do tipo e localização de mutações no
outro alelo. Assim, dependendo da mutação associada à variante p.Arg229Gln, esta pode sim ter
um papel fundamental no desenvolvimento da doença.
No caso da paciente 10 aqui descrita, a outra mutação identificada foi a p.Ala284Val, no
éxon 7, e com base nos achados reportados pelo grupo de Tory, podemos concluir que o genótipo
deste paciente condiz plenamente com seu fenótipo. A mutação p.Ala284Val foi descrita por
Karle et al. (Karle et al., 2002) em dois pacientes, sendo que nos dois, também havia sido
identificado o polimorfismo p.Arg229Gln no outro alelo. A associação destas duas mutações em
heterozigose composta também já foi descrita por Machuca et al. em 15 famílias, sendo 13
118
destas da América do Sul (Argentina e Chile) e com origem hispânica (Machuca et al., 2009). A
média de idade de desenvolvimento de GESF foi de aproximadamente 14 anos entre os
indivíduos afetados destas 15 famílias.
Tanto a alanina como a valina são aminoácidos hidrofóbicos. Porém a alanina é um
aminoácido pequeno e comum, enquanto que a valina, por sua vez, é um aminoácido grande e
possui uma estrutura peculiar, é ramificado em seu carbono beta (Cβ branched), o que influencia
na rigidez das proteínas e faz com que este aminoácido “prefira” folhas beta e não consiga se
inserir em regiões de alfa hélice (figura 38). Somente três aminoácidos possuem este tipo de
ramificação no carbono β: a valina, a isoleucina e a treonina.
O alinhamento de múltiplas sequências realizado pelo ClustalW mostra como o
aminoácido alanina é conservado ao longo da evolução entre as espécies de vertebrados
analisadas (figura 39).
Figura 38: Aminoácidos alanina e valina envolvidos na mutação p.Ala284Val. Acima está representada a
estrutura tridimensional e abaixo a fórmula estrutural. O círculo amarelo mostra a ramificação no carbono β.
Vermelho: átomo de oxigênio; azul: átomo de nitrogênio; preto: átomo de carbono; branco: átomo de hidrogênio
Adaptado de http://biochemunclassified.wordpress.com/ e http://www.explicatorium.com/quimica/Aminoacido.
119
HOMEM
CAMUNDONGO
RATO
SAPO
PEIXE ZEBRA
LPAGLQHSLAVEAEAQRQAKVRMIAAEAEKAASESLRMAAEILSG
LPAGLQHSLAVEAEAQRQAKVRVIAAEGEKAASESLRMAAEILSG
LPAGLQHSLAVEAEAQRQAKVRVIAAEGEKAASESLRMAAEILSG
LPEEVKQSIAVEAEAQRHAKVKVIAAEGEKTVSEYIKLAAEKLSG
LPPELQHNFAVEAEARRQAQVKVIAAEGEKAACEALKASVESVSG
** :::.:******:*:*:*::****.**:..* :: :.* :**
Figura 39: Alinhamento das sequências contendo a região vizinha do resíduo 284 (posições 270 a 314) da
proteína podocina humana com as de quatro espécies de vertebrados. A análise foi realizada pelo programa
Clustal W. O aminoácido alanina na posição 284 está ressaltado em amarelo. O número de acesso Uniprot das
sequências é: homem (Q9NP85), rato (Q8K4G9), camundongo (Q91X05), peixe zebra (Q52TF2) e sapo (C8C505).
Asteriscos (*) indicam posições que possuem um resíduo conservado e único; Dois pontos (:) indicam conservação
entre grupos de aminoácidos com propriedades similares. Um ponto (.) indica conservação entre grupos de
aminoácidos com propriedades diferentes.
Na tabela 21 estão apresentados os resultados das predições in silico, sendo que em todos
os programas a mutação p.Ala284Val foi avaliada como potencialmente prejudicial para a
função da podocina.
Tabela 21: Predição in silico para a substituição missense p.Ala284Val.
SUBSTITUIÇÕES DE
SIFT
PolyPhen
Align-GVGD
Deletéria
Provavelmente
deletéria
Class C65
AMINOÁCIDOS
p.Ala284Val
(0,00)
(1,000)
GV = 0.00; GD = 65.28
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
b
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas de
podocina. GD (Grantham distance) = pontuação da diferença química entre os resíduos normal e mutante. Quanto
maior a pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class
35 = probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
120
A paciente 10, atualmente com 18 anos, evoluiu para insuficiência renal e já foi
transplantada. A idade de início da SN na paciente foi aos 12 anos, próxima à média de 14 anos
encontrada pelo grupo de Machuca (Machuca et al., 2009), nos quais os pacientes apresentavam
a mesma associação [p.Arg229Gln]:[p.Ala284Val] que a paciente aqui descrita. Porém,
diferentemente dos pacientes identificados por esse grupo, que eram em sua maioria córtico
sensíveis e cuja idade média de início de diálise foi de 22 anos, a paciente 10 do nosso trabalho
apresentou-se resistente ao tratamento com os corticoides e teve uma evolução mais rápida para
insuficiência renal.
A segunda paciente na qual foram identificadas duas mutações em heterozigose no gene
NPHS2 foi a paciente 81. Esta paciente teve idade de início da SN aos dois anos e dois meses de
idade, com diagnóstico histológico de proliferação mesangial difusa e apresentou resistência aos
corticoides. A paciente 81 apresentou a mesma variante p.Arg229Gln identificada na paciente 10
e já discutida anteriormente, além da mutação p.Glu310Lis, no éxon 8 (figura 40, tabela 18). A
variante p.Arg229Gln foi herdada da mãe, e a p.Glu310Lis, muito provavelmente do pai
(material não disponível para análise).
121
Figura 40: Heredograma da família da paciente 81 e eletroferogramas das alterações identificadas
identificadas no gene NPHS2. Do lado esquerdo visualiza-se o heredograma da família 81. Não foi possível
verificar a segregação alélica pois o material do pai não estava disponível para análise. Quadrado com ponto de
interrogação: sem análise molecular. À direita estão ilustrados os eletroferogramas das alterações identificadas: as
substituições foram encontradas em heterozigose, ambas sendo a troca de uma guanina por uma adenina (G/A) e
encontram-se ressaltadas em amarelo nas sequências acima dos picos dos eletroferogramas e abaixo estão denotadas
em vermelho, onde se indicam as alterações c.686G>A (p.Arg229Gln) e c.928G>A (p.Glu310Lis), respectivamente.
O ácido glutâmico na posição 310 localiza-se em uma região rica em alanina e glutamato,
extremamente conservada entre as proteínas da família da estomatina-7 (figura 41). Algumas
substituições já foram descritas para esta posição: p.Glu310Val (Ruf et al., 2004; Tsukaguchi et
al., 2002), p.Glu310Ala e, inclusive a p.Glu310Lis (Machuca et al., 2009).
122
HOMEM
CAMUNDONGO
RATO
SAPO
PEIXE ZEBRA
AVEAEAQRQAKVRMIAAEAEKAASESLRMAAEILSGTPAAVQLRYL
AVEAEAQRQAKVRVIAAEGEKAASESLRMAAEILSGTPAAVQLRYL
AVEAEAQRQAKVRVIAAEGEKAASESLRMAAEILSGTPAAVQLRYL
AVEAEAQRHAKVKVIAAEGEKTVSEYIKLAAEKLSGSPTAIQLRYL
AVEAEARRQAQVKVIAAEGEKAACEALKASVESVSGSPLVMHLRLL
******:*:*:*::****.**:..* :: :.* :**:* .::** *
Figura 41: Alinhamento das sequências contendo a região vizinha do resíduo 310 (posições 279 a 324) da
proteína podocina humana com as de quatro espécies de vertebrados. A análise foi realizada com o programa
Clustal W. O aminoácido glutamina na posição 310 está ressaltado em amarelo. Esta região é conservada e rica em
alanina e glutamato (ressaltado em cinza). O número de acesso Uniprot das sequências é: homem (Q9NP85), rato
(Q8K4G9), camundongo (Q91X05), peixe zebra (Q52TF2) e sapo (C8C505). Asteriscos (*) indicam posições que
possuem um resíduo conservado e único; Dois pontos (:) indicam conservação entre grupos de aminoácidos com
propriedades similares. Um ponto (.) indica conservação com entre grupos de aminoácidos com propriedades
distintas.
Os aminoácidos ácido glutâmico e lisina possuem algumas propriedades em comum:
ambos são polares, situam-se em regiões de superfície das proteínas e estabelecem pontes
salinas. Porém o ácido glutâmico é carregado negativamente e a lisina é um aminoácido básico, o
que resulta em uma substituição com efeitos drásticos, já que os contatos estabelecidos devem
ser bem diferentes (figura 42).
Os resultados das predições in silico que estão detalhados na tabela 22, indicam que
provavelmente esta substituição tenha um efeito prejudicial para a proteína podocina, já que dois
programas a avaliaram como deletéria. O programa SIFT a classificou como tolerável, porém há
uma margem de erro para as predições in silico que, na ferramenta SIFT é de 31% para
resultados falso-negativos.
123
Figura 42: Aminoácidos ácido glutâmico e lisina envolvidos na mutação p.Glu310Lis. Acima está representada
a estrutura tridimensional e abaixo a fórmula estrutural. Estes aminácidos possuem propriedades diferentes, sendo
um carregado negativamente e o outro positivamente (grupamentos circulados em vermelho e azul, respectivamente.
Vermelho: átomo de oxigênio; azul: átomo de nitrogênio; preto: átomo de carbono; branco: átomo de hidrogênio.
Adaptado de http://biochemunclassified.wordpress.com/ e http://www.explicatorium.com/quimica/Aminoacido.
.
Tabela 22: Predição in silico para a substituição missense p.Glu310Lis.
SUBSTITUIÇÕES DE
SIFT
PolyPhen
Align-GVGD
AMINOÁCIDOS
p.Glu310Lis
Tolerável
(0,20)
Provavelmente
deletéria
(0,992)
Classe C55
GV = 0.00; GD = 56.87
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
b
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas de
podocina. GD (Grantham distance) = pontuação da diferença química entre os resíduos normal e mutante. Quanto
maior a pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class
35 = probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
124
Realizamos análise desta mutação em grupo controle com 107 indivíduos (214 alelos) e
nenhuma variante p.Glu310Lis foi encontrada (figura 43).
Figura 43: Análise de indivíduos controle para a alteração p.Glu310Lis. Foto de gel de agarose corado com
brometo de etídio e visualizado com luz UV após PCR alelo específico 107 indivíduos controle (214 alelos) para a
alteração p.Glu310Lis. Somente parte da análise está ilustrada. M: marcador de peso molecular 1 kb plus
(Invitrogen®). Poços 19 e 33: controle heterozigoto. Poço 32: controle negativo. Nenhum indivíduo controle
apresentou a alteração.
Atualmente a paciente 81 está com seis anos de idade, é CR e apresenta proteinúria, mas
sem edema e com a função renal estável.
As outras duas pacientes com duas mutações no gene NPHS2 são irmãs e foram
encaminhadas pelo serviço de nefrologia pediátrica da UNIFESP, SP. A paciente 131 teve início
de proteinúria aos três meses de idade e GESF como resultado de biópsia. A irmã mais velha,
paciente 132, iniciou quadro de SN com um ano e dois meses de idade e o resultado da biópsia
também foi de GESF. Ambas são resistentes ao tratamento com corticoides.
125
Identificamos duas mutações em heterozigose nestas duas pacientes (figura 44): a deleção
de um G na posição 714 do cDNA (ID do transcrito: ENST00000367615), que resulta em uma
proteína modificada a partir do aminoácido lisina na posição 239 e é interrompida quando
encontra um código de parada prematura após 13 resíduos (p.Lis239Argfx13*) e a mutação
missense p.Val260Glu. A mutação p.Lis239Argfx13* já foi anotada no banco de dados
www.ensembl.org com a referência “CM042097” porém no HGMD público só consta o registro
de uma deleção de nove nucleotídeos na mesma região, identificada pelo grupo de Weber et al.
(Weber et al., 2004). No HGMD professional consta uma mutação indel, porém não tivemos
acesso a esta descrição. A mutação p.Val260Glu já foi descrita anteriormente pelo mesmo grupo
(Weber et al., 2004).
Não foi realizada a análise molecular dos pais, portanto não podemos afirmar que estas
mutações estejam em heterozigose composta no paciente, apesar de ser um caso familiar de
SNCR fortemente sugestivo de herança autossômica recessiva.
126
Figura 44: Heredograma da família das irmãs 131 e 132 e eletroferogramas das alterações identificadas no
gene NPHS2. Do lado esquerdo visualiza-se o heredograma das irmãs 131 e 132, sendo que não foi possível
verificar a segregação alélica pois o material dos pais não estavam disponíveis para análise. Os pontos de
interrogação significam: sem análise molecular À direita estão ilustrados os eletroferogramas das mutações
identificadas. Acima, a deleção de um G denotado em vermelho na sequência normal (c.714delG) em heterozigose,
que causa o desalinhamento na análise da sequência, como pode ser visto pelas letras ressaltadas em amarelo após a
deleção e que leva à mutação frameshift p.Lis239Argfs*13; abaixo, a heterozigose T/A está ressaltada em amarelo
na sequência acima do pico do eletroferograma e denotada em vermelho na sequência abaixo, referente à alteração
c.779T>A (p.Val260Glu).
A mutação p.Lis239Argfx13* gera uma proteína modificada a partir do resíduo 239,
sendo interrompida no aminoácido 252, quando encontra um código de parada prematura.
Assim, o domínio PHB não é completamente formado (resíduos 128 a 284) em um dos alelos
(figura 7). Já foi demonstrado que o domínio PHB é responsável pela interação entre a podocina
e a nefrina (Huber et al., 2003). Deleções deste domínio ou mutações na nefrina (tais como a Fin
minor no aminoácido 1109) interrompem completamente esta interação, indicando que o
domínio PHB da podocina e a região mais próxima à porção C-terminal da nefrina são
127
necessárias para que a ligação entre estas duas proteínas ocorra. Estudos realizados com a
mutação p.Arg140Gln de camundongo e que corresponde à mutação missense p.Arg138Gln
(p.R138Q) humana, uma das mais frequentes em pacientes com SNCR, mostraram que esta não
prejudicou a ligação da podocina com a nefrina, nem a sua habilidade em realizar interações
homofílicas. Porém, de forma contrária, a mutação p.Arg138* (p.R138*) na mesma posição e
que causa a perda quase total do domínio PHB, resultou na perda da interação com a nefrina
(Huber et al., 2003). Huber et al., nesse mesmo trabalho, realizaram experimentos com
transfecção transiente de células HEK293T (células derivadas das células HEK293, mais
estáveis) com várias podocinas construídas mutantes e demonstraram que a podocina é
responsável em recrutar a nefrina para os “rafts” de lipídios. Construções tanto com a mutação
missense p.Arg138Gln como com a p.Arg138* interferiram na capacidade da podocina de
recrutar a nefrina para os “rafts” de lipídios. Através de estudos com microscopia confocal para
verificar a colocalização subcelular destas duas podocinas mutantes comparando-as com a
podocina normal, esse grupo demonstrou que a proteína normal foi inserida na membrana
plasmática dos podócitos, enquanto que a p.Arg140Gln (os experimentos foram realizados com a
podocina ortóloga de camundongos) colocalizou com a calnexina, indicando que este mutante
não deixou o retículo endoplasmático; em contraste, a p.Arg140* alcançou a membrana
plasmática mas não foi corretamente colocalizada nos “rafts” de lipídios. Esses resultados
demonstraram que as mutações mencionadas podem prejudicar dois níveis de organização
celular: o transporte intracelular entre as organelas e a membrana plasmática e o direcionamento
entre as frações da membrana plasmática (de regiões sem “rafts” para regiões com “rafts” de
lipídios).
128
A partir desses achados, pode-se inferir que a mutação p.Lis239Argfx13* identificada
neste trabalho deva prejudicar a interação entre as duas proteínas e o recrutamento da nefrina
para os “rafts” de lipídios. Porém, somente estudos funcionais de expressão e colocalização
subcelular com a nefrina, tais como os realizados pelo grupo de Huber, poderão indicar o real
impacto desta mutação na função da podocina.
A substituição missense de uma valina por um ácido glutâmico na posição 260 da
proteína, codificada pelo éxon 6 (c.779T>A, p.Val260Glu) foi identificada nas pacientes 131 e
132 (figura 44). O éxon 6 codifica a porção do domínio PHB localizado mais próximo da região
C-terminal da podocina (figura 7). A posição que a valina ocupa é conservada ao longo da
evolução em todas as espécies de vertebrados analisados pelo programa ClustalW (figura 45).
CAMUNDONGO
RATO
HOMEM
SAPO
PEIXE ZEBRA
SIAQDVKVALDAVTCIWGIKVERTEIKDVRLPAGLQHSLAVEAEA
SIAQDVKVALDSVTCVWGIKVERTEIKDVRLPAGLQHSLAVEAEA
SIAQDAKVALDSVTCIWGIKVERIEIKDVRLPAGLQHSLAVEAEA
SIGEEVKVALDAATCHWGIKVERTEIKDVKLPEEVKQSIAVEAEA
RIAQEIQVTLDSGTCRWGIKVEKAEIEEINLPPELQHNFAVEAEA
*.:: :*:**: ** ******: **:::.** :::.:******
Figura 45: Alinhamento das sequências contendo a região vizinha do resíduo 260 (posições 240 a 284) da
proteína podocina humana com as de quatro espécies de vertebrados. A análise foi realizada com o programa
Clustal W. O aminoácido valina na posição 260 está ressaltado em amarelo e é conservado entre as espécies
analisadas. O número de acesso Uniprot das sequências é: homem (Q9NP85), rato (Q8K4G9), camundongo
(Q91X05), peixe zebra (Q52TF2) e sapo (C8C505). Asteriscos (*) indicam posições que possuem um resíduo
conservado e único; Dois pontos (:) indicam conservação entre grupos de aminoácidos com propriedades similares;
Um ponto (.) indica conservação entre grupos de aminoácidos com propriedades diferentes.
Os aminoácidos valina e ácido glutâmico possuem propriedades bem diferentes: enquanto
a valina é um aminoácido pequeno, neutro e prefere ficar oculto no interior das proteínas, o ácido
glutâmico é um aminoácido grande, carregado negativamente e prefere a superfície das proteínas
(figura 46).
129
Figura 46: Aminoácidos valina e ácido glutâmico envolvidos na substituição missense p.Val260Glu. Acima
está representada a estrutura tridimensional e abaixo a fórmula estrutural. Estes aminácidos possuem propriedades
diferentes, sendo um neutro e o outro carregado positivamente (grupamento circulado em vermelho). Vermelho:
átomo de oxigênio; azul: átomo de nitrogênio; preto: átomo de carbono; branco: átomo de hidrogênio. Adaptado de
http://biochemunclassified.wordpress.com/ e http://www.explicatorium.com/quimica/Aminoacido.
.
Realmente, confirmando os dados anteriores, tanto o de conservação, como o das
propriedades dos aminoácidos, os três sites de predição in silico indicaram que esta substituição
de aminoácidos é potencialmente prejudicial para a função da proteína (tabela 23).
130
Tabela 23: Predição in silico para substituição p.Val260Glu.
SUBSTITUIÇÕES DE
SIFT
PolyPhen
Align-GVGD
Deletéria
Provavelmente
deletéria
Classe C65
AMINOÁCIDOS
p.Val260Glu
(0,00)
(1,00)
GV = 0.00; GD = 121,34
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
b
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas de
podocina. GD (Grantham distance) = pontuação da diferença química entre os resíduos normal e mutante. Quanto
maior a pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class
35 = probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
A mutação p.Val260Glu foi identificada pela primeira vez em homozigose em quatro
pacientes com SNCR (Weber et al., 2004); esse grupo avaliou 320 alelos controle e não
identificou a mutação p.Val260Glu em nenhum.
Roselli et al. (Roselli et al., 2004) realizaram estudos in vitro de tráfego intracelular com
moléculas de podocina mutantes, das quais 11 eram derivadas de mutações missense e uma de
mutação frameshift e sugeriram que a gravidade e a idade de início da doença são determinados
pela localização destas substituições de aminoácidos em domínios específicos da podocina e pelo
impacto que estas exercem no tráfego intracelular da podocina nos podócitos. Esse grupo
demonstrou que proteínas portadoras de algumas mutações missense, entre elas a p.Val260Glu,
são retidas no retículo endoplasmático (RE), não sendo transportadas para a membrana
plasmática. Através de uma correlação genótipo-fenótipo, o mesmo grupo concluiu que os
pacientes com mutações missense que produzem proteínas retidas no RE, as quais denominaram
“mais graves”, juntamente com as mutações frameshift ou ainda nonsense, desenvolviam SN
131
mais cedo se comparadas com pacientes com mutações missense que permitiam a correta
inserção da podocina na membrana plasmática, sendo a idade média de início da doença de um
ano e oito meses, versus dez anos e oito meses, respectivamente. Isto sugere que as moléculas de
podocina mutantes que conseguem se inserir corretamente na membrana plasmática ainda
realizam parcialmente sua função, o suficiente para manter a barreira de filtração glomerular
funcionando por um tempo maior de vida.
É importante mencionar o avanço da farmacogenômica nesta área que, com novas
estratégias terapêuticas, pode revolucionar o tratamento de pacientes com determinadas
mutações no gene NPHS2. Alguns agentes farmacológicos vêm sendo pesquisados e
desenvolvidos que corrigem o enovelamento incorreto de proteínas mutantes para que sigam o
tráfego normal de direcionamento para a membrana plasmática (Egan et al., 2002; Ohashi et al.,
2003).
2.1.2. Alterações em heterozigose simples
2.1.2.1 Alterações missense em heterozigose simples
Em 14 pacientes identificamos somente uma alteração em heterozigose no gene NPHS2
(14/147) correspondendo a 9,52% (figura 33). Destes 14 pacientes, 10 apresentaram alterações
missense (tabela 24).
A alteração p.Pro20Leu, identificada em heterozigose nas pacientes 4, 8 e 91, resulta da
transição da citosina para a timina na posição 59 do cDNA (c.59C>T) no éxon 1 (figura 47A).
Esta mutação foi descrita pela primeira vez por Boute et al. em 2000. Nesse trabalho esse grupo
mapeou o gene NPHS2 e descreveu algumas mutações sugerindo associação com SNCR. Para se
132
verificar se a troca de nucleotídeos é frequente foi realizado o sequenciamento do éxon 1 de 100
indivíduos controle saudáveis nos quais a variante p.Pro20Leu não foi identificada (Figura 47B).
Tabela 24: Pacientes com uma alteração missense no NPHS2.
Pacientes
Mutação
Éxon
Idade de início
Resposta ao tratamento
4*
p.Pro20Leu
1
5a
Proteinúria nefrótica
5
p.Ala242Val
5
4a
CS,RF
6
p.Arg229Gln
5
9a
CS,RF
7
p.Glu264Gln
6
7a
CS,RF
8
p.Pro20Leu
1
4a
CS,RF
9
p.Glu310Lis
8
1a
CS
87
p.Arg229Gln
5
10 a
CR
91*
p.Pro20Leu
1
11 a
Proteinúria nefrótica
102
p.Ala242Val
5
3a
CS,RF
133
p.Arg229Gln
5
2a
CS,RF
(n = 10)
*As pacientes 4 e 91 são irmãs; a = anos.
133
A figura 48 demonstra que não há conservação da prolina no aminoácido 20 da
podocina, mesmo entre mamíferos, comparando-se as ortólogas entre as espécies de vertebrados
ao longo da evolução.
Figura 47: Eletroferograma e análise de grupo controle da alteração p.Pro20Leu identificada nas pacientes 4,
8 e 91. A. A heterozigose C/T correpondente à alteração c.59C>T está ressaltada em amarelo na sequência acima do
pico do eletroferograma e denotada em vermelho na sequência abaixo. B. Alinhamento realizado com o auxílio do
programa CLC Sequence Viewer 6.3 após sequenciamento de 100 indivíduos controle, mostrando que esta troca
C>T (Y) identificada nos pacientes (primeira linha) não foi identificada em nenhum dos controles. Somente parte do
alinhamento destes controles está ilustrada (15 controles).
HUMANO
CAMUNDONGO
RATO
MERRARSSSRESRGRGGRTPHKENKRAKAERSGGGRGRQEAGP
MDSRARSSSREAHGRSSRSSSRDDKKAKAGRGSRGRARPDAGA
MDSRARSSSRKTHGRGSRSSSRDDKKSKAGRGNRGRARPDAGA
*: *******:::**..*:. :::*::** *.. **.* :**.
Figura 48: Alinhamento das sequências contendo a região vizinha do resíduo 20 (posições 1 a 43) da proteína
podocina humana com as de duas espécies de mamíferos. A análise foi realizada com o programa Clustal W. O
aminoácido prolina, na posição 20, está ressaltado em amarelo e não é conservado entre as espécies de mamíferos
analisados. O número de acesso Uniprot das sequências é: homem (Q9NP85), rato (Q8K4G9), camundongo
(Q91X05). Asteriscos (*) indicam posições que possuem um resíduo conservado e único; Dois pontos (:) indicam
conservação entre grupos de aminoácidos com propriedades similares; Um ponto (.) indica conservação entre grupos
de aminoácidos com propriedades diferentes.
134
Se levarmos em consideração as propriedades dos aminoácidos prolina e leucina, estes
possuem diferenças críticas tais como o volume e a estrutura: a prolina é pequena, possui um
anel cíclico e geralmente situa-se na superfície, causando dobras abruptas nas moléculas
proteicas; a leucina, por sua vez, ocupa um volume maior e “prefere” ficar situada no interior das
proteínas. Ambas são apolares (figura 49).
Pela avaliação dos sites de predição in silico (tabela 25), observa-se que o programa
Align-GVGD, que baseia sua análise em características tais como composição e volume,
classifcou a variante como grave. Porém, o programa PolyPhen, que interpreta os resultados a
partir da análise de alinhamentos com relação à estrutura ao longo da evolução, classificou a
variante p.Pro20Leu como benigna. O programa SIFT classificou a variante como grave, no
entando o resultado de 0,05 é bem limítrofe (> 0,05 = benigno; ≤ 0,05 = prejudicial).
Figura 49: Aminoácidos prolina e leucina envolvidos na substituição missense p.Pro20Leu. Acima estão
representadas as estruturas tridimensionais e abaixo as fórmulas estruturais. A região circulada em laranja na prolina
corresponde às cadeias laterais ligadas ao esqueleto de proteína tanto pelo carbono quanto pelo nitrogênio.
Vermelho: átomo de oxigênio; azul: átomo de nitrogênio; preto: átomo de carbono; branco: átomo de hidrogênio
Adaptado de http://biochemunclassified.wordpress.com/ e http://www.explicatorium.com/quimica/Aminoacido.
135
. Tabela
25: Predição in silico para a substituição missense p.Pro20Leu.
SUBSTITUIÇÕES DE
SIFT
PolyPhen
Align-GVGD
Deletéria
Benigna
(0,098)
Classe C65
AMINOÁCIDOS
p.Pro20Leu
(0,05)
GV = 0,00; GD = 97,78
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
b
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas de
podocina. GD (Grantham distance) = pontuação da diferença química entre os residuos normal e mutante. Quanto
maior a pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class
35 = probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
Os dados com relação à frequência da p.Pro20Leu em populações controle são
conflitantes e parecem variar com a etnia: Esta variante não foi identificada na análise de mais de
400 indivíduos controle saudáveis europeus (Caridi et al., 2005; Di Duca et al., 2006; Weber et
al., 2004), porém foi identificada em heterozigose, na mesma proporção, tanto em pacientes
como em indivíduos controle de origem afro-americana (Dusel et al., 2005). A variante
p.Pro20Leu é mais frequente entre pacientes da Itália e da Turquia, tendo sido identificada em 11
de 468 crianças italianas com SN e em cinco de 295 crianças turcas com SNCR, em heterozigose
composta ou heterozigose simples (Berdeli et al., 2007; Caridi et al., 2003, 2009). Weber et al.
também identificaram a alteração p.Pro20Leu em heterozigose tanto em casos familiares como
em casos esporádicos (Weber et al., 2004) e a ela atribuíram um papel funcional secundário,
visto que foi identificada juntamente com a mutação patogênica p.Arg168His, em homozigose,
em dois irmãos afetados. Ruf et al. (Ruf et al., 2004) descreveram a variante p.Pro20Leu como
um polimorfismo baseando-se nos seguintes achados: foi identificada em heterozigose
136
juntamente com mais duas mutações patogênicas, a p.Arg138Gln e a p.Arg168His em um
paciente com SNCS e em dois indivíduos controle saudáveis.
O grupo de Nishibori (Nishibori et al., 2004), após experimentos de tráfego e
colocalização intracelular da nefrina com a podocina mutante p.Pro20Leu, demonstrou que esta
comportou-se como a podocina normal, ou seja, foi corretamente inserida na membrana
plasmática, além de ter colocalizado com a nefrina. No entanto, o grupo de Di Duca, em 2006
(Di Duca et al., 2006), estudando algumas variantes na região promotora do gene NPHS2,
demonstrou que a variante p.Pro20Leu no éxon 1 está em desequilíbrio de ligação com a variante
rs146791300 (c.-535insCTTTTTT), localizada na região promotora e que a presença deste
haplótipo está associada com diminuição da expressão da podocina em estudos realizados in
vitro. Esse haplótipo foi identificado em pacientes com doença renal, principalmente em
pacientes com SN e menos frequentemente em pacientes com nefropatia IgA (Di Duca et al.,
2006).
Neste trabalho a variante p.Pro20Leu foi identificada em três pacientes (8, 4 e 91) os
quais apresentaram também a alteração c.-535insCTTTTTT (rs146791300) na região promotora,
assim como o observado pelo grupo de Di Duca (Di Duca et al., 2006). A paciente 8 é CS,RF e a
idade de início da SN foi de três anos e oito meses. As outras duas pacientes, 4 e 91, são irmãs,
ambas apresentando de proteinúria nefrótica isolada e com idade de início de 5 e 11 anos de
idade, respectivamente. Estas duas irmãs foram incluídas neste trabalho pois a irmã mais velha, a
paciente 90, atualmente com 30 anos de idade, foi acompanhada desde criança por SNCR, com
biópsia de GESF com clara indicação de mutação no gene NPHS2. Curiosamente, na paciente
90 não foi identificada nenhuma alteração no gene NPHS2, mas sim nas irmãs 4 e 91 (figura 50).
Além das irmãs 4 e 91, mais dois irmãos desta família (pacientes 28 e 92) foram analisados, pois
137
apresentavam proteinúria não nefrótica. Não identificamos a associação p.Pro20Leu + c.535insCTTTTTT nestes dois pacientes e em nenhum dos 100 indivíduos controle analisados.
A partir destes resultados podemos concluir, que, embora dois pacientes tenham
apresentado as variantes p.Pro20Leu e c.-535insCTTTTTT em heterozigose, deve haver outra
causa genética nesta família que não alterações no gene NPHS2 que de alguma forma manifeste
quadros clínicos variáveis, já que à apresentação mais grave da doença não se associou nenhuma
variação neste gene, sendo a associação verificada somente nos casos menos graves. Isto só
confirma a já conhecida heterogeneidade desta doença e que outros fatores, ambientais, ou
mesmo outros genes, podem estar atuando e contribuindo para que a doença se desenvolva. Além
disto, não há como afirmarmos que estas variantes estejam em um mesmo alelo, segregando
juntas, pois não foi possível a análise os pais.
Figura 50: Heredograma da família das irmãs 4 e 91, que apresentaram a alteração p.Pro20Leu. Indivíduo II1 = paciente 90; II-2 = paciente 4; II-3 = paciente 28; II-4 = paciente 92 e II-5 paciente 91. Circulo preenchido de
preto: CR com GESF - sem mutação. Preenchimento gradual: proteinúria nefrótica, com variante p.Pro20Leu
associada à alteração c.-535insCTTTTTT na região 5`reguladora. Quadrados com listras: proteinúria não nefrótica,
sem mutação.
138
A próxima variante identificada em heterozigose simples foi a p.Ala242Val, em dois
pacientes, 5 e 102 (tabela 24, figura 51). Na análise de 107 indivíduos controle (214 alelos) esta
alteração foi identificada em um indivíduo, em heterozigose simples (1/214, ~ 0.5%).
A figura 52 ilustra que o resíduo alanina na posição 242 da podocina humana é
conservado entre as proteínas das espécies analisadas, com exceção da podocina do sapo. Além
disso, não se obseva um grau de conservação elevado na região vizinha ao resíduo.
Como já mencionado anteriormente, na figura 38, a alanina e a valina, apesar de serem
apolares, possuem tamanho e estrutura diferentes, a alanina sendo menor e a valina possui uma
estrutura característica, com ramificação em seu carbono beta (Cβ branched), o que resulta em
maior rigidez na região do esqueleto proteico ocupada por este resíduo.
Figura 51: Eletroferograma e análise em grupo controle da alteração p.Ala242Val, identificada nos pacientes
5 e 102. A. A heterozigose C/T referente à alteração c.725C>T (p.Ala242Val) identificada no gene NPHS2 está
ressaltada em amarelo na sequência acima dos picos do eletroferograma e abaixo está denotada em vermelho. B.
Análise realizada por PCR alelo específico mostrando que a variante foi identificada em um controle (poço 36). M:
marcador de peso molecular 1 kb Plus (Invitrogen®); poço 37: controle negativo; poço 38: controle heterozigoto.
Somente parte da análise dos indivíduos controle esta apresentada.
139
HOMEM
CAMUNDONGO
RATO
SAPO
PEIXE ZEBRA
RSLTEILLERKSIAQDAKVALDSVTCIWGIKVERIE
RSLTEILLERKSIAQDVKVALDAVTCIWGIKVERTE
RSLTEILLERKSIAQDVKVALDSVTCVWGIKVERTE
RAFLDILLERKSIGEEVKVALDAATCHWGIKVERTE
HAFNDILLDRKRIAQEIQVTLDSGTCRWGIKVEKAE
::: :***:** *.:: :*:**: ** ******: *
Figura 52: Alinhamento das sequências contendo a região vizinha do resíduo 242 (posições 229 a 264) da
proteína podocina humana com as de quatro espécies de vertebrados. A análise foi realizada com o programa
Clustal W. O aminnoácido alanina na posição 242 está ressaltado em amarelo. O número de acesso Uniprot das
sequências é: homem (Q9NP85), rato (Q8K4G9), camundongo (Q91X05), peixe zebra (Q52TF2) e sapo (C8C505).
Asteriscos (*) indicam posições que possuem um resíduo conservado e único; Dois pontos (:) indicam conservação
entre grupos de aminoácidos com propriedades similares. Um ponto (.) indica conservação entre aminoácidos com
propriedades diferentes.
A substituição da alanina pela valina na posição 242 foi considerada potencialmente
prejudicial pelos três sites de predição in silico (tabela 26).
Tabela 26: Predição in silico da substituição missense p.Ala242Val.
SUBSTITUIÇÕES DE
SIFT
PolyPhen
Align-GVGD
Deletéria
Provavelmente
deletéria
Classe C65
AMINOÁCIDOS
p.Ala242Val
(0,04)
(0,992)
GV = 0,00; GD = 65,28
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
b
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas de
podocina. GD (Grantham distance) = pontuação da diferença química entre os residuos normal e mutante. Quanto
maior a pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class
35 = probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
140
Caridi et al. (Caridi et al., 2003) descreveram esta variante pela primeira vez em um
paciente com SN córtico-dependente, idade de início da doença de 25 anos e padrão histológico
de lesões mínimas. Posteriormente, Weber et al. identificaram a variante p.Ala242Val em sete
pacientes: dois com SNCR familiar, quatro casos esporádicos com SNCR e um com esclerose
mesangial difusa, no entanto, nesse estudo dois em 160 indivíduos controle apresentavam a
alteração em heterozigose simples; como a maioria dos pacientes e ambos os indivíduos controle
eram de origem africana, investigaram 74 indivíduos adicionais, todos descendentes de africanos
e detectaram mais seis indivíduos heterozigotos para a variante p.Ala242Val, demonstrando que
esta variante representa um polimorfismo na população africana, com uma frequência alélica de
4% (Weber et al., 2004). Em oposição aos dados reportados por Weber, Ruf et al. (Ruf et al.,
2004) descreveram a variante p.Ala242Val em heterozigose em um paciente com SNCR e GESF
sendo a idade de início de um ano e seis meses, e não a identificaram em nenhum dos 80
indivíduos controle analisados. Os pacientes estudados por esse grupo eram da Europa central,
Turquia e India, o que, considerando o trabalho descrito acima, indica que essa variante deva ser
frequente apenas na população africana. Os dados de frequência alélica fornecidos pelo site
www.ensembl.org baseados no projeto de variação humana 1000 genomas, fase 1 (Abecasis et
al., 2012) confirmam estes dados: a frequência do alelo mais raro T é de 0% nas populações
asiática e europeia, enquanto que na população africana é de 5%.
No caso da paciente 5 aqui estudada, pode-se dizer que é branca, porém não se sabe sua
ancestralidade. Por outro lado, o paciente 102, embora seja também branco, seu pai é mulato. O
que é interessante nestes dois casos é que a variante p.Ala242Val se associa à SNCS,RF, com
idade de início da síndrome aos quatro e aos três anos, respectivamente. Ainda, o indivíduo
controle no qual a variante foi identificada é também branco, apesar de que possa haver origem
141
africana nos ancestrais deste indivíduo devido ao alto grau de miscigenação na população
brasileira.
Considerando-se os resultados dos programas de predição in silico e de conservação deste
resíduo entre os mamíferos durante a evolução esta variante pode ser considerada
funcionalmente prejudicial para a podocina. Vale lembrar também que a posição 242 localiza-se
no domínio PHB, que atua na formação de complexos lipídicos importantes para a sinalização
celular realizada pela nefrina nos “rafts” de lipídios. Porém todo o contexto deve ser analisado: o
fato desta variante ter sido encontrada em 1% de nossa população indica que possa ser um
polimorfismo; e, o fato dos pacientes serem sensíveis ao tratamento indica que a alteração deve
permitir que a podocina consiga exercer parcialmente sua função, levando a um quadro clínico
menos grave. Ainda, é extremamente importante observar o fato desta variante ter sido
identificada em heterozigose, sendo que nenhuma outra alteração foi identificada no gene
NPHS2, nem na análise dos genes NPHS1 e WT1, que serão discutidos nas próximas seções.
Talvez a variante p.Ala242Val possa ter um efeito deletério quando em heterozigose composta,
assim como a p.Arg229Gln. Desta forma, para se investigar se a SN dos pacientes 5 e 102 tem
origem genética, outros genes devem ser analisados. No entanto, são necessários ensaios
funcionais de tráfego intracelular e colocalização com a nefrina tais como os já realizados para
outras mutações para a confirmação do que esta variante representa para função biológica da
podocina.
A variante p.Glu264Gln foi identificada em heterozigose no paciente 7 e não foi
identificada em nenhum dos 114 indivíduos controle analisados (figura 53). Esta variante
(rs369697947)
está
depositada
no
banco
de
dados
http://www.ncbi.nlm.nih.gov/projects/SNP/snp, como uma SNV (single nucleotide variation),
142
porém sem dados de frequência alélica populacional. O grupo de Santin et al. (Santín et al.,
2011) a identificou também em heterozigose em um estudo realizado com pacientes de origem
hispânica, porém não forneceu dados sobre o paciente na qual esta variante foi identificada. Esse
grupo a classificou como “variante com significado clínico desconhecido”, pois, apesar de ter
sido considerada com um efeito possivelmente deletério e de ser um aminoácido conservado ao
longo da evolução, a troca de aminoácidos é conservativa e foi identificada em heterozigose em
dois dos de 360 cromossomos controle por eles analisados.
A figura 54 mostra que o ácido glutâmico na posição 264 da podocina é conservado ao
longo da evolução nas espécies analisadas.
A substituição de um ácido glutâmico por uma glutamina é uma troca conservativa, já
que a diferença entre estes aminoácidos é de somente um grupamento amino na glutamina no
lugar da hidroxila no glutamato (figura 55).
Figura 53: Eletroferograma e análise de grupo controle para a alteração p.Glu264Gln identificada no
paciente 7. A. A heterozigose G/C referente à alteração c.790G>C (p.Glu264Gln) está ressaltada em amarelo na
sequência acima dos picos do eletroferograma e abaixo está denotada em vermelho. B. A variante p.Glu264Gln não
foi identificada após análise de 114 indivíduos controle, realizada por PCR alelo específico. M: marcador de peso
molecular 1 kb Plus (Invitrogen®). Poços 18 e 31: controle negativo; Poços 19 e 32: controle heterozigoto (paciente
7). Somente parte da análise está ilustrada.
143
HOMEM
CAMUNDONGO
RATO
SAPO
PEIXE ZEBRA
SIAQDAKVALDSVTCIWGIKVERIEIKDVRLPAGLQHSLAVEAEAQRQAK
SIAQDVKVALDAVTCIWGIKVERTEIKDVRLPAGLQHSLAVEAEAQRQAK
SIAQDVKVALDSVTCVWGIKVERTEIKDVRLPAGLQHSLAVEAEAQRQAK
SIGEEVKVALDAATCHWGIKVERTEIKDVKLPEEVKQSIAVEAEAQRHAK
RIAQEIQVTLDSGTCRWGIKVEKAEIEEINLPPELQHNFAVEAEARRQAQ
*.:: :*:**: ** ******: **:::.** :::.:******:*:*:
Figura 54: Alinhamento das sequências contendo a região vizinha do resíduo 264 (posições 240 a 289) da
proteína podocina humana com as de quatro espécies de vertebrados. A análise foi realizada com o programa
Clustal W. O aminoácido ácido glutâmico na posição 264 está ressaltado. O número de acesso Uniprot das
sequências é: homem (Q9NP85), rato (Q8K4G9), camundongo (Q91X05), peixe zebra (Q52TF2) e sapo (C8C505).
Asteriscos (*) indicam posições que possuem um resíduo conservado e único; Dois pontos (:) indicam conservação
entre grupos de aminoácidos com propriedades similares Um ponto (.) indica conservação entre grupos de
aminoácidos com propriedades diferentes.
Figura 55: Aminoácidos ácido glutâmico (glutamato) e glutamina envolvidos na substituição missense
p.Glu264Gln. Acima estão representadas as estruturas tridimensionais e abaixo as fórmulas estruturais. Circulado
em vermelho está o oxigênio do ácido glutâmico, que é substituído por um grupamento amino (cículo azul) na
glutamina. Vermelho: átomo de oxigênio; azul: átomo de nitrogênio; preto: átomo de carbono; branco: átomo de
hidrogênio.
Adaptado
de
http://biochemunclassified.wordpress.com/
e
http://www.explicatorium.com/quimica/Aminoacido.
144
A tabela 27 mostra que dois dos programas de predição in silico retornaram um resultado
potencialmente deletério para a função da proteína após esta substituição.
Tabela 27: Predição in silico da substituição missense p.Glu264Gln
SUBSTITUIÇÕES DE
SIFT
PolyPhen
Align-GVGD
Deletéria
Provavelmente
deletéria
Classe C25
AMINOÁCIDOS
p.Glu264Gln
(0,00)
a
(0,993)
GV = 0,00; GD = 29,27
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
b
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas de
podocina. GD (Grantham distance) = pontuação da diferença química entre os residuos normal e mutante. Quanto
maior a pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class
35 = probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
Como esta alteração está localizada no quinto nucleotídeo antes do final do éxon 6,
realizamos a predição in silico com o programa SpliceAid2 para verificar se a substituição
localiza-se em uma região regulatória de splicing. Pode-se constatar na figura 56 que esta
alteração interrompe uma sequência exonic splicing silencer (ESS) de reconhecimento para a
proteína hnRNP A1.
A variante p.Glu264Gln parece ser rara na população brasileira, já que não a
identificamos nos 228 cromossomos controle analisados. Somente após estudos funcionais, seja
com a técnica de minigene e de ensaios pull down ou para verificar se a variante atrapalha o
tráfego de nefrina, poder-se-á confirmar o efeito biológico desta variante para a função da
podocina.
145
O paciente 7 teve início da SN com sete anos de idade, é CS,RF e atualmente, com 18
anos, está com sua função renal preservada.
Figura 56: Predição in silico da alteração c.790G>C no final do éxon 6 (p.Glu264Gln). realizada através do
programa SpliceAid2 (http://www.íntroni.it/splicing.html). As barras acima da sequência correspondem a
proteínas que reconhecem sequências facilitadoras ou inibidoras de definição do éxon, ou seja, sequências exonic
splicing enhancer (ESEs) ou íntronic splicing silencer (ISSs), respectivamente. As barras abaixo correspondem a
sequências que facilitam ou inibem a definição dos íntrons, isto é, sequências exonic splicing silencer (ESSs) ou
íntronic splicing enhancer (ISEs), respectivamente. As barras possuem largura e altura variáveis, o que está
relacionado ao tamanho da sequência e ao grau de afinidade de ligação, respectivamente. As sequências normal e
mutante foram avaliadas e estão ilustradas abaixo do histograma, e as bases G e C envolvidas na substituição estão
denotadas em vermelho. A alteração interrompe uma ESS de ligação à proteína nuclear hnRNP A1, como pode ser
visualizado com o retângulo em roxo e o círculo em roxo, nas sequências normal e mutante, respectivamente.
A variante p.Arg229Gln foi identificada em heterozigose simples nos pacientes 6, 87 e
133. Como já foi discutido anteriormente, esta variante atua em associação com outra mutação
deletéria em heterozigose composta causando o desenvolvimento de SNCR tardia (Tsukaguchi et
146
al., 2002). A paciente 6 teve idade de início da SN com nove anos e é CS,RF. O paciente 87 teve
idade de início da SN com 10 anos e é CR. A paciente 133 é CS,RF, e teve idade de início da SN
aos dois anos de idade. Os paciente 6 e 87, além da variante p.Arg229Gln no gene NPHS2,
também apresentaram alterações no gene NPHS1 (descritas mais adiante).
A variante p.Glu310Lis (Machuca et al., 2009), já discutida anteriormente, foi
identificada em heterozigose simples na paciente 9. Esta paciente teve início da SN com um ano
de idade, é CS e teve diagnóstico histológico de proliferação mensangial difusa.
As primeiras mutações no gene NPHS2 foram descritas em famílias com SNCR com
herança autossômica recessiva e idade de início entre os três meses e os cinco anos de idade
(Fuchshuber et al., 1995). Nesses casos a doença evolui rapidamente para insuficiência renal e
geralmente não ocorre recidiva após transplante. No entanto, há descrições de formas diferentes
de apresentação da doença em diversos trabalhos, nos quais os pacientes apresentam somente
uma mutação deletéria no gene NPHS2. Características em comum a esses pacientes são a idade
de desenvolvimento da SN, que de forma geral se apresenta de forma tardia, em alguns casos,
inclusive, a idade de início é na fase adulta, e os pacientes apresentam sensibildade ao tratamento
com corticoides (Caridi et al., 2003; Karle et al., 2002; Weber et al., 2004).
Ao compararmos a média da idade de início da SN nos pacientes da nossa casuística que
são portadores de uma alteração em heterozigose com a dos pacientes que apresentam duas
mutações que são, respectivamente, cinco anos e seis meses e três anos e seis meses, estes dados
estão de acordo com a literatura, que apontam para idade de início mais tardia em pacientes com
somente uma mutação no gene NPHS2 (Weber et al., 2004). Weber et al. analisaram 338
pacientes, sendo 147 casos familiares e 172 casos esporádicos, e identificaram mutações em
homozigose, heterozigose composta e heterozigose simples em 43% dos casos familiares e
147
10,5% dos casos esporádicos. Com relação somente às mutações identificadas em heterozigose
simples, identificaram seis mutações consideradas deletérias em seis pacientes não aparentados
além de variantes com efeito desconhecido em quatro pacientes. Também identificaram a
alteração p.Ala242Val em heterozigose simples em sete pacientes e a consideraram como um
polimorfismo não-sinônimo. Fizeram uma correlação com a idade de desenvolvimento e
verificaram que os pacientes com apenas uma mutação no gene NPHS2 manifestavam a doença
tardiamente se comparados com os que apresentavam duas mutações. Outro grupo que
identificou alterações em heterozigose simples foi o grupo de Caridi et al. (2003) que analisaram
179 crianças com SN, dos quais 120 eram CR e 59 sensíveis ao tratamento com corticoides,
porém com recidivas frequentes, ou até mesmo dependentes da corticoterapia. Oito pacientes,
sendo cinco CS,RF, apresentaram somente uma mutação em heterozigose simples no gene
NPHS2. Entre os 10 pacientes da nossa casuística heterozigotos para somente uma alteração
missense, somente o paciente 87 é CR. Todos os outros apresentaram sensibilidade aos
corticoides, sendo cinco CS,RF (5, 6, 7, 8 e 133) tais como os pacientes descritos por Caridi et
al..
2.1.2.2 Mutações em heterozigose simples na região 5’ reguladora
Dentre os 14 pacientes que apresentaram somente uma mutação em heterozigose simples
no gene NPHS2, quatro apresentaram alterações na região 5’reguladora (tabela 28).
148
Tabela 28: Pacientes com alteração na região promotora do gene NPHS2.
Pacientes
Alteração
Idade início
Resposta ao tratamento
12
c.-267C>G
<1a
CS
82
c.-196C>G
4a
CR
113
c.-53C>T
nd
CS,RF
140
c.-164C>T
3a
CR
Atualmente somente duas alterações na região 5’reguladora do gene NPHS2 estão
descritas como mutações relacionadas a uma expressão reduzida do gene no banco de dados
público HGMD http://www.hgmd.cf.ac.uk/ac/index.php e há poucos artigos que tratam sobre a
região regulatória deste gene. Em 2002 Moeller et al. (2002) criaram um cassete de expressão
contendo a região 5’reguladora correspondente a um fragmento de 2,5 kb de DNA, com toda a a
região regulatória da transcrição do gene NPHS2 humano. O objetivo do referido estudo foi o de
identificar a região 5’regulatória mínima que direcionasse a expressão seletiva (isto é, somente
nas células desejadas, no caso, os podócitos) de transgenes e que pudesse ser utilizado como
ferramenta de expressão in situ em estudos de doenças glomerulares, tais como experimentos de
avaliação de ganho de função de uma proteína em animais transgênicos ou no desenvolvimento
de camundongos knockout. O fragmento de 2,5 kb atingiu seu objetivo, contendo todas as
sequências necessárias para a expressão do gene repórter da β-galactosidase, exclusivamente em
podócitos (Moeller et al., 2002). Alguns experimentos com esta região 5’regulatória mínima já
foram realizados. Um desses, realizado pelo próprio grupo de Moeller (Moeller et al., 2003) foi o
desenvolvimento de uma linhagem de camundongos transgênicos que expressam a enzima
149
recombinase Cre somente em podócitos, permitindo a utilização do sistema Cre-loxP de geração
de “knockout condicional”, o que significa que somente genes específicos nas células do tecido
de interesse serão silenciados (Quaggin, 2002); experimentos recentes utilizaram a região 5’
reguladora de 2,5 kb do gene NPHS2 para expressar a podocina com GFP (green fluorescent
protein) em peixe zebra, um experimento que abre uma gama de possibilidades de estudos
funcionais e de visualização de proteínas expressas nas células glomerulares (He et al., 2011;
Zhou & Hildebrandt, 2012).
A partir do sequenciamento de aproximadamente 700 pb da região 5’reguladora do gene
NPHS2 nos pacientes deste trabalho, identificamos quatro alterações em heterozigose em quatro
pacientes (tabela 28). Como nenhuma das quatro alterações está anotada no banco de dados
http://www.hgmd.cf.ac.uk/ac/index.php, um grupo controle de 278 indivíduos saudáveis (556
alelos) foi analisado. Ainda, para cada uma destas alterações foram realizadas predições in silico
através
do
programa
Alibaba2
gene
regulation
(http://www.gene-
regulation.com/pub/programs/alibaba2/index.html) (Grabe, 2002), que identifica sítios de
reconhecimento de fatores de transcrição, TFBS (transcription factor binding sites) na região
regulatória de interesse.
A paciente 12 teve início da SN muito cedo, com menos de um ano de idade, sendo
classificada com síndrome nefrótica infantil. É CS, já que teve boa resposta ao tratamento com
corticoides. Após análise molecular do gene NPHS2 foi identificada a alteração c.-267C>G
(figura 57A).
150
Figura 57: Eletroferograma, análise de grupo controle e predição in silico da alteração c.-267C>G
identificada na região 5’reguladora do gene NPHS2 no paciente 12. A. A heterozigose C/G referente à alteração
c.-267C>G está representada pela letra S em rosa na sequência acima dos picos do eletroferograma e abaixo está
denotada em vermelho. B. Alinhamento realizado com o auxílio do programa CLC Sequence Viewer 6.3 após
sequenciamento de 278 indivíduos controle, mostrando que esta troca C>G (S) que foi identificada no paciente 12
(primeira linha) não foi apresentada por nenhum dos controles. Somente parte do alinhamento destes controles está
ilustrada (17 controles). C. Predição in silico realizada com o programa Alibaba2 gene regulation (http://www.generegulation.com/pub/programs/alibaba2/index.html). Acima a sequência normal analisada, com o C denotado em
verde e a região reconhecida para o fator de transcrição SP1 sublinhada. Ao lado, o sítio de reconhecidmento para o
fator de transcrição Adf-1 (retângulo vermelho) que foi criado após substituição para um G, denotado em vemelho
na sequência acima. Este sítio não foi reconhecido na sequência normal (retângulo verde com ponto de
interrogação).
151
Na análise de 278 indivíduos controle, nenhum com da alteração c.-267C>G foi
identificado (57B). A predição in silico indica que a troca de um C para um G nesta posição
criou uma sequência de reconhecimento “ctctGccacc” para o fator de transcrição Adf-1 (figura
57C). O fator de transcrição Adf-1 é específico da mosca Drosophila melanogaster, portanto não
é expresso nas células renais humanas. Com esta informação é possível prever que
provavelmente não interfira na regulação tecido específica para a expressão do gene NPHS2 em
podócitos. Porém, somente através de estudos funcionais tais como o ensaio de retardo de
mobilidade eletroforética (EMSA - electrophoretic mobility shift assay) poderemos verificar se
realmente o fator de transcrição Sp1 interage com o sítio de reconhecimento predito pelo
programa Alibaba2 e se esta interação é prejudicada pela mutação. Após ensaios de expressão
com gene repórter da região 5’reguladora com e sem a mutação poderemos averiguar se esta
variante prejudica a expressão da podocina.
O paciente 82 teve início da SN aos quatro anos e é CR. A variante c.-196C>G foi
identificada na região promotora do gene NPHS2, em heterozigose. Após análise de 278
indivíduos controle (556 alelos) a variante c.-196C>G foi identificada em três indivíduos (0,5%,
3/556). Após predição in silico para a região regulatória, não houve alteração do sítio de
reconhecimento para o fator de transcrição Sp1 (figura 58), o que indica que provavelmente esta
alteração não prejudica a função da podocina. No entanto, somente após estudos funcionais de
expressão com gene repórter que poderemos confirmar quais as consequências desta alteração na
transcrição da podocina.
152
Figura 58: Eletroferograma, análise de grupo controle e predição in silico da alteração c.-196C>G
identificada na região 5’reguladora do gene NPHS2 no paciente 82. A. A heterozigose C/G referente à alteração
c.-196C>G está representada pela letra S em rosa na sequência acima dos picos do eletroferograma e abaixo está
denotada em vermelho. B. Alinhamento realizado com o auxílio do programa CLC Sequence Viewer 6.3 após
sequenciamento de 278 indivíduos controle, mostrando que esta troca C>G (S) que foi identificada no paciente 82
(primeira linha), foi identificada também em um dos controles (linha 10). Somente parte do alinhamento destes
controles está ilustrada (18 controles). C. Predição in silico realizada com o programa Alibaba2 gene regulation
(http://www.gene-regulation.com/pub/programs/alibaba2/index.html). Acima a sequência normal analisada, com o C
denotado em verde e a região reconhecida para o fator de transcrição SP1 (retângulo verde) está sublinhada. Ao
lado,não houve alteração para o sítio de reconhecidmento para o fator de transcrição SP1 (retângulo vermelho) após
substituição por um G (denotado em vermelho).
A paciente 140 teve início da SN aos três anos de idade e é CR. Após análise molecular a
alteração c.-164C>T
foi identificada na região promotora do gene NPHS2 (figura 59A).
Nenhuma variante c.-164C>T foi identificada após análise dos 278 indivíduos controle (556
alelos) (figura 59B). Após predição in silico com o programa Alibaba2 gene regulation, três
153
sítios de reconhecimento para os fatores de transcrição foram abolidos, dois para o ETF e um
para o GLI3, e no lugar foi criado um sítio para o fator de transcrição WT1 (figura 59C).
Figura 59: Eletroferograma, análise de grupo controle e predição in silico da alteração c.-267C>G
identificada na região 5’reguladora do gene NPHS2 na paciente 140. A. A heterozigose C/T referente à alteração
c.-164C>T está representada pela letra Y ressaltada em amarelo na sequência acima dos picos do eletroferograma e
abaixo está denotada em vermelho. B. Alinhamento realizado com o auxílio do programa CLC Sequence Viewer 6.3
após sequenciamento de 278 indivíduos controle, mostrando que esta troca C>T (Y) que foi identificada na paciente
140 (primeira linha) não foi apresentada por nenhum dos controles. Somente parte do alinhamento destes controles
está ilustrada (12 controles). C. Predição in silico realizada com o programa Alibaba2 gene regulation
(http://www.gene-regulation.com/pub/programs/alibaba2/index.html). Acima a sequência normal analisada, com o C
denotado em verde. Abaixo, dois fatores de transcrição, ETF e GLI3, (retângulos amarelo e verde, respectivamente)
reconhecem esta região. Ao lado, após a substituição para um T, denotado em vemelho na sequência acima, foi
criado um sítio de reconhecidmento para o fator de transcrição WT1 (retângulo vermelho) e abolidos os sítios para
os fatores ETF e GLI3.
154
O ETF (também denominado TEAD2) é um fator de transcrição expresso principalmente
em tecidos embrionários de mamíferos, liga-se em regiões ricas em GC (“GC boxes”) de
promotores sem TATA box, característicos de genes constitutivos (“housekeeping”) (Kageyama
et al., 1989; Yasunami et al., 1995) e possui papel no desenvolvimento neuronal (Kaneko et al.,
2007). O GLI3 é um fator de transcrição que participa da via de sinalização Sonic Hedgehog e
atua no desenvolvimento do tubo neuronal em vertebrados. Aparentemente, nenhum dos dois é
expresso ou possui algum tipo de papel nas células renais. Já o WT1, cujo sítio de
reconhecimento foi criado após a alteração c.-164C>T, é um fator de transcrição do tipo dedo de
zinco que possui papel fundamental no desenvolvimento do trato genito urinário (Pritchard-Jones
et al., 1990); além da função já bem estabelecida no desenvolvimento embrionário, o WT1
também é expresso em podócitos humanos adultos e regula diversos genes que codificam
proteínas tais como a nefrina (Guo et al., 2004), a podocalixina (Morrison et al., 2008) - proteína
transmembrana codificada pelo gene PODXL e recentemente indicado como gene responsável
por causar GESF (Barua et al., 2013) - além de proteínas importantes envolvidas na arquitetura
do citoesqueleto destas células renais tais como filamentos de actina (Viney et al., 2007) e
nestina (Wagner et al., 2006). Não existem descrições da regulação do gene NPHS2 pelo WT1,
portanto somente após estudos funcionais com EMSA e ensaios de expressão com gene repórter
que o efeito desta alteração poderá ser confirmado.
O paciente 113, classificado como CS,RF, apresentou a alteração c.-53C>T em
heterozigose na região 5’reguladora do gene NPHS2 (figura 60A). Não foi verificada nenhuma
variante c.-53C>T na análise da região 5’reguladora dos 278 indivíduos controle (556 alelos)
(figura 60B). Após predição in silico realizada para a região afetada, verificou-se que esta
alteração aboliu um sítio para reconhecimento do fator de transcrição c-ETS-1 (figura 60C). O
155
fator de transcrição ETS-1 faz parte de uma família de fatores de transcrição extremamente
conservada ao longo da evolução, a qual regula vários genes envolvidos em células normais e
tumorais. Mais especificamente, o ETS-1 foi recentemente relacionado com algumas funções no
sistema imune (Garrett-Sinha, 2013). No entanto, assim como para as outras alterações
identificadas na região 5’reguladora, as consequências desta alteração somente poderão ser
efetivamente confirmadas após ensaios funcionais, apesar de que como não existem descrições
na literatura de que o fator c-ETS-1 tenha algum papel na regulação de células renais e como a
principal função deste fator é no sistema imune, provavelmente esta alteração não prejudique a
expressão da podocina.
Ainda com relação à região 5’reguladora, identificamos três alterações previamente já
descritas na literatura (Di Duca et al., 2006): a variante c.-535insCTTTTTT anotada no banco de
dados www.ensembl.org (rs146791300) e as variantes c.-116C>T (rs1079291) e c.-51G>T
(rs12406197) anotadas como mutações no HGMD sob os registros CR068161 e CR068160,
respectivamente (http://www.hgmd.cf.ac.uk/ac/index.php) (figura 61).
156
Figura 60: Eletroferograma, análise de grupo controle e predição in silico da alteração c.-53C>T identificada
na região 5’reguladora do gene NPHS2 na paciente 113. A. A heterozigose C/T referente à alteração c.-164C>T
está representada pela letra Y ressaltada em amarelo na sequência acima dos picos do eletroferograma e abaixo está
denotada em vermelho. B. Alinhamento realizado com o auxílio do programa CLC Sequence Viewer 6.3 após
sequenciamento de 278 indivíduos controle, mostrando que esta troca C>T (Y) que foi identificada na paciente 113
(primeira linha) não foi apresentada por nenhum dos controles. Somente parte do alinhamento destes controles está
ilustrada (17 controles). C. Predição in silico realizada com o programa Alibaba2 gene regulation (http://www.generegulation.com/pub/programs/alibaba2/index.html). Acima a sequência normal analisada, com o C denotado em
verde. Abaixo o sítio de reconhecimento para o fator de transcrição c-Ets-1 (retângulo lilás) está sublinado. Ao lado,
após a substituição para um T, denotado em vemelho na sequência acima, o sítio para o fator c-Ets-1 foi abolido
(retângulo vermelho com ponto de interrogação).
157
Figura 61: Eletroferograma das variantes c.-535CTTTTTT, c.-116C>T e c.-51G>T identificadas em
heterozigose em pacientes e em controles na região 5’reguladora do gene NPHS2. Acima, a inserção CTTTTTT
inserida está denotada em vermelho na sequência mutante abaixo do eletroferograma. A análise da sequência a partir
do ponto da inserção fica desalinhada, como pode ser visualizado pelas letras ressaltadas em amarelo na sequência
acima dos picos do eletroferograma. Abaixo estão representadas as heterozigoses C/T e G/T referentes às alterações
c.-116C>T e c.-51G>T, respectivamente, que estão ressaltadas em amarelo na sequência acima dos picos dos
eletroferogramas e estão denotadas em vermelho na sequência abaixo.
Di Duca et al. demonstraram que, quando as isoformas c.-535insCTTTTTT, c.-116T e c.51T foram expressas em podócitos, houve a redução na expressão da podocina em 89% , 59% e
73%, respectivamente. Além disto, esse grupo demonstrou que a variante c.-535insCTTTTTT
(rs146791300) encontra-se em desequilíbrio de ligação (LD) com a variante p.Pro20Leu (já
discutido anteriormente na seção 2.1.2) e que esse haplótipo está associado com
desenvolvimento de síndrome nefrótica (Di Duca et al., 2006). Na nossa casuística identificamos
158
a variante c.-535insCTTTTTT em heterozigose simples em seis pacientes (35, 57, 85, 121, 124 e
143), quatro CS, um CR e um com SNC. Em outros três pacientes, (duas são irmãs),
identificamos a variante c.-535insCTTTTTT em associação com a variante p.Pro20Leu. Não
existem dados de frequência alélica da variante c.-535insCTTTTTT depositadas nos sites
www.ensembl.org e http://www.ncbi.nlm.nih.gov. Não identificamos esta variante sozinha, nem
a associação desta variante com a p.Pro20Leu em nenhum dos 278 (556 alelos) indivíduos
controle analisados.
Com relação às variantes c.-116C>T (rs1079291) e c.-51G>T (rs12406197), estas foram
identificadas tanto nos pacientes como nos controles, em homozigose e em heterozigose.
Fizemos um cálculo das frequências alélicas identificadas na nossa casuística e nos nossos
indivíduos controle. Entre os 147 pacientes analisados para o gene NPHS2, 13 são parentes de
primeiro grau, portanto fizemos os cálculos das frequências alélicas entre os 134
não
aparentados, para não haver uma superestimação destas frequências. A tabela 28 mostra as
frequências alélicas e genotípicas das três variações, a c.-535CTTTTTT, c.-116C>T, c.-51G>T
identificadas nos 134 pacientes triados para o gene NPHS2, nos 278 controles analisados neste
trabalho e as descritas pelo projeto 1000 genomes, www.ensembl.org.
Para verificar se a frequência destas variantes estava se comportando de maneira
semelhante entre os pacientes, controles brasileiros e projeto 1000 genomes, calculamos o pvalue para cada uma das combinações. Realizamos este cálculo somente para as variantes c.116C>T e c.-51G>T, pois eram as únicas para as quais possuíamos os dados completos. Os
resultados encontram-se na tabela 29. A frequência alélica da variante c.-116C>T apresentou-se
em equilíbrio nas três comparações analisadas: pacientes x controles brasileiros, pacientes x
1000 genomes e controles brasileiros x 1000 genomes. Já a frequência da variante c.-51G>T
159
apresentou-se em equilíbrio somente entre os pacientes x controles brasileiros e entre os
pacientes x 1000 genomes.
Tabela 28: Frequências alélicas e genotípicas de algumas alterações da região
5`reguladora entre pacientes, controles e projeto 1000 genomesa
1000
genomes
Pacientes (n = 134)
Pacientes (n = 134)
Frequência Alélica
Genótipo
rs146791300
c. -535insCTTTTTT
CTTTTTT2= 97,38%
CTTTTTT3 = 2,62%
127 (CTTTTTT2/ CTTTTTT2)
7 (CTTTTTT2/CTTTTTT3)
0 (CTTTTTT3/ CTTTTTT3)
rs1079291b
c.-116C>T
C = 74,63%
T = 25,37%
77 (CC)
46 (CT)
11 (TT)
C = 71%
T = 29%
551 (C/C)
438 (C/T)
103 (T/T)
rs12406197
c.-51G>T
G = 77,24%
T = 22,76
78 (GG)
51 (GT)
5 (TT)
G = 81%
T = 19%
717 (C/C)
328 (C/T)
47 (T/T)
Controles
brasileiros
Controles brasileiros
( n = 278)
1000
genomes
1000 genomes
Genótipo
(n = 1092)
-
Alterações
Alterações
(n = 278)
Frequência Alélica
(n = 1092)
1000 genomes
Genótipos
-
Genótipos
rs146791300
c. -535insCTTTTTT
CTTTTTT2= 100%
CTTTTTT3 = 0%
CTTTTTT2/ CTTTTTT2 = 278
CTTTTTT2/CTTTTTT3= 0
CTTTTTT3/ CTTTTTT3 = 0
rs1079291
c.-116C>T
C = 70,5%
T = 29,55
138 (C/C)
116 (C/T)
24 (T/T)
C = 71%
T = 29%
551 (C/C)
438 (C/T)
103 (T/T)
rs12406197
c.-51G>T
G = 75,18%
T = 24,82%
149 (G/G)
120 (G/T)
9 (T/T)
G = 81%
T = 19%
717 (C/C)
328 (C/T)
47 (T/T)
a
-
Fonte: site www.ensembl.org, projeto 1000 genomes (Abecasis et al., 2012).
160
Tabela 29: Cálculo p-Value comparando pacientes/controles/1000 genomes
PACIENTES (n = 134)
Controles brasileiros (n= 134a)
Alterações
Observado
Esperadob
Observado
Esperadob
p-Valuec
rs1079291
c.-116C>T
77 (C/C)
46 (C/T)
11 (T/T)
74,63 (C/C)
50,75 (C/T)
8,63 (T/T)
66 (C/C)
56 (C/T)
12 (T/T)
65,94 (C/C)
56,12 (C/T)
11,94 (T/T)
0.0433
rs12406197
c.-51G>T
78 (G/G)
51 (G/T)
5 (T/T)
79,94 (G/G)
47,12 (G/T)
6,94 (T/T)
72 (G/G)
58 (G/T)
4 (T/T)
76,13 (G/G)
49,75 (G/T)
8,13 (T/T)
0.3501
PACIENTES (n = 134)
1000 GENOMES (n= 134a)
Alterações
Observado
Esperadob
Observado
Esperadob
p-Valuec
rs1079291
c.-116C>T
77 (C/C)
46 (C/T)
11 (T/T)
74,63 (C/C)
50,75 (C/T)
8,63 (T/T)
68 (C/C)
54 (C/T)
67,35 (C/C)
55,3 (C/T)
0,1180
12 (T/T)
11,35 (T/T)
rs12406197
c.-51G>T
78 (G/G)
51 (G/T)
5 (T/T)
79,94 (G/G)
47,12 (G/T)
6,94 (T/T)
88 (G/G)
40 (G/T)
87,04(G/G)
41,91 (G/T)
6 (T/T)
5,04 (T/T)
a
0,0434
Valor corrigido (de 278 controles, para 134 e de 1092 – 1000 genomes para 134), para realização dos cálculos.
Controles brasileiros (n = 134a)
1000 GENOMES (n= 134a)
Alterações
Observado
Esperadob
Observado
Esperadob
p-Valuec
rs1079291
c.-116C>T
66 (C/C)
56 (C/T)
12 (T/T)
65,94 (C/C)
56,12 (C/T)
11,94 (T/T)
68 (C/C)
54 (C/T)
67,35 (C/C)
55,3 (C/T)
0,9024
12 (T/T)
11,35 (T/T)
rs12406197
c.-51G>T
72 (G/G)
58 (G/T)
4 (T/T)
76,13 (G/G)
49,75 (G/T)
8,13 (T/T)
88 (G/G)
40 (G/T)
87,04(G/G)
41,91 (G/T)
6 (T/T)
5,04 (T/T)
0,0002d
b
Cálculos realizados com o auxílio do programa OEGE - Online Encyclopedia for Genetic Epidemiology studies
(Santiago Rodriguez et al., 2009);
c
Correção de Bonferroni: α/n, portanto 0,05/2 = 0,025. Assim, p-value ≤ 0,025.
d
p-value ≤ 0,025, diferença significativa para este polimorfismo entre os controles e o 1000 genomes.
161
Porém, ao compararmos os controles brasileiros x 1000 genomes o p-value ficou abaixo
do limite de 0,025 (após correção de Bonferroni), portanto esta variante não está em equilíbrio de
Hardy-Weinberg com a população 1000 genomes. Observando-se a tabela 28, o alelo T é mais
frequente na população dos controles brasileiros (24,82%) do que na população 1000 genomes
(19%). A frequência do alelo T também é maior entre os pacientes (22%), mas talvez não o
suficiente para ter causado desequilíbrio quando comparamos com o 1000 genomes.
Provavelmente esta variante deve ser mais frequente na população brasileira e se um maior
número de pacientes fosse analisado esta também apareceria em desequilíbrio na comparação de
pacientes x 1000 genomes.
Finalizando esta seção sobre as variações missense ou na região 5’reguladora que foram
identificadas em heterozigose simples no gene NPHS2, algumas considerações podem ser
abordadas. Em primeiro lugar, podemos assumir que as variações nucleotídicas, mesmo em
heterozigose simples no gene NPHS2 exerçam um papel na doença destes pacientes, como
discutido neste e em outros trabalhos citados anteriormente. Deve-se também abordar aqui a
possibilidade destes alelos mutantes causarem a diminuição da função da podocina (alelos
hipomórficos) e que predisponham os indivíduos a uma disfunção glomerular na presença de
outros eventos renais, como já proposto por Kim et al. para a haploinsuficiência do gene CD2AP
(Kim et al., 2003). Uma segunda hipótese que surge para esses casos é que, considerando-se a
SN como uma doença de herança autossômica recessiva, uma segunda mutação pode não ter sido
detectada, seja na região 5’reguladora ou em regiões intrônicas. Embora a triagem tenha sido
abrangente e minuciosa, foi realizado o sequenciamento de aproximadamente 700 pb upstream
do códon de início de leitura e entre 120 a 180 pb dos íntrons, nos limites íntron-éxon. Porém, há
elementos de ação cis ainda mais upstream e elementos cis intrônicos que ficaram fora da região
162
incluída na análise. Ainda uma terceira hipótese é a de que estas variantes da podocina possam
atuar como modificadores em associação com variantes mutantes de outros genes, nos moldes de
uma herança digênica, como já foi descrito por Koziell et al. (Koziell et al., 2002). Por este
motivo, para os pacientes nos quais identificamos apenas uma alteração em heterozigose no gene
NPHS2 também realizamos o sequenciamento do gene NPHS1.
Para finalizar o tópico relativo ao estudo do gene NPHS2, a figura 62 ilustra a
distribuição das 12 alterações identificadas no gene NPHS2 (podocina) nos 18 pacientes (quatro
com alterações em heterozigose composta e 14 com alterações em heterozigose simples) deste
trabalho. Estas alterações estão presentes tanto na região reguladora como na região codificante
do gene, porém observa-se uma maior concentração de alterações missense na região C-terminal
da podocina, o que está de acordo com os dados da literatura, de que o acúmulo de mutações
nesta região está correlacionado com o maior prejuízo no papel funcional deste domínio,
principalmente de interação com outras proteínas das fendas do diafragma, tais como a nefrina, a
CD2AP e a NEPH (Huber et al., 2001; Huber et al., 2003; Sellin et al., 2003)
163
Figura 62. Esquema das alterações identificadas na região 5’reguladora do gene NPHS2 e na região
codificante da podocina. A. Alterações identificadas na região 5’reguladora do gene NPHS2. B. Distribuição das
alterações missense ao longo da podocina. Observar maior concentração de alterações na região C-terminal.
164
2.2 Gene NPHS1
Para os 14 pacientes nos quais identificamos somente uma alteração em heterozigose no gene
NPHS2, foi realizado o sequenciamento de todos os 29 éxons, região promotora
(aproximadamente 700 pb) e junções íntron-éxon do gene NPHS1. Destes, três apresentaram
alterações no gene NPHS1 (tabela 30, figura 63).
Tabela 30: Pacientes com alterações em heterozigose nos genes NPHS2 e NPHS1
Pacientes
6
NPHS2
p.Arg229Gln
(éxon 5)
12
c.-267C>G
(região 5`reguladora)
87
p.Arg229Gln
(éxon 5)
NPHS1
p.Glu117Lis
(éxon 3)
p.Arg408Gln
(éxon 10)
p.Asn1077Ser
(éxon 24)
P.Glu117Lis
(éxon 3)
p.Asn1077Ser
(éxon 24)
p.Arg408Gln
(éxon 10)
Idade Início
Resposta ao
tratamento
9a
CS,RF
<1a
CS
10 a
CR
165
Figura 63: Heredogramas das famílas da paciente 6 e do paciente 87 e eletroferogramas das alterações
identificadas nos genes NPHS2 e NPHS1 dos pacientes 6, 12 e 87. As heterozigoses identificadas estão
ressaltadas em amarelo nas sequências acima dos picos dos eletroferogramas e denotadas em vermelho nas
sequências abaixo. A paciente 6 apresenta quatro alterações, uma no gene NPHS2, que foi herdada do pai e três no
gene NPHS1 (uma herdada do pai e outra da mãe, a terceira não foi analisada nos pais). O paciente 12 apresenta três
alterações, uma no NPHS2 e duas no NPHS1; não foi realizada análise molecular dos pais. O paciente 87 apresenta
duas alterações, uma no NPHS2, proveniente do pai e a outra no NPHS1; não foi realizada análise molecular da
mãe.
166
A paciente 6 apresentou três variantes no gene NPHS1 (figura 63). A variante c.349G>A
(p.Glu117Lis) foi identificada no éxon 3 e é um polimorfismo frequente na população em geral,
e seg o projeto 1000 genomes, com uma frequência do alelo raro (MAF) de 0,34
(www.ensembl.org/).
Não
está
depositada
no
banco
de
dados
de
mutações
http://www.hgmd.cf.ac.uk/ac/index.php. A outra alteração identificada no gene NPHS1 da
paciente 6 foi a p.Asn1077Ser que já foi descrita na seção 1.2 pois também foi identificada na
paciente 143, com SNC. Esta variante representa um polimorfismo na população brasileira, já
que foi identificada em heterozigose e em homozigose nos indivíduos controle triados neste
trabalho (15/93, 16%) e está depositada no banco de dados com frequência de 8% dentro do
projeto 1000 genomes (www.ensembl.org/). A outra alteração identificada na paciente 6 foi a
p.Arg408Gln. Lenkkeri et al.. descreveram esta variante pela primeira vez e a identificaram em
heterozigose composta com outras mutações em três pacientes e também em heterozigose em
quatro de 30 controles analisados (13%) (Lenkkeri et al., 1999). Lahdenkari et al. (Lahdenkari et
al., 2004) identificaram esta variante em 4/50 alelos em pacientes com doença de lesões mínimas
e em 3/50 alelos de indivíduos controle analisados. Em 2004, Schultheiss et al. (Schultheiss et
al., 2004) realizaram o sequenciamento do gene NPHS1 em busca de mutações que pudessem
estar agindo em sinergia com um padrão de herança digênica em três grupos de pacientes: grupo
I = 48 pacientes, sendo 11 com SNC, nos quais previamente haviam sido identificadas duas
mutações no gene NPHS2; grupo II = 14 pacientes, sendo quatro com SNC, nos quais havia sido
identificada uma única mutação no gene NPHS2; grupo III = 14 pacientes com SNC sem
mutações prévias no gene NPHS2. Além de outras mutações no gene NPHS1, identificaram a
variante p.Arg408Gln em heterozigose em 5/48 (10%) pacientes do grupo I, 1/14 (7%) pacientes
do grupo II e em 2/14 (14%) pacientes do grupo III, além de identificarem esta variante em 10/83
167
(12%) indivíduos controle. Esta variante foi considerada portanto como um polimorfismo sem
função deletéria. Na nossa casuística esta variante foi identificada em 3/200 (1,5%) alelos
controles analisados.
Na análise de conservação pelo programa ClustalW pode-se verificar que o resíduo
arginina na posição 408 é conservado ao longo da evolução nas espécies de vertebrados
analisadas (figura 64). Este resíduo, codificado pelo éxon 10 do gene NPHS1, localiza-se no
domínio extracelular do tipo Ig4 da proteína nefrina. As predições in silico para esta substituição
missense estão ilustradas na tabela 31.
Assim como o observado para a troca p.Arg229Gln (discutida na seção 2.1.1), os
resultados de predição foram similares (tabela 20), já que os aminoácidos que foram substituídos
foram os mesmos, arginina por uma glutamina. Com relação às propriedades físico químicas
destes aminoácidos, a arginina é um aminoácido polar e com carga positiva e a glutamina
também é polar, porém não tem carga, o que pode influenciar nas interações que estes
aminoácidos estabelecem dentro da proteína (figura 36).
A paciente 6 apresentou ainda uma variante no gene NPHS2, a p.Arg229Gln (figura 63).
HUMANO
CAMUNDONGO
RATO
SAPO
PEIXE ZEBRA
ETVMDGLHGGHISMSNLTFLARREDNGLTLTCEAFSEAFTKETFKKSLIL
ETVMDGLHGGHISMSNLTLLVKREDNGLSLTCEAFSDAFSKETFKKSLTL
ETVMDGLHGGHISMSNLTFLVRREDNGLPLTCEAFSDAFSKETFKKSLTL
VTITDAAHGGKVTMSNLTMVALREEHGMSLTCEAFNEVILYTK-SNSVTL
VTFAEGDNGGMMTVSNLTHKVSREDNGLSLQCEAFNKGTRFSS-VRAAEL
*. :. :** :::**** . **::*:.* ****..
. .: *
Figura 64: Alinhamento das sequências contendo a região vizinha do resíduo 408 (posições 386 a 485) da
proteína nefrina humana com as de quatro espécies de vertebrados. A análise foi realizada com o programa
Clustal W. O aminoácido arginina na posição 408 está ressaltado em amarelo e é conservado entre os animais
analisados ao longo da evolução. O número de acesso Uniprot das sequências é: homem (O60500), rato (Q9R044),
camundongo (Q9QZS7), peixe zebra (Q1KMS5) e sapo (Q58QC3). Asteriscos (*) indicam posições que possuem
um resíduo conservado e único; dois pontos (:) indicam conservação entre grupos de aminoácidos com propriedades
similares; um ponto (.) indica conservação entre grupos de aminoácidos com propriedades diferentes.
168
Tabela 31: Predições in silico para a substituição missense p.Arg408Gln
SUBSTITUIÇÕES DE
SIFT
PolyPhen
Align-GVGD
Tolerável
(0,24)
Provavelmente
deletéria
(1,00)
Classe 35
GV = 0,00; GD = 42,81
AMINOÁCIDOS
p.Arg408Gln
a
SIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
b
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas. GD
(Grantham distance) = pontuação da diferença química entre os residuos normal e mutante. Quanto maior a
pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class 35 =
probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
A paciente 12 apresentou as alterações p.Glu117Lis (éxon 3) e p.Asn1077Ser (éxon 24)
no gene NPHS1, além da variante c.-267G>C na região 5’reguladora (figura 63).
O paciente 87 apresentou a variante p.Arg408Gln no gene NPHS1 e a p.Arg229Gln no
gene NPHS2.
Koziell et al. em 2002 (Koziell et al., 2002), ao analisarem os genes NPHS1 e NPHS2 em
pacientes com GESF congênito com fenótipo menos grave do que a SNC clássica, identificaram
mutações em ambos os genes em quatro pacientes, sugerindo uma interrelação entre as duas
proteínas nos podócitos, seguindo um modelo de herança digênico. Dois anos mais tarde,
Schultheiss et al. (Schultheiss et al., 2004), ao avaliarem o gene NPHS1 de pacientes com SNC
apresentando uma ou duas mutações já descritas previamente no gene NPHS2, concluiram que
não há evidências de herança digênica: esse grupo alega que mesmo havendo identificado a
alteração p.Arg229Gln em dois de quatro pacientes com duas mutações no gene NPHS1, esta
169
alteração não modificou o fenótipo desses pacientes e atribuem a doença às mutações no gene
NPHS1. Ainda, em um paciente no qual haviam sido identificadas duas mutações no gene
NPHS2, observaram a presença de uma mutação no gene NPHS1. No entanto, pelo quadro
clínico desse paciente não se justificava a ação da alteração no NPHS1 e atribuíram o fenótipo às
mutações recessivas no gene NPHS2. O grupo em questão não identificou
pacientes
apresentando uma mutação em heterozigose no gene NPHS1 e uma mutação em heterozigose no
gene NPHS2.
No caso da paciente 6, que apresentou as variantes p.Glu117Lis, p.Asn1077Ser e
p.Arg408Gln no gene NPHS1 e p.Arg229Gln no gene NPHS2, tivemos a oportunidade de
analisar o DNA dos pais e verificamos que duas (p.Arg408Gln e p.Arg229Gln) foram herdadas
da mãe e uma (p.Asn1077Ser) do pai, ambos sem histórico de doença renal (não avaliamos a
variante p.Glu117Lis). Não podemos descartar a hipótese de que o conjunto destas quatro
alterações estejam atuando em sinergia, colaborando para o quadro clínico da paciente 6, que é
de SNCS,RF.
Também analisamos os genes NPHS2 e NPHS1 do pai do paciente 87. Como o pai
também é afetado, com SNCR, e é portador da variante p.Arg229Gln do gene NPHS2 mas não
apresenta a variante p.Arg408Gln do gene NPHS1, provavelmente esta associação não se
correlaciona com o fenótipo do filho, que apresenta também a SNCR. Assim, com estes
resultados, não há evidências de que as variantes p.Arg229Gln e p.Arg408Gln nos genes NPHS2
e NPHS1, respectivamente, estejam agindo nos moldes de uma herança digênica conforme
relatado pelo grupo de Schultheiss (2004). Infelizmente não foi possível analisar o DNA dos pais
da paciente 12, o que torna-se imprescindível para realizarmos conclusões sobre o papel das
alterações em conjunto nesta paciente.
170
2.3 Polimorfismos identificados nos genes NPHS2 e NPHS1
Os polimorfismos de nucleotídeos únicos (SNPs, do inglês single nucleotide
polymorphism) são as variações genéticas mais comuns entre os seres humanos. Estima-se que a
cada 300 nucleotídeos um seja polimórfico, o que representa aproximadamente 10 milhões de
SNPs no genoma humano. A maioria dos polimorfismos encontra-se em regiões intrônicas e
regulatórias, mas muitos também podem estar presentes nas regiões codificantes. A definição de
polimorfismo é meramente uma questão de frequência na população: acima de 1%, considera-se
a variante como polimorfismo. O estudo dos SNPs é extremamente importante já que podem
atuar como SNP tags, ou seja, marcadores biológicos que segregam junto com alguma
característica individual, tal como resposta a certas drogas, susceptibilidade a fatores ambientais
e risco de desenvolver doenças.
Foi feita uma análise mais detalhada para 18 SNPs no gene NPHS2 que apresentaram
heterozigose ou homozigose do alelo mais raro, nos 134 pacientes não aparentados estudados
para este gene. Os dados deste estudo foram comparados aos dos decorrentes do projeto 1000
genomes depositados nos bancos de dados (tabela 32). Para os 11 SNPs identificados em
heterozigose ou homozigose do alelo mais raro no gene NPHS1 entre os 13 pacientes não
aparentados (para um dos 14 pacientes não foi realizada a análise de polimorfismos) com SN
infantil, na infância, juvenil e com proteinúria isolada e entre os três pacientes com SNC, foi
realizada também a comparação com as frequências alélicas registradas do banco de dados 1000
genomes (www.ensembl.org). Estes dados estão apresentados na tabela 33.
171
Tabela 32: Comparação das frequências alélicas dos SNPs/NPHS2 /134 pacientes não
aparentados com SN infantil, infância, juvenil e proteinúria isolada e 1000 genomesa
Pacientes
Pacientes
1000 genomes
1000 genomes
(n = 134)
(n=134)
(n=1092)
(n =1092)
genótipo
Freq. Alélica
genótipo
Freq. Alélica
rs78541594 (C/G)
5’UTR
122 (C/C)
12 (C/G)
0 (G/G)
C = 95,5%
G = 4,5%
rs111306764 (C/T)
p.Ser48Ser Éxon 1
132 (C/C)
2 (C/T)
0 (T/T)
C = 99,25%
0,75%
rs3738423 (C/T)
p.Ser96Ser Éxon 2
118 (C/C)
15 (C/T)
1 (T/T)
C = 93,7 %
T = 6,3%
rs73051838 (T/C)
Íntron 3
116 (T/T)
18 (T/C)
0 (C/C)
T = 93,3%
C = 6,7%
rs41267604 (C/T)
Íntron 3
124 (C/C)
9 (C/T)
1 (T/T)
C = 95,9%
T = 4,1%
rs16854341 (T/C)
Íntron 3
113 (T/T)
21 (T/C)
0 (C/C)
T = 92,2%
C = 7,8%
rs12401708 (C/T)
Íntron 3
81 (C/C)
46 (C/T)
7 (T/T)
C= 77,6%
T = 22,4%
rs2274626 (C/A)
Íntron 6
53 (C/C)
63 (C/A)
18 (A/A)
C = 63,1%
A = 36,9%
rs71630230 (C/T)
Íntron 6
132 (C/C)
2 (C/T)
0 (T/T)
C = 99,25%
0,75%
rs11808359 (A/G)
Íntron 7
129 (A/A)
5 (A/G)
0 (G/G)
A = 98,2%
G = 1,8%
rs72704483 (T/C)
Íntron 7
130 (T/T)
4 (T/C)
0 (C/C)
T = 98,5%
C = 1,5%
Polimorfismos
(SNPs)
1010 (C/C)
71 (C/G)
11 (G/G)
ND
932 (C/C)
152 (C/T)
8 (T/T)
1059 (T/T)
30 (T/C)
3 (C/C)
1025 (C/C)
63 (C/T)
4 (T/T)
1020 (T/T)
66 (T/C)
6 (C/C)
807(C/C)
250 (C/T)
35 (T/T)
350 (C/C)
532 (C/A)
210 (A/A)
ND
1033 (A/A)
56 (A/G)
3 (G/G)
1061 (T/T)
30 (T/C)
1 (C/C)
C = 96%
G = 4%
ND
C = 92%
T = 8%
T = 98%
C = 2%
C = 97%
A = 3%
T = 96%
C = 4%
C = 85%
T = 15%
C = 56%
A = 44%
ND
A = 97%
G = 3%
T = 99%
C = 1%
172
Continuação da Tabela 32
rs79569192 (C/A)
Íntron 7
131 (C/C)
3 (C/A)
0 (A/A)
C = 98,9%
A = 1,1%
1037 (C/C)
51 (C/A)
4 (A/A)
C = 97%
A = 3%
rs1410592 (T/C)
p.Ala318Ala Éxon 8
36 (T/T)
49 (T/C)
49 (C/C)
T = 45,1%
C = 54,9%
362 (T/T)
531 (T/C)
199 (T/T)
T = 57%
C = 43%
rs1410591 (G/C)
3’UTR
26(G/G)
58 (G/C)
50 (C/C)
G = 41%
C = 59%
199 (G/G)
531 (G/C)
362 (C/C)
G= 43%
C = 57%
rs1410590 (G/A)
3’UTR
121 (G/G);
13 (G/C);
0 (C/C)
G = 95,1%
C = 4,9%
900 (G/G)
173 (G/C)
19 (C/C)
G = 90%
C = 10%
rs2274623 (G/A)
3’UTR
93 (G/G);
34 (G/A);
7 (A/A)
G = 82,1%
A = 17,9%
729 (G/G)
315 (G/A)
48 (A/A)
G = 81%
A = 19%
rs2274622 (A/G)
3’UTR
80 (A/A);
45 (A/G);
9 (G/G)
A = 76,5%
G = 23,5%
565 (A/A)
428 (A/G)
99 (G/G)
A = 71%
G = 29%
rs1060775 (A/G)
3’UTR
120 (A/A);
13(A/G);
1 (G;G)
A = 94,4%
G =5,6%
897 (A/A)
175 (A/G)
20 (G/G)
A = 90%
G = 10%
a
Dados obtidos através do site: www.ensembl.org (1000 genomes, Phase I).
Para todos estes SNPs nos genes NPHS2 e NPHS1 foram realizados cálculos de quiquadrado para verificar se encontram-se em equilíbrio de Hardy-Weinberg (tabelas 34 e 35,
respectivamente).
173
Tabela 33: Frequências alélicas 11 SNPs/NPHS1 entre 16 pacientesa e 1000 genomesb
Polimorfismos
(SNPs)
Pacientes
(n = 16)
genótipo
Pacientes
(n=16)
Freq. Alélica
1000 genomes
(n=1092)
genótipo
1000 genomes
(n =1092)
Freq. Alélica
rs401824 (T/C)
5’UTR
13(T/T)
3 (T/C)
0 (C/C)
T = 91%
C = 9%
795 (T/T)
249 (T/C)
48 (C/C)
T = 84%
rs3814995 (G/A)
p.Glu117Lis Éxon 3
9 (G/G)
6 (G/A)
1 (A/A)
G = 76%
A = 24%
514 (G/G)
418 (G/A)
160 (A/A)
G = 66%
A = 34%
rs412175 (A/G)
Íntron 3
13(A/A)
3 (A/G)
0 (G/G)
A = 91%
G = 9%
782 (A/A)
258 (A/G)
52 (G/G)
A = 83%
G = 17%
rs396178 (G/A)
Íntron 3
15 (G/G)
1 (G/A)
0 (A/A)
G = 97%
A = 3%
1063 (G/G)
29 (G/A)
0 (A/A)
G = 99%
A = 1%
rs417786 (C/T)
Íntron 9
13(C/C)
3 (C/T)
0 (T/T)
A = 91%
G = 9%
963 (C/C)
123 (C/T)
6 (T/T)
G = 94%
T = 6%
rs144353637 (AAC/-)
Íntron 14
13 (AAC/AAC)
3 (AAC/-)
0 (-/-)
A = 91%
G = 9%
1001 (AAC/AAC)
88 (AAC/ - )
3 (- / -)
AAC = 96%
- = 4%
rs437168 (C/T)
Éxon 17
14 (C/C)
2 (C/T)
0 (T/T)
C = 94%
T = 6%
782 (C/C)
252 (C/T)
58 (T/T)
C = 83%
T = 17%
rs466452 (C/T)
Íntron 24
6 (C/C)
8 (C/T)
2 (T/T)
C = 62%
T = 38%
483 (C/C)
431 (C/T)
178 (T/T)
C = 64%
T = 36%
rs2071327 (G/A)
p.Ser1105Ser - Éxon 26
14 (G/G)
2 (G/A)
0 (A/A)
A = 91%
G = 9%
476 (G/G)
385 (G/A)
231 (A/A)
G = 61%
A = 39%
rs731934 (C/T)
Íntron 27
13 (C/C)
3 (C/T)
0 (T/T)
C = 88%
T = 12%
478 (C/C)
385(C/T)
229 (T/T)
C = 61%
T = 39%
rs71354105 (G/A)
3’UTR
15 (G/G)
1 (G/A)
0 (A/A)
C = 94%
T = 6%
980 (G/G)
110 (G/A)
2 (A/A)
G = 95%
A = 5%
C = 16%
a
(13 com SN infantil, infância, juvenil e proteinúria isolada e três com SNC) - não foi realizada a análise para a
paciente 133. bDados obtidos através do site: www.ensembl.org.
174
Tabela 34: Qui-quadrado (x2) e p-value dos 18 SNPs / NPHS2 nos 134 pacientes não
aparentados com SN infantil, na infância, juvenil e proteinúria isolada, para verificar
equilíbrio de Hardy-Weinberg.
Polimorfismos Localização
(SNPs)
no gene
Observado
Esperadoa
(n = 134)
(n = 134)
QuiQuadradoa
(x2)
p-valueb
rs78541594
5’UTR
122
12
0
122,27
11,46
0,27
0,29
0,8624
rs111306764
Éxon 1
132
2
0
132,01
1,99
0,01
0,01
0,9949
rs3738423
Éxon 2
118
15
1
117,54
15,92
0,54
0,45
0,7997
rs73051838
Íntron 3
116
18
0
116,6
16,79
0,6
0,69
0,7081
rs41267604
Íntron 3
124
9
1
123,23
10,55
0,23
2,89
0,2453
rs16854341
Íntron 3
113
21
0
113,82
19,35
0,82
0,97
0,6167
rs12401708
Íntron 3
81
46
7
80,72
46,57
6,72
0,02
0,9902
rs2274626
Íntron 6
53
63
18
53,29
62,43
18,29
0,27
0,9943
rs71630230
Íntron 6
132
2
0
132,01
1,99
0,01
0.01
0,9949
rs11808359
Íntron 7
129
5
0
129,05
4,91
0,05
0,05
0,9745
rs72704483
Íntron 7
130
4
0
130,03
3,94
0,03
0,03
0,9846
rs79569192
Íntron 7
131
3
0
131,02
2,97
0,02
0,02
0,9899
rs1410592
Éxon 8
36
49
49
27,32
66,37
40,32
9,18C
0,0102C
rs1410591
3’UTR
26
58
50
22,57
64,85
46,57
1,5
0,473
rs1410590
3’UTR
121
13
0
121,32
12,37
0,32
0,35
0,8382
rs2274623
3’UTR
93
34
7
90,3
39,4
4,3
2,52
0,2842
rs2274622
3’UTR
80
45
9
78,4
48,19
7,4
0,59
0,7446
rs1060775
3’UTR
120
13
1
119,42
14,16
0,42
0,9
0,6380
a
Cálculos realizados com o auxílio do programa OEGE - Online Encyclopedia for Genetic Epidemiology
studies(Santiago Rodriguez et al., 2009).bP-value ≤ 0,05; cálculos realizados pela função qui-quadrado do Excel.
C
Polimofismo fora de equilíbrio de Hardy-Weinberg.
175
Tabela 35: Qui-quadrado (x2) e p-value dos 11 SNPs identificados no gene NPHS1
entre os 16 pacientes (13 SN infantil, infância, juvenil e proteinúria isolada e três SNC),
para verificar equilíbrio de Hardy-Weinberg.
Polimorfismos Localização
(SNPs)
no gene
Observado
Esperadoa
(n = 16)
( n = 16)
Qui-Quadradoa
(x2)
p-valueb
rs401824
5’UTR
13
3
0
13,14
2,72
0,14
0,17
0,9323
rs3814995
Éxon 3
9
6
1
9
6
1
0
1
rs412175
Íntron 3
13
3
0
13,14
2,72
0,14
0,17
0,9323
rs396178
Íntron 3
15
1
0
15,02
0,97
0,02
0,02
0,9905
rs417786
Íntron 9
13
3
0
13,14
2,72
0,14
0,17
0,9323
rs144353637
Íntron 14
13
3
0
13,14
2,72
0,14
0,16
0,9323
rs437168
Éxon 17
14
2
0
14,06
1,88
0,06
0,07
0,9704
rs466452
Íntron 24
6
8
2
6,25
7,5
2,25
0,07
0,9650
rs2071327
Éxon 26
14
2
0
14,06
1,88
0,06
0,07
0,9704
rs731934
Íntron 27
13
3
0
13,14
2,72
0,14
0,17
0,9323
rs71354105
3’UTR
15
1
0
15,02
0,97
0,02
0,02
0,9905
a
Cálculos realizados com o auxílio do programa OEGE - Online Encyclopedia for Genetic Epidemiology studies
(http://www.oege.org/software/hwe-mr-calc.shtml) (Santiago Rodriguez, 2009).
b
P-value ≤ 0,05; cálculos realizados pela função qui-quadrado do Excel.
Hardy e Weinberg, ao estudarem Genética de Populações, perceberam que “se não
existissem fatores evolutivos atuando sobre uma população, as frequências gênicas
permaneceriam inalteradas e as proporções genotípicas atingiriam um equilíbrio estável,
mostrando a mesma relação constante entre si ao longo do tempo” (Beiguelman, 2008). Os
fatores evolutivos a que esses pesquisadores se referiam são: mutações, seleção natural, deriva
genética e fluxo gênico de populações migrantes ou apenas a frequência genotípica pelo aumento
176
da homozigose (efeito dos casamentos consanguíneos ou da subdivisão da população em grandes
isolados).
Hardy e Weinberg postularam algumas premissas para que uma população teórica
permanecesse em equilíbrio genético ao longo do tempo (retirado do livro de Beiguelman, 2008):
1. A população é infinita.
2. Existe o mesmo número de homens e de mulheres na população.
3. A população está em panmixia, isto é, todos se casam e os casamentos ocorrem
aleatoriamente, não existindo, por conseguinte, casamentos preferenciais entre indivíduos por
causa de seu genótipo, fenótipo, estratificação social ou consanguinidade. Aliás, por serem os
casamentos realizados aleatoriamente, os casamentos consanguíneos podem existir, desde que
ocorram aleatoriamente.
4. Todos os casais da população são igualmente férteis e geram o mesmo número de
filhos.
5. Não há sobreposição de gerações na população, isto é, elas não se imbricam ao longo
do tempo, porque todos os indivíduos devem ter a mesma idade ao casar.
6. Os genes da população não sofrem mutação.
7. A população não está sob pressão da seleção natural, porque todos os indivíduos são
igualmente viáveis, não existindo fatores que aumentem ou diminuam a sobrevivência de
indivíduos com determinado genótipo.
8. A população não recebe nem emite um fluxo gênico capaz de alterar a sua composição
gênica original, porque ela não sofre miscigenação com uma população imigrante que apresenta
frequências gênicas diferentes da dela, nem há emigração diferencial, isto é, a saída de grupos de
indivíduos com frequência gênica distinta do resto da população.
177
Apesar de nenhuma população humana real conseguir satisfazer estas oito premissas, o
que permite que ocorra a evolução, pode parecer um paradoxo, mas a maioria dos SNPs
encontra-se em equilíbrio de Hardy-Weinberg. Isto pode acontecer devido ao fato de que
casamentos consanguíneos favorecem doenças monogênicas com herança autossômica recessiva
o que acaba gerando certa homogeneidade genotípica. E realmente, entre os 18 SNPs
identificados no gene NPHS2 e entre os 11 no gene NPHS1 em nossa casuística (tabelas 34 e 35),
somente um, o rs1410592, que representa a substituição sinônima p.Ala318Ala, no éxon 8 do
gene NPHS2, não encontra-se em equilíbrio de Hardy-Weinberg (tabela 34), o que pode ser
causado pela presença de casamentos não aleatórios na população brasileira, favorecendo um dos
alelos e levando ao desequilíbrio.
Comparando-se as frequências alélicas observadas para os SNPs em ambos os genes
(tabelas 32 e 33) entre os pacientes e a população em geral (1000 genomes) nota-se que alguns
SNPs apresentam frequência semelhante entre estas populações e outras aparentemente bem
diferentes. Mas como saber se a diferença encontrada é significativa? Para verificar estes dados,
fizemos os cálculos do p-value para as combinações pacientes x 1000 genomes para as
frequências observadas e esperadas em cada SNP dos genes NPHS2 e NPHS1 e, com isto,
verificamos se a diferença observada entre as frequências alélicas dos SNPs para ambos os genes
eram ou não significativas. Observa-se que houve diferença significativa em sete dos 16 SNPs e
em dois dos 10 SNPs para os quais foi possível a realização do cálculo do p-value nos gene
NPHS2 e NPHS1, respectivamente (tabelas 36 e 37). Estes resultados foram inesperados e
claramente indicam que o comportamento de vários SNPs, principalmente no gene NPHS2, está
bem discrepante quando comparado com a população em geral.
178
Tabela 36: P-value da comparação dos SNPs / NPHS2 entre 134 pacientes e 1000 genomes
PACIENTES (n = 134)
Alterações
rs78541594
rs111306764
rs3738423
rs73051838
rs41267604
rs16854341
rs12401708
rs2274626
rs71630230
rs11808359
rs72704483
rs79569192
rs1410592
rs1410591
rs1410590
rs2274623
rs2274622
rs1060775
1000 GENOMES (n= 134a)
Observado
Esperadob
Observado
Esperadob
p-Valuec
122 CC
12 CG
0 GG
132 CC
2 CT
0 TT
118 CC
15 CT
1 TT
116 TT
18 TC
0 CC
124 CC
9 CT
1 TT
113 TT
21 TC
0 CC
81 CC
46 CT
7 TT
53 CC
63 CA
18 AA
132 CC
2 CT
0 TT
129 AA
5 AG
0 GG
130 TT
4 TC
0 CC
131 CC
3 CA
0 AA
36 TT
49 TC
49 CC
26 GG
58 GC
50 CC
121 GG
13 GC
0 CC
93 GG
34 GA
7 AA
80 AA
45 AG
9 GG
122,27 CC
11,46 CG
0,27 GG
132,01 CC
1,99 CT
0,01 TT
117,54 CC
15,92 CT
0,54 TT
116,6 TT
16,79 TC
0,6 CC
123,23 CC
10,55 CT
0,23 TT
123,82 TT
19,35 TC
0,82 CC
80,72 CC
46,57 CT
6,72 TT
53,29 CC
62,43 CA
18,29 AA
132,01 CC
1,99 CT
0,01 TT
129,05 AA
4,91 AG
0,05 GG
130,03 TT
3,94 TC
0,03 CC
131,02 CC
2,97 CA
0,02 AA
27,32 TT
66,37 TC
40,32 CC
22,57 GG
64,85 GC
46,57 CC
121,32 GG
12,37 GC
0,32 CC
90,3 GG
39,4 GA
4,3 AA
78,4 AA
48,19 AG
7,4 GG
124 CC
9 CG
1 GG
123,23 CC
10,55 CG
0,23 GG
0,3455
ND
ND
-
114 CC
19 CT
1 TT
129,95 TT
3,68 TC
0,37 CC
125,78 CC
7,73 CT
0,5 TT
125,16 TT
8,1 TC
0,73 CC
99,02 CC
30,68 CT
4,3 TT
42,95 CC
65,28 CA
25,77 AA
113,82 CC
19,35 CG
0,82 GG
129,62 TT
4,35 TC
0,04 CC
125,43 CC
8,43 CT
0,14 TT
124,6 TT
9,23 TC
0,17 CC
97,6 CC
33,52 CT
2,88 TT
42,64 CC
65,9 CA
25,46 AA
ND
ND
126,76 AA
6,87 AG
0,37 GG
130,19 TT
3,68 TC
0,12 CC
127,25 CC
6,26 CA
0,49 AA
44,42 TT
65,16 TC
24,25 CC
24,25 GG
65,16 GC
44,42 CC
110,44 GG
21,23 GC
2,33 CC
89,46 GG
38,65 GA
5,89 AA
69,33 AA
52,52 AG
12,15 GG
126,5 AA
7,39 AG
0,11 GG
130,11 TT
3,86 TC
0,03 CC
126,86 CC
7,04 CA
0,1 AA
44,25 TT
65,51 TC
24,42 CC
24,42 GG
65,51 GC
44,25 CC
109,36 GG
23,39 GC
1,25 CC
88,31 GG
40,95 GA
4,75 AA
68,19 AA
54,8 AG
11,01 GG
120 AA
13 AG
1 GG
119,42 AA
14,16 AG
0,42 GG
110,07 AA
21,47 AG
2,45 GG
108,93 AA
23,78 AG
1,3 GG
0,4050
6,06343E-23d
0,5057
9,37883E-09 d
2,56804E-06 d
0,0080 d
0,3995
0,9278
0,0884
4,37429E-11 d
0,3901
0,0010 d
0,5901
0,0295
0,0050 d
a
Valor corrigido (1092 para 134) para realização dos cálculos. bCálculos realizados com o auxílio do programa OEGE - Online Encyclopedia for
Genetic Epidemiology studies (Santiago Rodriguez et al., 2009); cCorreção de Bonferroni: α/n, portanto 0,05/2 = 0,025. Assim, p-value ≤ 0,025.
d
p-value ≤ 0,025, diferença significativa para este polimorfismo entre os pacientes e o 1000 genomes.
179
Tabela 37: P-value da comparação SNPs/NPHS1 entre 16 pacientes e 1000 genomes
PACIENTES (n = 16)
1000 GENOMES (n= 16a)
Alterações
Observado
Esperadob
Observado
Esperadob
13 TT
13,14 TT
11,65 TT
11,35 TT
rs401824
13 TC
2,72 TC
3,65 TC
4,25 TC
0 CC
0,14 CC
0,7 CC
0,4 CC
9 GG
9 GG
7,53 GG
7,02 GG
6 GA
6 GA
6,13 GA
7,16 GA
1 AA
1 AA
2,34 AA
1,83 AA
13 AA
13,14 AA
11,46 AA
11,14 AA
13 AG
2,72 AG
3,78 AG
4,42 AG
0,44 GG
rs3814995
rs412175
rs396178
rs417786
rs144353637
rs437168
rs466452
rs2071327
rs731934
rs71354105
0 GG
0,14 GG
0,76 GG
15 GG
15,02 GG
15,57GG
15,57 GG
1 GA
0,97 GA
0,43 GA
0,42 GA
0 AA
0, 02 AA
0 AA
0 AA
13 CC
13,14 CC
14,11 CC
14,08 CC
13 CT
2,72 CT
1,8 CT
1,86 CT
0 TT
0,14 TT
0,09 TT
0,06 TT
13 AAC/AAC
13,14 AAC/AAC
14,67 AAC/AAC
14,66 AAC/AAC
3 AAC/ -
2,72 AAC/ -
1,29 AAC/ -
1,31 AAC/ -
0-/-
0 -/-
0,04 -/ -
0,03 -/ -
14 CC
14,06 CC
11,46 CC
11,06 CC
2 CT
1,88 CT
3,69 CT
4,48 CT
0 TT
0,06 TT
0,85 TT
0,45 TT
6 CC
6,25 CC
7,08 CC
6,55 CC
8 CT
7,5 CT
6,32 CT
7,37 CT
2 TT
2,25 TT
2,6 TT
2,07 TT
14 GG
14,06 GG
6,98 GG
6 GG
2 GA
1,88 GA
5,64 GA
7,6 GA
0 AA
0,06 AA
3,38 AA
2,4 AA
13 CC
13,14 CC
7 CC
6,03 CC
13 CT
2,72 CT
5,64 CT
7,59 CT
0 TT
0,14 TT
3,36 TT
2,39 TT
15 GG
15,02 GG
14,36 GG
14,38 GG
1 GA
0,97 GA
1,61 GA
1,57 GA
0 AA
0, 02 AA
0,02 AA
0,04 AA
p-Valuec
0,3726
0,3362
0,3095
-d
0,4620
0,1015
0,0885
0,6765
2,7634E-07e
8,44027E-06 e
0,7605
a
Valor corrigido (1092 para 16) para realização dos cálculos. bCálculos realizados com o auxílio do programa
OEGE - Online Encyclopedia for Genetic Epidemiology studies (Santiago Rodriguez et al., 2009); cCorreção de
Bonferroni: α/n, portanto 0,05/2 = 0,025. Assim, p-value ≤ 0,025.
d
Não foi possível calcular o p-value. ep-value ≤ 0,025, diferença significativa para este polimorfismo entre os
pacientes e o 1000 genomes.
180
Outra análise realizada para estes SNPs foi a do estudo de desequilíbrio de ligação (LD)
através do programa Haploview 4.2. O desequilíbrio de ligação (LD) ocorre quando alelos
vizinhos estão muito próximos e são segregados juntos ao longo das gerações, formando
haplótipos que são descendentes de um único cromossomo ancestral. O mapa de ligação dos 18
SNPs analisados para o gene NPHS2 está ilustrado na figura 65. Os mesmos SNPs identificados
com a frequência diferente após comparação com a população 1000 genomes encontram-se em
desequilíbrio de ligação (ressaltados em vermelho), o que sugere que estes alelos que estão mais
frequentes na casuística de indivíduos com SN encotram-se ligados, formando haplótipos que
aparentemente estão segregando. O mapa de ligação dos 11 SNPs analisados para o gene NPHS1
está ilustrado na figura 66. Os dois SNPs identificados com diferença estatisticamente
significativa entre os 16 pacientes se comparados com a população de outros países depositada
no banco de dados 1000 genomes encontram-se em LD, indicando que provavelmente estes
alelos mais frequentes segregam juntos nos indivíduos analisados. Também foi formado um
haplótipo com um grande bloco de SNPs segregando juntos, que abrange desde a região
5’reguladora até o éxon 17 (SNPs ressaltados em azul na figura 66).
181
Figura 65: Mapa de desequilíbrio de ligação realizado pelo programa Haploview 4.2 para os 18 polimorfismos
identificados no gene NPHS2 entre os 134 pacientes com SN infantil, na infância, juvenil e com proteinúria
isolada. Os blocos de haplótipos estão ilustrados. Dentro dos quadrados estão representados os valores de r 2.
Quadrados em cinza escuro são considerados em DL, portanto os SNPs ressaltados em vermelho estão segregando
juntos entre os 134 pacientes analisados. O SNP rs1410592 encontra-se em desequilíbrio de
Hardy-Weinberg
(observar tabela 33). Quadrados em cinza claro e em branco indicam recombinação.
182
Figura 66: Mapa de desequilíbrio de ligação realizado pelo programa Haploview 4.2 para os 11 SNPs
identificados no gene NPHS1 entre os 16 pacientes analisados. Os blocos de haplótipos estão ilustrados.
Quadrados pretos significam 100% de chance de desequilíbrio de ligação, valor de r 2 = 1. Quadrados cinza claro e
branco = recombinação. SNPs ressaltados em vermelho são os mesmos que foram identificados com frequência
diferente entre 16 pacientes e 1000 genomes. SNPs em azul formam um bloco em LD na nossa casuística.
Uma possível causa para a formação destes blocos de haplótipos entre os pacientes
estudados é a já mencionada maior frequência alélica de alguns SNPs entre os mesmos e que
difere da população em geral depositada nos bancos de dados. Estes resultados podem sugerir
que estes haplótipos estejam associados com a doença na casuística brasileira estudada, porém
183
para um estudo de associação será necessário um grupo controle maior, com poder estatístico
suficiente para validar ou não esta hipótese.
2.4 Gene WT1
Para o gene WT1, foi realizada a triagem molecular somente do hot spot compreendendo
os éxons 8 e 9, já que somente mutações nesse fragmento foram descritas até o momento que
estejam relacionadas à SN isolada (Benetti et al., 2010; Bockenhauer et al., 2009; Chernin et al.,
2010; Fujita et al., 2010; Mucha et al., 2006). Foram triados os mesmos 147 pacientes com SN
infantil, SN na infância e juvenil e com proteinúria isolada. Foram identificadas mutações em
heterozigose (herança autossômica dominante) em três pacientes.
2.4.1 Mutação c.1227+5G>A, paciente 1
A paciente 1 iniciou acompanhamento no ambulatório de nefrologia pediátrica da
Unicamp quando tinha cinco anos de idade apresentando edema e proteinúria persistentes. Não
há registro de dados histológicos, pois, como apresentava rins em ferradura, não foi possível a
realização de biópsia. Após várias tentativas de tratamento, uma amostra desta paciente, que já
estava com sete anos, foi encaminhada para nosso laboratório para pesquisa genética. Após
análise dos genes NPHS2 e WT1, a mutação c.1227+5G>A foi identificada no íntron 9 do gene
WT1, mutação esta indicativa de síndrome de Frasier (Barbaux et al., 1997; Klamt et al., 1998)
(figura 67). A análise do cariótipo foi imediatamente requisitada, com resultado 46,XY. Devido
ao alto risco de desenvolvimento de gonadoblastoma, a gonadectomia bilateral foi realizada
184
pelos médicos e o resultado da histologia foi de disgerminoma bilateral. Aos 10 anos, a paciente
1 desenvolveu insuficiência renal o que a levou à diálise peritoneal durante um mês e ao
transplante renal logo em seguida. Este caso foi descrito em uma publicação no ano de 2012
(Guaragna et al., 2012).
Figura 67: Eletroferograma da mutação c.1227+5G>A identificada no íntron 9 do gene WT1 da paciente 1. A
heterozigose G/A está ressaltada em amarelo na sequência acima dos picos do eletroferograma e denotada em
vermelho na sequência abaixo. Os sítios de splicing para as isoformas KTS (-) e KTS (+) estão denotados em azula e
verde, respecitamente. K = lisina; T = treonina; S = serina.
Este achado confirma o fato de que, como na grande maioria dos casos de FS a genitália
externa é feminina, não há suspeita de sexo reverso até a adolescência, quando estas pacientes
queixam-se de puberdade atrasada e/ou amenorreia primária (Bache et al., 2010; Santín et al.,
2011). Deste modo, este resultado só vem a reforçar que em casos com SNCR e puberdade
atrasada é fundamental a análise tanto do cariótipo quanto do fragmento 8-9 do gene WT1, única
forma de se confirmar o diagnóstico, possibilitando a retirada do tratamento ineficiente e
realização de gonadectomia preventiva, de preferência antes do transplante renal.
185
2.4.2 Mutação c.1227+4C>T, paciente 2
A paciente 2, com cariótipo 46,XX, tem histórico de diabetes mellitus tipo 1 desde os
dois anos de idade, mesma idade na qual foi diagnosticada proteinúria nefrótica isolada, sem
edema ou hipoalbuminemia. Aos três anos de idade foi realizada uma biópsia renal, com
resultado de glomerulonefrite mesangial proliferativa. Como sua função renal piorou muito, aos
oito anos de idade foi realizada uma segunda biópsia, com resultado histológico de GESF. Sua
mãe já teve seus rins transplantados e desenvolveu disgerminoma após administração de
ciclosporina. Assim, as amostras da paciente 2 e de sua mãe foram encaminhadas para análise
molecular no nosso laboratório. Após análise do fragmento 8-9 do gene WT1, foi identificada a
mutação c.1227+4C>T em heterozigose no íntron 9 de ambas (figura 68).
Figura 68: Heredograma da família da paciente 2 e eletroferograma da mutação c.1227+4C>T identificada no
gene WT1. À esquerda, o heredograma mostra que a mutação c.1227+4C>T foi herdada da mãe. Do lado direito, o
eletroferograma da mutação c.1227+4C>T, com a heterozigose C/T ressaltada em amarelo na sequência acima dos
picos do eletroferograma. Os sítios de splicing para as isoformas KTS (-) e KTS (+) estão denotados em azula e
verde, respecitamente. K = lisina; T = treonina; S = serina.
186
Esta transição de uma citosina para uma timina na posição +4 do íntron 9 prejudica o
splicing no seg sítio doador (sítio verde na figura 68). Tanto esta mutação, como a identificada
na posição +5 na paciente 1, impedem a formação da isoforma KTS (+) e a proporção correta de
isoformas KTS (+): KTS (-) (2:1) é alterada. Esta mutação já foi bem descrita e correlacionada
com FS (Barbaux et al., 1997). A descrição clássica de Síndrome de Frasier é de pacientes
46,XY, com gonadoblastoma e GESF e geralmente as mutações no gene WT1 são mutações de
novo. No entanto, a paciente 2 é 46,XX, apresenta como sintoma somente proteinúria isolada e
sua mutação é herdada. Casos de mutações em pacientes do sexo feminino e mutações herdadas
já foram descritas anteriormente (Benetti et al., 2010; Demmer et al., 1999; Denamur et al.,
1999; Mucha et al., 2006; Ruf et al., 2004; Tsuda et al., 1999).
Após várias tentativas de tratamento, em 2013, com 10 anos de idade, a paciente 2 atingiu
insuficiência renal, quando então foi iniciada diálise.
2.4.3 Mutação c.1278C>T - p.Ser393Fen, paciente 3
A paciente 3, com cariótipo 46,XX, foi diagnosticada aos sete anos com proteinúria não
nefrótica. Em seu histórico não há dados histológicos de GESF, embora haja relato de que seu tio
paterno teve diagnóstico de GESF. Seu pai tem insuficiência renal crônica. Após análise do
fragmento 8-9 foi identificada a troca de uma citosina por uma timina na posição 1278 do éxon 9
do gene WT1, que leva a uma substituição missense, a p.Ser393Fen (figura 69); esta mutação é
ainda não havia sido descrita e, juntamente com os resultados da paciente 2, foi recentemente
publicada (Guaragna et al., 2013). Analisamos o fragmento 8-9 do gene WT1 em 100 indivíduos
controle (200 alelos) e a substituição p.Ser393Fen não foi identificada em nenhum dos controles.
187
Realizamos algumas predições in silico para esta mutação. A figura 70 ilustra a análise de
conservação realizada pelo programa ClustalW, aonde observa-se que a serina na posição 393 é
um resíduo conservado ao longo da evolução. Este resíduo localiza-se no terceiro dedo de zinco
do fator de transcrição WT1, domínio de ligação ao DNA (figura 8).
Após predições in silico realizadas através dos programas SIFT, PolyPhen e Align-GVGD
para verificar o potencial efeito da substituição destes aminoácidos na proteína, todos os
programas retornaram um resultado deletério para esta mutação (tabela 38).
Figura 69: Heredograma da família da paciente 3 e eletroferograma da mutação c.1278C>T identificada no
éxon 9 do gene WT1. À esquerda o heredograma mostra que a mutação foi herdada do pai. Do lado direito, o
eletroferograma da mutação c.1278C>T (p.Ser393Fen), com a heterozigose ressaltada em amarelo na sequência
acima dos picos do eletroferograma e abaixo denotada em vermelho.
188
Homem
Rato
Camundongo
Sapo
Javali
LKRHQRRHTGVKPFQCKTCQRKFSRSDHLKTHTRTHTGKTSEKPFSCRW
LKRHQRRHTGVKPFQCKTCQRKFSRSDHLKTHTRTHTGKTSEKPFSCRW
LKRHQRRHTGVKPFQCKTCQRKFSRSDHLKTHTRTHTGKTSEKPFSCRW
LKRHQRRHTGIKPFQCKTCQRKFSRSDHLKTHTRTHTGKTSEKPFSCRW
LKRHQRRHTGVKPFQCKTCQRKFSRSDHLKTHTRTHTGKTSEKPFSCRW
**********:**************************************
Figura 70: Alinhamento das sequências contendo a região vizinha do resíduo 393 (resíduos 370 a 409) do
fator de trasncrição WT1 humano com as de quatro espécies de vertebrados. A análise foi realizada com o
programa Clustal W., O aminoácido serina na posição 393 está ressaltado em amarelo. O número de acesso Uniprot
das sequências é: homem (P19544), rato (P49952), camundongo (P22561), sapo (B5DE03) e javali (O62651).
Asteriscos (*) indicam posições que possuem um resíduo conservado e único.
Tabela 38: Predições in silico para a mutação p.Ser393Fen.
SUBSTITUIÇÕES DE
SIFT
PolyPhen
Align-GVGD
AMINOÁCIDOS
p.Ser393Fen
Deletéria
(0,00)
Provavelmente
deletéria
(1,00)
Classe 65
GV = 0,00; GD = 154,81
(aSIFT: ≤ 0.05 = deletéria; > 0.05 = tolerável
b
PolyPhen: 1.000 = provavelmente deletéria; 0.5000 = possivelmente deletéria; 0.000 = benigna
c
Align-GVGD: GV: Grantham variation = pontuação de acordo com a diferença química entre 11 ortólogos
(abrangendo desde os chimpanzés até o peixe zebra). Valor zero = conservação completa entre as ortólogas de
podocina. GD (Grantham distance) = pontuação da diferença química entre os residuos normal e mutante. Quanto
maior a pontuação, maior a diferença. Classe 45, classe 55 e classe 65 = grande probabilidade de ser deletéria; Class
35 = probabilidade média de ser deletéria; Class 25 = não classificada; Class 15 e C0 = benigna.
189
O fator de transcrição WT1, diferentemente das proteínas nefrina e podocina, já teve parte
da sua estrutura protéica resolvida - quatro dedos de zinco complexados com o DNA (Stoll et al.,
2007) - tanto por cristalografia de raio-X quanto por espectometria de ressonância magnética
MR). A estrutura obtida por MR (PDB: 2PJA) foi utilizada para modelamento estrutural, tanto da
proteína normal como da proteína mutante, através do programa disponível on-line SWISS
MODEL (http://www.swissmodel.expasy.org). Este programa gerou arquivos que foram
enviados para análise de contatos internos da proteína pelo programa de acesso livre STING
Milleninum Blue Star Sting (http://www.cnptia.embrapa.br/) e editados pelo programa
PyMOL® (figura 71). Como pode ser visto na figura 71, a substituição p.Ser393Fen cria novas
interações com os aminoácidos Lis381 e His397, o que pode causar maior rigidez na
conformação proteica.
190
Figura 71: Análise in silico da proteína WT1 normal e mutante. A. Modelo estrutural dos dedos de zinco 2 e 3
do WT1 gerados pelo programa PyMOL®. As linhas pontilhadas indicam a distância em ângstrons (Ǻ) entre os
contatos internos realizados pelo resíduo normal (Ser393) e pelo resíduo mutante (Fen393). B. Análise dos contatos
internos gerados pelo programa Sting Millenium (www.cnptia.embrapa.br/): o resíduo normal Ser393 está em
contato com a Fen383, mantendo interações hidrofóbicas; a Fen393, por sua vez, realiza interações com a Lis381 e
His397, além das interações hidrofóbicas com a Fen383; C. Estrutura do complexo do WT1 com o DNA obtido por
espectrometria de ressonância magnética nuclear (NMR), após download do modelo PDB:2PJA. Os dados foram
editados pelo programa PyMOL®.
191
O WT1 pertence à família de dedos de zinco do tipo C2H2, na qual duas cisteínas e duas
histidinas coordenam um íon de zinco (figura 72).
Figura 72: Estrutura secundária dos quatro dedos de zinco (ZF-1 a ZF4) do fator de transcrição WT1. O
resíduo Ser393 no ZF3 (ressaltado em azul claro) está denotado em vermelho. O resíduo Fen383 com o qual a
Ser393 faz contato está ilustrado em laranja. As serinas 365 (ZF2) e 393 (ZF3) que sofrem fosforilação estão
ressaltadas em amarelo. O resíduo Fen392 (sublinhado), adjacente à Ser393, é importante para o enovelamento
estável do ZF3. Figura adaptada de (Kaltenis et al., 2004).
A seguinte organização descreve esse tipo de dedo de zinco: #-X-C-X[1–5]-C-X3-#-X5#-X2-H-X[3–6]-(H/C), onde X pode ser qualquer aminoácido e os números entre colchetes
indicam o número de resíduos. As posições marcadas com um # são importantes para o correto
enovelamento do dedo de zinco. A posição final de cada dedo pode ser tanto um resíduo Cis ou
um His. Os dedos de zinco do tipo C2H2 são compostos por duas folhas beta pequenas seguidas
192
por uma alfa hélice e ligam-se a fenda maior da dupla hélice do DNA (número de acesso
PF00096,
http://pfam.sanger.ac.uk/).
A
sequência
do
terceiro
dedo
de
zinco
é:
FQCKTCQRKFSRSDHLKTHTRTH ressaltado em cinza claro na figura 72. A Ser393, em
vermelho, localiza-se próxima a um aminoácido importante para o enovelamento estável do ZF3,
o resíduo Fen392 (sublinhado na figura 72). Esse resíduo (Fen392) já foi identificado mutado por
Kaltenis et al. (2004) e correlacionado ao desenvolvimento lento de GESF em um paciente do
sexo masculino, cariótipo 46,XY e presença de hipospadia e criptorquidia.
O WT1 sofre modificações pós traducionais, tais como fosforilação. Sakamoto et al.
(1997) demonstraram que a atividade de ligação ao DNA do WT1 é modulada in vivo pela
fosforilação dos resíduos Ser365 e Ser393 através da enzima quinase PKA, o que inibe a ligação
com o DNA e consequentemente diminui a atividade de repressão da transcrição exercida pelo
WT1 sobre alguns genes alvo. Esse grupo demonstrou que os dois sítios precisam estar
fosforilados para que ocorra a completa neutralização da atividade de repressão. Como o resíduo
Ser393 foi substituído por uma fenilalanina, a fosforilação não pode ocorrer na isoforma do WT1
mutante. Assim, o resultado pode ser um aumento da ligação do WT1 aos seus genes alvo no
podócito, já que sua atividade repressora não será neutralizada. Ainda, a p.Ser393Fen cria novos
contatos internos levando a diferentes interações intra-moleculares, como já demonstrado na
figura 71. Estes dados sugerem alguns possíveis mecanismos pelos quais esta mutação possa
estar causando o quadro clínico da paciente 3 e de seu pai. No entanto somente através de
ensaios funcionais que será possível avaliar o efeito real desta variante.
A tabela 39 resume os resultados identificados na casuística total nos três genes.
193
Tabela 39: Distribuição do número de indivíduos com mutação nas diversas formas da doença
TOTAL ANALISADOS: (n = 151)
Gene
N° de pacientes com alterações/número total (%)
Genótipo
NPHS1 ou NPHS2 ou WT1
9/151 (6)
heterozigoto de umaa ou duasb alterações
NPHS1 ou NPHS2
16/151 (10,6)
heterozigotoc
NPHS1 e NPHS2
3/151 (2)
heterozigotod
SÍNDROME NEFRÓTICA CONGÊNITA (n = 3)
Gene
N° alterações
N° de pacientes com alterações/número total (%)
Genótipo
NPHS1
2
2/4 (50)
heterozigotob
NPHS1
1 ou 2 em cis
2/4 (50)
heterozigotoc
SN INFANTIL/ NA INFÂNCIA/ JUVENIL E PROTEINÚRIA ISOLADA (n = 147)
Gene
N° alterações
N° de pacientes com
alterações/n° total (%)
N° de pacientes SNCR com
alterações/nº total de
pacientes SNCR (%)
NPHS2
2
4/147 (2,7%)
4/26 (15,6)
NPHS2
1
14/147 (9,5%)
NPHS2 e NPHS1
1
WT1
1
a
N° de pacientes SNCS,RF com
alterações/nº total de pacientes
SNCS,RF (%)
N° de pacientes SNCS com
alterações/nº total de pacientes
SNCS (%)
3/26 (11,5)
7/65 (10,8)
2/34 (5,9)
3/147 (2,04%)
1/26 (3,8)
1/65 (1,5)
1/34 (2,9)
3/147 (2,04%)
1/26 (3,8)
-
-
b
uma alteração para o gene WT1, compatível com herança autossômica dominante; duas alterações para os genes NPHS1 ou NPHS2 em possível heterozigose
composta compatível com herança autossômica recessiva; cheterozigotos simples; dheterozigotos de uma alteração em cada gene.
194
CONCLUSÕES
195
196
CONCLUSÕES
No estudo dos genes NPHS1, NPHS2 e WT1 para o grupo de 151 pacientes que reuniu
casos de SNC, SN infantil, SN na infância, SN juvenil e proteinúria isolada:
o
6% apresentaram alterações que podem se correlacionar com os respectivos
quadros clínicos clássicos, com um padrão de herança autossômico recessivo ou
autossômico dominante para os genes NPHS1 e NPHS2 ou para o gene WT1,
repectivamente.
o 8,6% apresentaram alterações em heterozigose simples, a maior parte no gene
NPHS2, sendo que a maioria se correlacionou com casos de SNCS.
o 2% dos pacientes apresentaram alterações nos genes NPHS1 e NPHS2, podendo
nestes casos haver a correlação genótipo-fenótipo dentro de um padrão de herança
digênico.
No estudo do gene NPHS1 dos casos de SNC (n=4) pode-se dizer que:
o Foram identificadas duas ou três alterações, pelo menos duas com ação deletéria,
em um genótipo com provável heterozigose composta em dois pacientes;
o Foram identificadas alterações em heterozigose simples em dois pacientes, porém
em um deles o gene NPHS1 não foi investigado por completo, devido a problemas
técnicos.
197
No estudo dos genes NPHS2 e WT1 dos casos com SN infantil, SN infância, SN juvenil e
proteinúria isolada (n=147) pode-se dizer que:
o
na análise molecular do gene NPHS2, foram identificadas alterações em
heterozigose em genótipos com heterozigose composta em quatro pacientes, todos
CR;
o
ainda na análise do gene NPHS2, 14 pacientes dos 147, sendo três CR, dois CS,
sete CS,RF e dois com proteinúria isolada, apresentaram alterações em
heterozigose simples, 10 missenses e quatro na região protomora;
o
foi realizada a análise do gene NPHS1 para estes 14 pacientes e em três deles,
sendo um CS, um CS,RF e um CR, foram identificadas alterações no gene
NPHS1, além das já encontradas no gene NPHS2.
o
Para os mesmos 147 pacientes para os quais os éxons 8 e 9 do gene WT1 foram
triados, identificamos mutações em heterozigose em três pacientes, confirmando o
quadro clínico dos mesmos e estando nos padrões de herança autossômica
dominante.
Em relação às frequências em cada uma das formas da doença, podemos fazer as
seguintes considerações:
o
Nos casos de SNC (n=4) 50% apresentaram duas alterações em heterozigose
sugerindo herança autossômica recessiva clássica para a doença, sendo que os
outros 50% apresentaram hereterozigose simples, porém para o caso cujo material
198
era proveniente de blocos de parafina a análise foi realizada em apenas oito dos
29 éxons do gene.
o
Nos casos de SN infantil, SN na infância, SN juvenil e pacientes com proteinúria
isolada, quatro pacientes, todos CR, correspondendo a 2,7% do total de 147,
apresentaram genótipos compatíveis com herança autossômica recessiva, padrão
clássico para a doença. Se considerarmos somente os pacientes do grupo de CR,
que totalizam 26, a frequência passa a ser 15,4%.
o Ainda, entre os 147 casos, 14 apresentaram heterozigose simples apenas no gene
NPHS2, correspondendo a 9,5%. Para estes casos, acredita-se que estas alterações
possam estar causando o desenvolvimento tardio da SN e com sensibilidade ao
tratamento com corticoides.
o Três casos apresentaram heterozigoses tanto no gene NPHS2 quanto no gene
NPHS1. Não foi possível estabelecer uma correlação das alterações identificadas
nos dois genes com o quadro clínico dos pacientes já que as variantes
identificadas no gene NPHS1, apesar de serem substituições de aminoácidos não
sinônimas, são polimorfismos já bem descritos e frequentes na população em
geral.
o Para o gene WT1 2% apresentaram mutações deletérias que se correlacionam
com o quadro clínico dos pacientes e segregam nos padrões de herança
autossômica dominante.
199
200
REFERÊNCIAS
201
202
REFERÊNCIAS
ABECASIS, G.R.; AUTON, A.; BROOKS, L.D.; DEPRISTO, M.A.; DURBIN, R.M.;
HANDSAKER, R.E.; MCVEAN, G.A. (2012). An integrated map of genetic variation from
1,092 human genomes. Nature, 491(7422), 56–65.
ADDIS T. (1948). Glomerulonephritis nephritis: diagnosis and treatment. New York, NY: The
McMillan Company.
ANDERSON, M.; KIM, E.Y.; HAGMANN, H.; BENZING, T. & DRYER, S.E. (2013).
Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by
membrane stretch or diacylglycerol. American journal physiology Cell physiology, 305(3),
C276–89.
ARIAS, L.F.; VIECO, B.E. & ARTETA, A.A. (2009). [Expression of nephrin, podocin and aactinine-4 in renal tissue of patients with proteinuria]. Nefrología : publicación oficial de la
Sociedad Española Nefrologia, 29(6), 569–75.
ARNOLD, K.; BORDOLI L.; KOPP, J. & SCHWEDE, T. (2006). The SWISS-MODEL
workspace: a web-based environment for protein structure homology modelling.
Bioinformatics 22(2), 195–201.
AUCELLA, F.; BISCEGLIA, L.; DE BONIS, P.; GIGANTE, M.; CARIDI, G.; BARBANO, G.;
GHIGGERI, G.M. (2006). WT1 mutations in nephrotic syndrome revisited. High prevalence
in young girls, associations and renal phenotypes. Pediatric nephrology, 21(10), 1393–8.
BACHE, M.; DHEU, C.; DORAY, B.; FOTHERGILL, H.; SOSKIN, S.; PARIS, F.;
FISCHBACH, M. (2010). Frasier syndrome, a potential cause of end-stage renal failure in
childhood. Pediatric nephrology, 25(3), 549–52.
BAIROCH, A.; APWEILER, R.; WU, C.H.; BARKER, W.C.; BOECKMANN, B.; FERRO, S;
YEH, L.S.L. (2005). The Universal Protein Resource (UniProt). Nucleic acids research, 33
(Database issue), D154–9.
BALLERMANN, B.J. & STAN, R.V. (2007). Resolved: capillary endothelium is a major
contributor to the glomerular filtration barrier. Journal of the American Society of
Nephrology : JASN, 18(9), 2432–8.
BARBAUX, S.; NIAUDET, P.; GUBLER, M.C.; GRÜNFELD, J.P.; JAUBERT, F.;
KUTTENN, F. & MCELREAVEY, K. (1997). Donor splice-site mutations in WT1 are
responsible for Frasier syndrome. Nature genetics, 17(4), 467–70.
203
BARLETTA, G.M.; KOVARI, I.A.; VERMA, R.K.; KERJASCHKI, D. & HOLZMAN, L. B.
(2003). Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction
and form cis hetero-oligomers. The Journal of biological chemistry, 278(21), 19266–71.
BARRETT, J.C.; FRY, B.; MALLER, J. & DALY, M.J. (2005). Haploview: analysis and
visualization of LD and haplotype maps. Bioinformatics, 21(2), 263–5.
BARUA, M.; SHIEH, E.; SCHLONDORFF, J.; GENOVESE, G.; KAPLAN, B.S. & POLLAK,
M.R. (2013). Exome sequencing and in vitro studies identified podocalyxin as a candidate
gene for focal and segmental glomerulosclerosis. Kidney international; 85(1), 124-33.
BEIGUELMAN, B. (2008). Genética de Populacões Humanas. Editora SBG, Ed. Ribeirão Preto.
BELTCHEVA, O.; MARTIN, P.; LENKKERI, U. & TRYGGVASON, K. (2001). Mutation
spectrum in the nephrin gene (NPHS1) in congenital nephrotic syndrome. Human mutation,
17(5), 368–73.
BELTCHEVA, O.; HJORLEIFSDOTTIR, E.E.; KONTUSAARI, S. & TRYGGVASON, K.
(2010). Sp1 specifically binds to an evolutionarily conserved DNA segment within a region
necessary for podocyte-specific expression of nephrin. Nephron Experimental nephrology,
114(1), e15–22.
BENETTI, E.; CARIDI, G.; MALAVENTURA, C.; DAGNINO, M.; LEONARDI, E.;
ARTIFONI, L.; GHIGGERI, G.M.; TOSATTO, S.C. & MURER, L. (2010). A novel WT1
gene mutation in a three-generation family with progressive isolated focal segmental
glomerulosclerosis. Clinical journal of the American Society of Nephrology : CJASN, 5(4),
698–702.
BENOIT, G.; MACHUCA, E. & ANTIGNAC, C. (2010). Hereditary nephrotic syndrome: a
systematic approach for genetic testing and a review of associated podocyte gene mutations.
Pediatric nephrology, 25(9), 1621–32.
BENZING, T. (2004). Signaling at the slit diaphragm. Journal of the American Society of
Nephrology : JASN, 15(6), 1382–91.
BERDELI, A.; MIR, S.; YAVASCAN, O.; SERDAROGLU, E.; BAK, M.; AKSU, N.; ONER,
A.; ANARAT, A.; DONMEZ, O.; YILDIZ, N.; SEVER, L.; TABEL, Y.; DUSUNSEL, R.;
SONMEZ, F. & CAKAR, N. (2007). NPHS2 (podicin) mutations in Turkish children with
idiopathic nephrotic syndrome. Pediatric nephrology , 22(12), 2031–40.
BERGET, S.M. (1995). Exon recognition in vertebrate splicing. The Journal of biological
chemistry, 270(6), 2411–4.
BERKOVIC, S.F.; DIBBENS, L.M.; OSHLACK, A.; SILVER, J.D.; KATERELOS, M.;
VEARS, D.F.; LÜLLMANN-RAUCH, R..; BLANZ, J.; ZHANG, K.W.; STANKOVICH,
204
J., KALNINS, R.M.; DOWLING, J.P.; ANDERMANN, E.; ANDERMANN, F.; FALDINI,
E.; D'HOOGE, R.; VADLAMUDI, L.; MACDONELL, R.A.; HODGSON, B.L.; BAYLY,
M.A.; SAVIGE, J.; MULLEY, J.C.; SMYTH, G.K.; POWER, D.A.; SAFTIG, P. &
BAHLO, M. (2008). Array-based gene discovery with three unrelated subjects shows
SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. American
journal of human genetics, 82(3), 673–84.
BETTS, M.J., & RUSSELL, R.B. (2003). Amino Acid Properties and Consequences of
Substitutions, in Bioinformatics for Geneticists; (eds M.J. Betts & I.C. Gray), John Willey
& Sons Ltd., ch 14.
BLACKALL, J. (1818). Observations on the nature and cure of dropsies, and particularly on the
presence of the coagulable part of the blood in dropsical urine. (3rd editio.). London:
Longman Green.
BLACK, D.L. (1995). Finding splice sites within a wilderness of RNA. RNA, 1(8), 763–71.
BLENCOWE, B.J. (2000). Exonic splicing enhancers: mechanism of action, diversity and role
in human genetic diseases. Trends in biochemical sciences, 25(3), 106–10.
BOCKENHAUER, D.; VAN’T HOFF, W.; CHERNIN, G.; HEERINGA, S.F. & SEBIRE, N.J.
(2009). Membranoproliferative glomerulonephritis associated with a mutation in Wilms’
tumour suppressor gene 1. Pediatric nephrology, 24(7), 1399–401.
BOERKOEL, C.F.; TAKASHIMA, H.; JOHN, J.; YAN, J.; STANKIEWICZ, P.;
ROSENBARKER, L.; ANDRÉ, J.L.; BOGDANOVIC, R.; BURGUET, A.; COCKFIELD,
S.; CORDEIRO, I.; FRÜND, S.; ILLIES, F.; JOSEPH, M.; KAITILA, I.; LAMA,
G.; LOIRAT, C.; MCLEOD, D.R.; MILFORD, D.V.; PETTY, E.M.; RODRIGO,
F.; SARAIVA, J.M.; SCHMIDT, B.; SMITH, G.C.; SPRANGER, J.; STEIN, A.; THIELE,
H.; TIZARD, J.; WEKSBERG, R.; LUPSKI, J.R.; STOCKTON, D.W. (2002). Mutant
chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia.
Nature genetics, 30(2), 215–20.
BOLK, S.; PUFFENBERGER, E.G.; HUDSON, J.; MORTON, D.H. & CHAKRAVARTI, A.
(1999). Elevated frequency and allelic heterogeneity of congenital nephrotic syndrome,
Finnish type, in the old order Mennonites. American journal of human genetics, 65(6),
1785–90.
BOUTE, N.; GRIBOUVAL, O.; ROSELLI, S.; BENESSY, F.; LEE, H.; FUCHSHUBER, A.;
DAHAN, K.; GLUBER, M.C.; NIAUDET, P. & ANTIGNAC, C. (2000). NPHS2, encoding
the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant
nephrotic syndrome. Nature genetics, 24(4), 349–54.
205
BRIGHT R. (1827). Report of medical cases with a view of illustrating the symptoms and cure
of disease by a reference to morbid anatomy. L. Longman & and G. Rees, Orme, Brown,
Eds.
BROWN, E.J.; SCHLÖNDORFF, J. S.; BECKER, D. J.; TSUKAGUCHI, H.; TONNA, S. J.;
USCINSKI, A.L.; HIGGS, H.N.; HENDERSON, J.M. & POLLAK, M.R. (2010).
Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nature
genetics, 42(1), 72–6.
BRUENING, W.; BARDEESY, N.; SILVERMAN, B.L.; COHN, R.A.; MACHIN, G.A.;
ARONSON, A.J.; HOUSMAN, D. & PELLETIER, J. (1992). Germline íntronic and exonic
mutations in the Wilms’ tumour gene (WT1) affecting urogenital development. Nature
genetics, 1(2), 144–8.
BRUNEAU, S. & DANTAL, J. (2009). New insights into the pathophysiology of idiopathic
nephrotic syndrome. Clinical immunology), 133(1), 13–21.
BUTTA, N.; LARRUCEA, S.; ALONSO, S.; RODRIGUEZ, R. B.; ARIAS-SALGADO, E. G.;
AYUSO, M.S.; GONZÁLEZ-MANCHÓN, C. & PARRILLA, R. (2006). Role of
transcription factor Sp1 and CpG methylation on the regulation of the human podocalyxin
gene promoter. BMC molecular biology, 7, 17.
CALL, K.M.; GLASER, T.; ITO, C.Y.; BUCKLER, A.J.; PELLETIER, J.; HABER, D.A.;
ROSE, E.A.; KRAL, A.; YEGER, H.; LEWIS, W.H.; JONES, C. & HOUSMAN, DE.
(1990). Isolation and characterization of a zinc finger polypeptide gene at the human
chromosome 11 Wilms’ tumor locus. Cell, 60(3), 509–20.
CARA-FUENTES, G.; ARAYA, C.; WEI, C.; RIVARD, C.; ISHIMOTO, T.; REISER, J.;
JOHNSON, R.J. & GARIN, E.H. (2013). CD80, suPAR and Nephrotic Syndrome in a case
of NPHS2 mutation. Nefrologia, 33 (5): 727-31.
CARIDI, G.; BERTELLI, R.; DI DUCA, M.; DAGNINO, M.; EMMA, F.; ONETTI MUDA, A.;
SCOLARI
F.;
MIGLIETTI,
N.; MAZZUCCO,
G.; MURER,
L.; CARREA,
A.; MASSELLA, L.; RIZZONI, G.; PERFUMO, F. & GHIGGERI, G.M. (2003).
Broadening the spectrum of diseases related to podocin mutations. Journal of the American
Society of Nephrology : JASN, 14(5), 1278–86.
CARIDI, G.; GIGANTE, M.; RAVANI, P.; TRIVELLI, A.; BARBANO, G.; SCOLARI, F.;
DAGNINO M.; MURER, L.; MURTAS, C.; EDEFONTI, A.; ALLEGRI, L.; AMORE,
A.; COPPO, R.; EMMA, F.; DE PALO, T.; PENZA, R.; GESUALDO, L. & GHIGGERI,
G.M. (2009). Clinical features and long-term outcome of nephrotic syndrome associated
with heterozygous NPHS1 and NPHS2 mutations. Clinical journal of the American Society
of Nephrology : CJASN, 4(6), 1065–72.
206
CARIDI, G.; PERFUMO, F. & GHIGGERI, G.M. (2005). NPHS2 (Podocin) mutations in
nephrotic syndrome. Clinical spectrum and fine mechanisms. Pediatric research, 57(5 Pt 2),
54R–61R.
CATTRAN, D.C. (2011). Historical aspects of proteinuria. Advances in chronic kidney disease,
18(4), 224–32.
CHADWICK, J. & MANN, W. (n.d.). The medical works of Hippocrates. O. University, Ed. pp.
1950, 240, 244. London.
CHERNIN, G.; VEGA-WARNER, V.; SCHOEB, D.S.; HEERINGA, S.F.; OVUNC, B.;
SAISAWAT, P.; CLEPER, R.; OZALTIN, F.; HILDEBRANDT, F. & MEMBERS OF
THE GPN STUDY GROUP. (2010). Genotype/phenotype correlation in nephrotic
syndrome caused by WT1 mutations. Clinical journal of the American Society of
Nephrology : CJASN, 5(9), 1655–62.
COHEN, H.T.; BOSSONE, S.A.; ZHU, G.; MCDONALD, G.A. & SUKHATME, V.P. (1997).
Sp1 is a critical regulator of the Wilms’ tumor-1 gene. The Journal of biological chemistry,
272(5), 2901–13.
COTUGNO D. (1974). De Ischiade Nervosa Commentarius (Naples. 1765). Vienna. Austria:
Graffer; 1770:29 (In: Dock W. ed. Proteinuria: the story of 250 years of trials, errors and
rectifications). Bull NY Acad Med, 5, 659–666.
DE ARAÚJO, M. (1996). Estudo da organização molecular do “cluster” gênico CYP21 e C4 em
famílias com a forma clássica de deficiência da 21-hidroxilase no Brasil [tese de mestrado].
Campinas: Instituto de Biologia; Universidade Estadual de Campinas.
DE-ARAUJO, M.; SANCHES, M.R.; SUZUKI, L.A.; GUERRA, G.; FARAH, S.B. & DEMELLO, M.P. (1996). Molecular analysis of CYP21 and C4 genes in Brazilian families
with the classical form of steroid 21-hydroxylase deficiency. Brazilian journal of medical
and biological research = Revista brasileira de pesquisas médicas e biológicas / Sociedade
Brasileira de Biofísica, 29(1), 1–13.
DEMMER, L.; PRIMACK, W.; LOIK, V.; BROWN, R.; THERVILLE, N. & MCELREAVEY,
K. (1999). Frasier syndrome: a cause of focal segmental glomerulosclerosis in a 46,XX
female. Journal of the American Society of Nephrology : JASN, 10 (10), 2215–8.
DENAMUR, E.; BOCQUET, N.; MOUGENOT, B.; DA SILVA, F.; MARTINAT, L.; LOIRAT,
C.; ELION J.; BENSMAN, A. & RONCO, P.M. (1999). Mother-to-child transmitted WT1
splice-site mutation is responsible for distinct glomerular diseases. Journal of the American
Society of Nephrology : JASN, 10(10), 2219–23.
DENYS, P.; MALVAUX, P.; VAN DEN BERGHE, H.; TANGHE, W. & PROESMANS, W.
(1967).
[Association
of
an
anatomo-pathological
syndrome
of
male
207
pseudohermaphroditism, Wilms’ tumor, parenchymatous nephropathy and XX/XY
mosaicism]. Archives françaises de pédiatrie, 24(7), 729–39.
DESMET, F.O.; HAMROUN, D.; LALANDE, M.; COLLOD-BÉROUD, G.; CLAUSTRES, M.
& BÉROUD, C. (2009). Human Splicing Finder: an online bioinformatics tool to predict
splicing signals. Nucleic acids research, 37(9), e67.
DI DUCA, M.; OLEGGINI, R.; SANNA-CHERCHI, S.; PASQUALI, L.; DI DONATO, A.;
PARODI, S., BERTELLI, R.; CARIDI, G.; FRASCA, G.; CERULLO, G.; AMOROSO,
A.; SCHENA,
F.P.; SCOLARI,
F., GHIGGERI,
G.M.
& EUROPEAN
IGA
NEPHROPATHY CONSORTIUM. (2006). Cis and trans regulatory elements in NPHS2
promoter: implications in proteinuria and progression of renal diseases. Kidney
international, 70(7), 1332–41.
DRASH, A.; SHERMAN, F.; HARTMANN, W.H. & BLIZZARD, R.M. (1970). A syndrome of
pseudohermaphroditism, Wilms’ tumor, hypertension, and degenerative renal disease. The
Journal of pediatrics, 76(4), 585–93.
DREYER, S.D.; ZHOU, G.; BALDINI, A.; WINTERPACHT, A.; ZABEL, B.; COLE, W.;
JOHNSON, R.L. & LEE, B. (1998). Mutations in LMX1B cause abnormal skeletal
patterning and renal dysplasia in nail patella syndrome. Nature genetics, 19(1), 47–50.
DREYFUSS, G.; MATUNIS, M. J.; PIÑOL-ROMA, S. & BURD, C.G. (1993). hnRNP proteins
and the biogenesis of mRNA. Annual review of biochemistry, 62, 289–321.
DUSEL, J. A.E.; BURDON, K.P.; HICKS, P.J.; HAWKINS, G.A.; BOWDEN, D.W. &
FREEDMAN, B.I. (2005). Identification of podocin (NPHS2) gene mutations in African
Americans with nondiabetic end-stage renal disease. Kidney international, 68(1), 256–62.
EGAN, M.E.; GLÖCKNER-PAGEL, J.; AMBROSE, C.; CAHILL, P.A.; PAPPOE, L.;
BALAMUTH, N.; CHO, E.; CANNY, S; WAGNER, C.A.; GEIBEL, J.; CAPLAN, M. J.
(2002). Calcium-pump inhibitors induce functional surface expression of Delta F508-CFTR
protein in cystic fibrosis epithelial cells. Nature medicine, 8(5), 485–92.
FRASIER, S.D.; BASHORE, R.A. & MOSIER, H.D. (1964). Gonadoblastoma associated with
pure gonadal dysgenesis in monozygous twins. The Journal of pediatrics, 64, 740–5.
FRISHBERG, Y.; BEN-NERIAH, Z.; SUVANTO, M.; RINAT, C.; MÄNNIKKÖ, M.;
FEINSTEIN, S.; BECKER-COHEN, R.; JALANKO, H.; ZLOTOGORA, J. & KESTILÄ,
M. (2007). Misleading findings of homozygosity mapping resulting from three novel
mutations in NPHS1 encoding nephrin in a highly inbred community. Genetics in medicine :
official journal of the American College of Medical Genetics, 9(3), 180–4.
FUCHSHUBER, A.; JEAN, G.; GRIBOUVAL, O.; GUBLER, M.C.; BROYER, M.;
BECKMANN, J.S.; NIAUDET, P. & ANTIGNAC, C. (1995). Mapping a gene (SRN1) to
208
chromosome 1q25-q31 in idiopathic nephrotic syndrome confirms a distinct entity of
autosomal recessive nephrosis. Human molecular genetics, 4(11), 2155–8.
FUCHSHUBER, A.; NIAUDET, P.; GRIBOUVAL, O.; JEAN, G.; GUBLER, M.C.; BROYER,
M. & ANTIGNAC, C. (1996). Congenital nephrotic syndrome of the Finnish type: linkage
to the locus in a non-Finnish population. Pediatric nephrology, 10(2), 135–8.
FUJITA, S.; SUGIMOTO, K.; MIYAZAWA, T.; YANAGIDA, H.; TABATA, N.; OKADA, M.
& TAKEMURA, T. (2010). A female infant with Frasier syndrome showing splice site
mutation in Wilms’ tumor gene (WT1) íntron 9. Clinical nephrology, 73(6), 487–91.
GARRETT-SINHA, L.A. (2013). Review of Ets1 structure, function, and roles in immunity.
Cellular and molecular life sciences : CMLS, 70(18), 3375–90.
GBADEGESIN, R. & SMOYER, W. (2008). Nephrotic Syndrome. In Mosby Elsevier (Ed.),
Comprehensive Pediatric Nephrology (First., pp. 205–218).
GENOVESE, G.; FRIEDMAN, D.J.; ROSS, M.D.; LECORDIER, L.; UZUREAU, P.;
FREEDMAN, B.I.; BOWDEN, D.W.; LANGEFELD, C.D.; OLEKSYK, T.K.; USCINSKI
KNOB, A.L.; BERNHARDY, A.J.; HICKS, P.J.; NELSON, G.W.; VANHOLLEBEKE,
B.; WINKLER, C.A.; KOPP, J.B.; PAYS & POLLAK, M. R. (2010). Association of
trypanolytic ApoL1 variants with kidney disease in African Americans. Science (New York,
N.Y.), 329(5993), 841–5.
GESSLER, M.; POUSTKA, A.; CAVENEE, W.; NEVE, R. L.; ORKIN, S. H. & BRUNS, G. A.
(1990). Homozygous deletion in Wilms tumours of a zinc-finger gene identified by
chromosome jumping. Nature, 343(6260), 774–8.
GIGANTE, M.; PIEMONTESE, M.; GESUALDO, L.; IOLASCON, A. & AUCELLA, F.
(2011). Molecular and genetic basis of inherited nephrotic syndrome. International journal
of nephrology, 2011, 792195.
GIGANTE, M.; PONTRELLI, P.; MONTEMURNO, E.; ROCA, L.; AUCELLA, F.; PENZA,
R.; CARIDI, G.; RANIERI, E.; GHIGGERI, G.M. & GESUALDO, L. (2009). CD2AP
mutations are associated with sporadic nephrotic syndrome and focal segmental
glomerulosclerosis (FSGS). Nephrology, dialysis, transplantation : official publication of
the European Dialysis and Transplant Association - European Renal Association, 24(6),
1858–64.
GRABE, N. (2002). AliBaba2: context specific identification of transcription factor binding
sites. In silico biology, 2(1), S1–15.
GRANTHAM, R. (1974). Amino acid difference formula to help explain protein evolution.
Science, 185(4154), 862–4.
209
GUARAGNA, M.S.; LUTAIF, A.C.G.B.; PIVETA, C.S.C.; BELANGERO, V.M.S.; MACIELGUERRA, A.T.; GUERRA-JUNIOR, G. & DE MELLO, M.P. (2013). Two distinct WT1
mutations identified in patients and relatives with isolated nephrotic proteinuria.
Biochemical and biophysical research communications.
GUARAGNA, M.S.; LUTAIF, A.C.G.B.; BITTENCOURT, V.B.; PIVETA, C.S.C.; SOARDI,
F.C.; CASTRO, L.C.G.; BELANGERO, V.M.; MACIEL-GUERRA, A.T.; GUERRAJUNIOR, G. & DE MELLO, M.P. (2012). Frasier syndrome: four new cases with unusual
presentations. Arquivos brasileiros de endocrinologia e metabologia, 56(8), 525–32.
GUEZ, S.; GIANI, M.; MELZI, M.L.; ANTIGNAC, C. & ASSAEL, B.M. (1998). Adequate
clinical control of congenital nephrotic syndrome by enalapril. Pediatric nephrology, 12(2),
130–2.
GUO, G.; MORRISON, D.J.; LICHT, J.D. & QUAGGIN, S.E. (2004). WT1 activates a
glomerular-specific enhancer identified from the human nephrin gene. Journal of the
American Society of Nephrology : JASN, 15(11), 2851–6.
HABER, D.A.; SOHN, R.L.; BUCKLER, A.J.; PELLETIER, J.; CALL, K.M. & HOUSMAN,
D.E. (1991). Alternative splicing and genomic structure of the Wilms tumor gene WT1.
Proceedings of the National Academy of Sciences of the United States of America, 88(21),
9618–22.
HALTIA, A.; SOLIN, M.L.; HOLMBERG, C.; REIVINEN, J.; MIETTINEN, A. &
HOLTHÖFER, H. (1998). Morphologic changes suggesting abnormal renal differentiation
in congenital nephrotic syndrome. Pediatric research, 43(3), 410–4.
HAMED, R.M. & SHOMAF, M. (2001). Congenital nephrotic syndrome: a clinico-pathologic
study of thirty children. Journal of nephrology, 14(2), 104–9.
HARENDZA, S.; STAHL, R.A.K. & SCHNEIDER, A. (2009). The transcriptional regulation of
podocin (NPHS2) by Lmx1b and a promoter single nucleotide polymorphism. Cellular &
molecular biology letters, 14(4), 679–91.
HE, B.; EBARASI, L.; HULTENBY, K.; TRYGGVASON, K. & BETSHOLTZ, C. (2011).
Podocin-green fluorescence protein allows visualization and functional analysis of
podocytes. Journal of the American Society of Nephrology : JASN, 22(6), 1019–23.
HEERINGA, S.F.; MÖLLER, C.C.; DU, J.; YUE, L.; HINKES, B.; CHERNIN, G.;
VLANGOS, C.N.; HOYER, P.F.; REISER, J. & HILDEBRANDT, F. (2009). A novel
TRPC6 mutation that causes childhood FSGS. PloS one, 4(11), e7771.
HEERINGA, S.F.; VLANGOS, C.N.; CHERNIN, G.; HINKES, B.; GBADEGESIN, R.; LIU, J.;
HOSKINS, B.E.; OZALTIN, F.; HILDEBRANDT, F & MEMBERS OF THE APN
STUDY GROUP. (2008). Thirteen novel NPHS1 mutations in a large cohort of children
210
with congenital nephrotic syndrome. Nephrology, dialysis, transplantation : official
publication of the European Dialysis and Transplant Association - European Renal
Association, 23(11), 3527–33.
HINKES, B.G.; MUCHA, B.; VLANGOS, C.N.; GBADEGESIN, R.; LIU, J.;
HASSELBACHER, K.; HANGAN, D.; OZALTIN, F.; ZENKER, M.; HILDEBRANDT,
F. & ARBEITSGEMEINSCHAFT FÜR PAEDIATRISCHE NEPHROLOGIE STUDY
GROUP. (2007). Nephrotic syndrome in the first year of life: two thirds of cases are caused
by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics, 119(4), e907–19.
HINKES, B.; VLANGOS, C.; HEERINGA, S.; MUCHA, B.; GBADEGESIN, R.; LIU, J.;
HASSELBACHER, K.; OZALTIN, F.; HILDEBRANDT, F. & APN STUDY GROUP
(2008). Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic
syndrome. Journal of the American Society of Nephrology : JASN, 19(2), 365–71.
HINKES, B.; WIGGINS, R.C.; GBADEGESIN, R.; VLANGOS, C.N.; SEELOW, D.;
NÜRNBERG, G.; GARG, P.; VERMA, R.; CHAIB, H.; HOSKINS, B.E.; ASHRAF,
S.; BECKER, C.; HENNIES, H.C.; GOYAL, M.;WHARRAM, B.L.; SCHACHTER,
A.D.; MUDUMANA,
S.; DRUMMOND,
I.; KERJASCHKI,
D.; WALDHERR,
R.; DIETRICH, A.; OZALTIN, F.; BAKKALOGLU, A.; CLEPER, R.; BASELVANAGAITE, L.; POHL, M.; GRIEBEL, M.; TSYGIN, A.N.; SOYLU, A.; MÜLLER,
D.; SORLI, C.S.; BUNNEY, T.D.; KATAN, M.; LIU, J., ATTANASI, M.; O'TOOLE,
J.F.; HASSELBACHER, K.; MUCHA, B.; OTTO, E.A.; AIRIK, R.; KISPERT, A.;
KELLEY, G.G.; SMRCKA, A.V.; GUDERMANN, T.; HOLZMAN, L.B.; NÜRNBERG,
P. & HILDEBRANDT, F. (2006). Positional cloning uncovers mutations in PLCE1
responsible for a nephrotic syndrome variant that may be reversible. Nature genetics,
38(12), 1397–405.
HOGG, R.J.; PORTMAN, R.J.; MILLINER, D.; LEMLEY, K.V.; EDDY, A., &
INGELFINGER, J. (2000). Evaluation and management of proteinuria and nephrotic
syndrome in children: recommendations from a pediatric nephrology panel established at
the National Kidney Foundation conference on proteinuria, albuminuria, risk, assessment,
detection, and eliminati. Pediatrics, 105(6), 1242–9.
HONDA, K.; YAMADA, T.; ENDO, R.; INO, Y.; GOTOH, M.; TSUDA, H.; YAMADA,
Y.; CHIBA, H. & HIROHASHI, S. (1998). Actinin-4, a novel actin-bundling protein
associated with cell motility and cancer invasion. The Journal of cell biology, 140(6), 1383–
93.
HOYER, J.R.; VERNIER, R.L.; NAJARIAN, J.S.; RAIJ, L.; SIMMONS, R.L. & MICHAEL,
A.F. (1972). Recurrence of idiopathic nephrotic syndrome after renal transplantation.
Lancet, 2(7773), 343–8.
211
HUBER, T.B.; KOTTGEN, M.; SCHILLING, B.; WALZ, G. & BENZING, T. (2001).
Interaction with podocin facilitates nephrin signaling. The Journal of biological chemistry,
276(45), 41543–6.
HUBER, T.B.; HARTLEBEN, B.; KIM, J.; SCHMIDTS, M.; SCHERMER, B.; KEIL,
A.; EGGER, L.; LECHA, R.L.; BORNER, C.; PAVENSTÄDT, H.; SHAW, A.S.; WALZ,
G. & BENZING, T. (2003). Nephrin and CD2AP associate with phosphoinositide 3-OH
kinase and stimulate AKT-dependent signaling. Molecular and cellular biology, 23(14),
4917–28.
HUBER, T.B.; SCHERMER, B. & BENZING, T. (2007). Podocin organizes ion channel-lipid
supercomplexes: implications for mechanosensation at the slit diaphragm. Nephron.
Experimental nephrology, 106(2), e27–31.
HUBER, T.B.; SCHERMER, B.; MÜLLER, R.U.; HÖHNE, M.; BARTRAM, M.; CALIXTO,
A.; HAGMANN, H.; REINHARDT, C.; KOOS, F.; KUNZELMANN, K.; SHIROKOVA,
E.; KRAUTWURST,
D.; HARTENECK,
C.;
SIMONS,
M.; PAVENSTÄDT,
H.; KERJASCHKI, D.; THIELE, C.; WALZ, G.; CHALFIE, M. & BENZING, T. (2006).
Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels.
Proceedings of the National Academy of Sciences of the United States of America, 103(46),
17079–86.
HUBER, T.B.; SIMONS, M.; HARTLEBEN, B.; SERNETZ, L.; SCHMIDTS, M.;
GUNDLACH, E.; SALEEM, M.A.; WALZ, G. & BENZING, T. (2003). Molecular basis of
the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin
targeting to lipid raft microdomains. Human molecular genetics, 12(24), 3397–405.
HUTTUNEN, N.P.; RAPOLA, J.; VILSKA, J. & HALLMAN, N. (1980). Renal pathology in
congenital nephrotic syndrome of Finnish type: a quantitative light microscopic study on 50
patients. The International journal of pediatric nephrology, 1(1), 10–6.
ISKDC. (1981). The primary nephrotic syndrome in children. Identification of patients with
minimal change nephrotic syndrome from initial response to prednisone. A report of the
International Study of Kidney Disease in Children. The Journal of pediatrics, 98(4), 561–4.
JAAKKO PATRAKKA. (2001). Nephrin - Role in human glomerulogenesis and nephrotic
disorders. University of Helsinki, Helsinki, Finland.
JEAN-PHILIPPE, J.; PAZ, S., & CAPUTI, M. (2013). hnRNP A1: the Swiss army knife of gene
expression. International journal of molecular sciences, 14(9), 18999–9024.
JEANPIERRE, C.; DENAMUR, E.; HENRY, I.; CABANIS, M.O.; LUCE, S.; CÉCILLE, A.;
ELION, J.; PEUCHMAUR, M.; LOIRAT, C.; NIAUDET, P.; GUBLER, M.C. & JUNIEN,
C. (1998). Identification of constitutional WT1 mutations, in patients with isolated diffuse
212
mesangial sclerosis, and analysis of genotype/phenotype correlations by use of a
computerized mutation database. American journal of human genetics, 62(4), 824–33.
KAGEYAMA, R.; MERLINO, G.T. & PASTAN, I. (1989). Nuclear factor ETF specifically
stimulates transcription from promoters without a TATA box. The Journal of biological
chemistry, 264(26), 15508–14.
KALTENIS, P.; SCHUMACHER, V.; JANKAUSKIENE, A.; LAURINAVICIUS, A. &
ROYER-POKORA, B. (2004). Slow progressive FSGS associated with an F392L WT1
mutation. Pediatric nephrolog), 19(3), 353–6.
KAMBHAM, N.; TANJI, N.; SEIGLE, R. L.; MARKOWITZ, G.S.; PULKKINEN, L.; UITTO,
J. & D’AGATI, V. D. (2000). Congenital focal segmental glomerulosclerosis associated
with beta4 integrin mutation and epidermolysis bullosa. American journal of kidney
diseases : the official journal of the National Kidney Foundation, 36(1), 190–6.
KANEKO, K.J.; KOHN, M. J.; LIU, C. & DEPAMPHILIS, M.L. (2007). Transcription factor
TEAD2 is involved in neural tube closure. Genesis, 45(9), 577–87.
KAPLAN, J.M.; KIM, S.H.; NORTH, K.N.; RENNKE, H.; CORREIA, L.A.; TONG, H.Q.;
MATHIS, B.J.; RODRÍGUEZ-PÉREZ, J.C.; ALLEN, P.G.; BEGGS, A.H. & POLLAK,
M.R. (2000). Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental
glomerulosclerosis. Nature genetics, 24(3), 251–6.
KARLE, S.M.; UETZ, B; RONNER, V.; GLAESER, L.; HILDEBRANDT, F. &
FUCHSHUBER, A. (2002). Novel mutations in NPHS2 detected in both familial and
sporadic steroid-resistant nephrotic syndrome. Journal of the American Society of
Nephrology : JASN, 13(2), 388–93.
KESTILÄ, M.; LENKKERI, U.; MÄNNIKKÖ, M.; LAMERDIN, J.; MCCREADY, P.;
PUTAALA, H.; RUOTSALAINEN, V.; MORITA, T.; NISSINEN, M.; HERVA,
R.; KASHTAN, C.E.; PELTONEN, L.; HOLMBERG, C.; OLSEN, A. & TRYGGVASON,
K. (1998). Positionally cloned gene for a novel glomerular protein--nephrin--is mutated in
congenital nephrotic syndrome. Molecular cell, 1(4), 575–82.
KESTILÄ, M.; MÄNNIKKÖ, M.; HOLMBERG, C.; GYAPAY, G.; WEISSENBACH, J.;
SAVOLAINEN, E.R.; PELTONEN, L. & TRYGGVASON, K. (1994). Congenital
nephrotic syndrome of the Finnish type maps to the long arm of chromosome 19. American
journal of human genetics, 54(5), 757–64.
KIEFER, F., ARNOLD, K., KÜNZLI, M., BORDOLI, L., & SCHWEDE, T. (2009). The
SWISS-MODEL Repository and associated resources. Nucleic acids research, 37(Database
issue), D387–92.
213
KIM, E.Y.; ANDERSON, M.; WILSON, C.; HAGMANN, H.; BENZING, T. & DRYER, S. E.
(2013). NOX2 interacts with podocyte TRPC6 channels and contributes to their activation
by diacylglycerol: Essential role of podocin in formation of this complex. American journal
of physiology. Cell physiology.
KIM, J.M.; WU, H.; GREEN, G.; WINKLER, C.A; KOPP, J.B.; MINER, J.H.; UNANUE, E.R.
& SHAW, A.S. (2003). CD2-associated protein haploinsufficiency is linked to glomerular
disease susceptibility. Science, 300(5623), 1298–300.
KIM, M.S.; STABLEIN, D. & HARMON, W.E. (1998). Renal transplantation in children with
congenital nephrotic syndrome: a report of the North American Pediatric Renal Transplant
Cooperative Study (NAPRTCS). Pediatric transplantation, 2(4), 305–8.
KLAMT, B.; KOZIELL, A.; POULAT, F.; WIEACKER, P.; SCAMBLER, P.; BERTA, P. &
GESSLER, M. (1998). Frasier syndrome is caused by defective alternative splicing of WT1
leading to an altered ratio of WT1 +/-KTS splice isoforms. Human molecular genetics, 7(4),
709–14.
KOBAYASHI, Y.; MOMOI, M.Y.; TOMINAGA, K.; MOMOI, T.; NIHEI, K.;
YANAGISAWA, M.; KAGAWA, Y. & OHTA, S. (1990). A point mutation in the
mitochondrial tRNALeu(UUR) gene in melas (mitochondrial myopathy, encephalopathy,
lactic acidosis and stroke-like episodes). Biochemical and Biophysical Research
Communications, 173(3), 816–822.
KOPP, J.B.; SMITH, M.W.; NELSON, G.W.; JOHNSON, R.C.; FREEDMAN, B.I.; BOWDEN,
D.W.; OLEKSYK, T.; MCKENZIE, L.M.; KAJIYAMA, H.; AHUJA, T.S.; BERNS,
J.S.; BRIGGS, W.; CHO, M.E.; DART, R.A.; KIMMEL, P.L.; KORBET, S.M.; MICHEL,
D.M.; MOKRZYCKI,
M.H.; SCHELLING,
J.R.; SIMON,
E.; TRACHTMAN,
H.; VLAHOV, D. & WINKLER, C. A. (2008). MYH9 is a major-effect risk gene for focal
segmental glomerulosclerosis. Nature genetics, 40(10), 1175–84.
KOZIELL, A.; GRECH, V.; HUSSAIN, S.; LEE, G.; LENKKERI, U.; TRYGGVASON, K.; &
SCAMBLER, P. (2002). Genotype/phenotype correlations of NPHS1 and NPHS2 mutations
in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration.
Human molecular genetics, 11(4), 379–88.
KRAWCZAK, M.; REISS, J. & COOPER, D.N. (n.d.). The mutational spectrum of single basepair substitutions in mRNA splice junctions of human genes: causes and consequences.
Human genetics, 90(1-2), 41–54.
KREIDBERG, J.A.; DONOVAN, M.J.; GOLDSTEIN, S.L.; RENNKE, H.; SHEPHERD, K.;
JONES, R.C. & JAENISCH, R. (1996). Alpha 3 beta 1 integrin has a crucial role in kidney
and lung organogenesis. Development, 122(11), 3537–47.
214
LAHDENKARI, A.T.; KESTILÄ, M.; HOLMBERG, C.; KOSKIMIES, O. & JALANKO, H.
(2004). Nephrin gene (NPHS1) in patients with minimal change nephrotic syndrome
(MCNS). Kidney international, 65(5), 1856–63.
LAHDENPERÄ, J.; KILPELÄINEN, P.; LIU, X.L.; PIKKARAINEN, T.; REPONEN, P.;
RUOTSALAINEN, V. & TRYGGVASON, K. (2003). Clustering-induced tyrosine
phosphorylation of nephrin by Src family kinases. Kidney international, 64(2), 404–13.
LEEUWIS, J. W.; NGUYEN, T.Q.; DENDOOVEN, A.; KOK, R. J. & GOLDSCHMEDING, R.
(2010). Targeting podocyte-associated diseases. Advanced drug delivery reviews, 62(14),
1325–36.
LEHTONEN, S.; ZHAO, F. & LEHTONEN, E. (2002). CD2-associated protein directly interacts
with the actin cytoskeleton. American journal of physiology. Renal physiology, 283(4),
F734–43.
LEMLEY, K.V. (2009). Kidney disease in nail-patella syndrome. Pediatric nephrology (Berlin,
Germany), 24(12), 2345–54.
LENKKERI, U.; MÄNNIKKÖ, M.; MCCREADY, P.; LAMERDIN, J.; GRIBOUVAL, O.;
NIAUDET, P.M.; ANTIGNAC, C.K.; KASHTAN, C.E.; HOMBERG, C.; OLSEN,
A.; KESTILÄ, M. & TRYGGVASON, K. (1999). Structure of the gene for congenital
nephrotic syndrome of the finnish type (NPHS1) and characterization of mutations.
American journal of human genetics, 64(1), 51–61.
LIU, H.X.; CHEW, S.L.; CARTEGNI, L.; ZHANG, M.Q. & KRAINER, A.R. (2000). Exonic
splicing enhancer motif recognized by human SC35 under splicing conditions. Molecular
and cellular biology, 20(3), 1063–71.
LIU, L.; DONÉ, S.C.; KHOSHNOODI, J.; BERTORELLO, A.; WARTIOVAARA, J.;
BERGGREN, P.O. & TRYGGVASON, K. (2001). Defective nephrin trafficking caused by
missense mutations in the NPHS1 gene: insight into the mechanisms of congenital nephrotic
syndrome. Human molecular genetics, 10(23), 2637–44.
LOIRAT, C.; ANDRÉ, J. L.; CHAMPIGNEULLE, J.; ACQUAVIVA, C.; CHANTEREAU, D.;
BOURQUARD, R.; DENAMUR, E. (2003). WT1 splice site mutation in a 46,XX female
with minimal-change nephrotic syndrome and Wilms’ tumour. Nephrology, dialysis,
transplantation : official publication of the European Dialysis and Transplant Association European Renal Association, 18(4), 823–5.
LÖWIK, M.; LEVTCHENKO, E.; WESTRA, D.; GROENEN, P.; STEENBERGEN, E.;
WEENING, J.; LILIEN, M.; MONNENS, L. & VAN DEN HEUVEL, L. (2008). Bigenic
heterozygosity and the development of steroid-resistant focal segmental glomerulosclerosis.
Nephrology, dialysis, transplantation : official publication of the European Dialysis and
Transplant Association - European Renal Association, 23(10), 3146–51.
215
LÖWIK, M.M.; GROENEN, P.J.T.A.; PRONK, I.; LILIEN, M.R.; GOLDSCHMEDING, R.;
DIJKMAN, H.B.; LEVTCHENKO, E.N.; MONNENS, L.A. & VAN DEN HEUVEL, L. P.
(2007). Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP
mutation. Kidney international, 72(10), 1198–203.
MACHUCA, E.; HUMMEL, A.; NEVO, F.; DANTAL, J.; MARTINEZ, F.; AL-SABBAN, E.;
BAUDOUIN, V.; ABEL, L.; GRÜNFELD, J.P. & ANTIGNAC, C. (2009). Clinical and
epidemiological assessment of steroid-resistant nephrotic syndrome associated with the
NPHS2 R229Q variant. Kidney international, 75(7), 727–35.
MATHE, E.; OLIVIER, M.; KATO, S.; ISHIOKA, C.; HAINAUT, P. & TAVTIGIAN, S.V.
(2006). Computational approaches for predicting the biological effect of p53 missense
mutations: a comparison of three sequence analysis based methods. Nucleic acids research,
34(5), 1317–25.
MCKENZIE, L.M.; HENDRICKSON, S.L.; BRIGGS, W.A.; DART, R.A.; KORBET, S.M.;
MOKRZYCKI, M.H.; KIMMEL, P.L.; AHUJA, T.S.; BERNS, J.S.; SIMON, E.E.; SMITH,
M.C.; TRACHTMAN, H.; MICHEL, D.M.; SCHELLING, J.R.; CHO, M.; ZHOU,
Y.C.; BINNS-ROEMER, E.; KIRK, G.D.; KOPP, J.B. & WINKLER, C.A. (2007). NPHS2
variation in sporadic focal segmental glomerulosclerosis. Journal of the American Society of
Nephrology : JASN, 18(11), 2987–95.
MOELLER, M.J.; SANDEN, S.K.; SOOFI, A.; WIGGINS, R.C. & HOLZMAN, L.B. (2002).
Two gene fragments that direct podocyte-specific expression in transgenic mice. Journal of
the American Society of Nephrology : JASN, 13(6), 1561–7.
MOELLER, M.J.; SANDEN, S.K.; SOOFI, A.; WIGGINS, R.C. & HOLZMAN, L.B. (2003).
Podocyte-specific expression of cre recombinase in transgenic mice. Genesis, 35(1), 39–42.
MOFFETT, P.; BRUENING, W.; NAKAGAMA, H.; BARDEESY, N.; HOUSMAN, D.;
HOUSMAN, D.E. & PELLETIER, J. (1995). Antagonism of WT1 activity by protein selfassociation. Proceedings of the National Academy of Sciences of the United States of
America, 92(24), 11105–9.
MOLLET, J.; GIURGEA, I.; SCHLEMMER, D.; DALLNER, G.; CHRETIEN, D.;
DELAHODDE, A.; BACQ, D.; DE LONLAY, P.; MUNNICH, A. & RÖTIG, A. (2007).
Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase
(COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. The
Journal of clinical investigation, 117(3), 765–72.
MORRISON, A.A.; VINEY, R.L. & LADOMERY, M.R. (2008). The post-transcriptional roles
of WT1, a multifunctional zinc-finger protein. Biochimica et biophysica acta, 1785(1), 55–
62.
216
MORRISON, A.A.; VINEY, R.L.; SALEEM, M.A. & LADOMERY, M.R. (2008). New insights
into the function of the Wilms tumor suppressor gene WT1 in podocytes. American journal
of physiology. Renal physiology, 295(1), F12–7.
MUCHA, B.; OZALTIN, F.; HINKES, B.G.; HASSELBACHER, K.; RUF, R.G.;
SCHULTHEISS,
M.; HANGAN,
D.; HOSKINS,
B.E.; EVERDING,
A.S.; BOGDANOVIC, R.; SEEMAN, T.; HOPPE, B.; HILDEBRANDT, F. & MEMBERS
OF THE APN STUDY GROUP. (2006). Mutations in the Wilms ’ Tumor 1 Gene Cause
Isolated Steroid Resistant Nephrotic Syndrome and Occur in Exons 8 and 9, Pediatr Res.
59(2), 325–331.
MÜLLER F. (1905). Morbus Brightii. Verh Dtsch Ges Path, 9, 64–99.
MUNDEL, P.; HEID, H.W.; MUNDEL, T.M.; KRÜGER, M.; REISER, J. & KRIZ, W. (1997).
Synaptopodin: an actin-associated protein in telencephalic dendrites and renal podocytes.
The Journal of cell biology, 139(1), 193–204.
NASH, M.A. (1992). The nephrotic syndrome. In editor In Edelmann CMJ, Pediatric Kidney
Disease (5th editio.). Little, Brown, and Company.
NESHICH, G.; TOGAWA, R.C.; MANCINI, A. L.; KUSER, P.R.; YAMAGISHI, M.E.B.;
PAPPAS, G.; TORRES, W.V.; FONSECA, E.; CAMPOS, T.; FERREIRA, L.L.; LUNA,
F.M.; OLIVEIRA, A.G.; MIURA, R.T.; INOUE, M.K.; HORITA, L.G.; DE SOUZA,
D.F.; DOMINIQUINI, F.; ALVARO, A.; LIMA, C.S.; OGAWA, F.O.; GOMES,
G.B.; PALANDRANI, J.F.; DOS SANTOS, G.F.; DE FREITAS, E.M.; MATTIUZ,
A.R.; COSTA, I.C.; DE ALMEIDA, C.L.; SOUZA, S.; BAUDET, C. & HIGA, R.H.
(2003). STING Millennium: A web-based suite of programs for comprehensive and
simultaneous analysis of protein structure and sequence. Nucleic acids research, 31(13),
3386–92.
NEW, L.A.; KEYVANI CHAHI, A. & JONES, N. (2013). Direct regulation of nephrin tyrosine
phosphorylation by Nck adaptor proteins. The Journal of biological chemistry, 288(3),
1500–10.
NG, P.C. & HENIKOFF, S. (2001). Predicting deleterious amino acid substitutions. Genome
research, 11(5), 863–74.
NG, P.C. & HENIKOFF, S. (2006). Predicting the effects of amino acid substitutions on protein
function. Annual review of genomics and human genetics, 7, 61–80.
NIAUDET, P. (2004). Steroid-resistant idiopathic nephrotic syndrome in children. In Avner ED,
Harmon WE, Pediatric nephrology.
217
NIAUDET, P.; GAGNADOUX, M.F. & BROYER, M. (1998). Treatment of childhood steroidresistant idiopathic nephrotic syndrome. Advances in nephrology from the Necker Hospital,
28, 43–61.
NISHIBORI, Y.; LIU, L.; HOSOYAMADA, M.; ENDOU, H.; KUDO, A.; TAKENAKA, H.;
HIGASHIHARA,
E.; BESSHO,
F.; TAKAHASHI,
S.; KERSHAW,
D.; RUOTSALAINEN, V.; TRYGGVASON, K.; KHOSHNOODI, J. & YAN, K. (2004).
Disease-causing missense mutations in NPHS2 gene alter normal nephrin trafficking to the
plasma membrane. Kidney international, 66(5), 1755–65.
NORIO, R. (1966). Heredity in the congenital nephrotic syndrome. A genetic study of 57 finnish
families with a review of reported cases. Annales paediatriae Fenniae, 12, Suppl 27:1–94.
OHASHI, T.; UCHIDA, K.; UCHIDA, S.; SASAKI, S. & NIHEI, H. (2003). Intracellular
mislocalization of mutant podocin and correction by chemical chaperones. Histochemistry
and cell biology, 119(3), 257–64.
PALMER, R.E.; KOTSIANTI, A.; CADMAN, B.; BOYD, T.; GERALD, W. & HABER, D.A.
(2001). WT1 regulates the expression of the major glomerular podocyte membrane protein
Podocalyxin. Current biology : CB, 11(22), 1805–9.
PAVENSTÄDT, H.; KRIZ, W. & KRETZLER, M. (2003). Cell biology of the glomerular
podocyte. Physiological reviews, 83(1), 253–307.
PELLETIER, J.; BRUENING, W.; LI, F.P.; HABER, D.A.; GLASER, T. & HOUSMAN, D.E.
(1991). WT1 mutations contribute to abnormal genital system development and hereditary
Wilms’ tumour. Nature, 353(6343), 431–4.
PEREIRA, A.C.; PEREIRA, A.B.; MOTA, G.F.; CUNHA, R.S.; HERKENHOFF, F.L.;
POLLAK, M.R.; MILL, J.G. & KRIEGER, J.E. (2004). NPHS2 R229Q functional variant is
associated with microalbuminuria in the general population. Kidney international, 65(3),
1026–30.
PHILIPPE, A.; NEVO, F.; ESQUIVEL, E.L.; REKLAITYTE, D.; GRIBOUVAL, O.; TÊTE,
M.J.;
LOIRAT,
C.; DANTAL,
J.; FISCHBACH,
M.; POUTEIL-NOBLE,
C.; DECRAMER, S.; HOEHNE, M.; BENZING, T.; CHARBIT, M.; NIAUDET, P. &
ANTIGNAC, C. (2008). Nephrin mutations can cause childhood-onset steroid-resistant
nephrotic syndrome. Journal of the American Society of Nephrology : JASN, 19(10), 1871–
8.
PIVA, F.; GIULIETTI, M.; NOCCHI, L. & PRINCIPATO, G. (2009). SpliceAid: a database of
experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics,
25(9), 1211–3.
218
PRITCHARD-JONES, K.; FLEMING, S.; DAVIDSON, D.; BICKMORE, W.; PORTEOUS, D.;
GOSDEN, C.; BARD, J.; BUCKLER, A.; PELLETIER, J. & HOUSMAN, D. (1990). The
candidate Wilms’ tumour gene is involved in genitourinary development. Nature,
346(6280), 194–7.
PUTAALA, H.; SOININEN, R.; KILPELÄINEN, P.; WARTIOVAARA, J. & TRYGGVASON,
K. (2001). The murine nephrin gene is specifically expressed in kidney, brain and pancreas:
inactivation of the gene leads to massive proteinuria and neonatal death. Human molecular
genetics, 10(1), 1–8.
QIN, X.S.; TSUKAGUCHI, H.; SHONO, A.; YAMAMOTO, A.; KURIHARA, H. & DOI, T.
(2009). Phosphorylation of nephrin triggers its internalization by raft-mediated endocytosis.
Journal of the American Society of Nephrology : JASN, 20(12), 2534–45.
QUACK, I.; RUMP, L. C.; GERKE, P.; WALTHER, I.; VINKE, T.; VONEND, O.;
GRUNWALD, T. & SELLIN, L. (2006). beta-Arrestin2 mediates nephrin endocytosis and
impairs slit diaphragm integrity. Proceedings of the National Academy of Sciences of the
United States of America, 103(38), 14110–5.
QUAGGIN, S.E. (2002). A “molecular toolbox” for the nephrologist. Journal of the American
Society of Nephrology : JASN, 13(6), 1682–5.
RAMENSKY, V.; BORK, P. & SUNYAEV, S. (2002). Human non-synonymous SNPs: server
and survey. Nucleic acids research, 30(17), 3894–900.
REDDY, J.C.; MORRIS, J.C.; WANG, J.; ENGLISH, M.A.; HABER, D.A.; SHI, Y. & LICHT,
J.D. (1995). WT1-mediated transcriptional activation is inhibited by dominant negative
mutant proteins. The Journal of biological chemistry, 270(18), 10878–84.
REISER, J.; POLU, K.R.; MÖLLER, C.C.; KENLAN, P.; ALTINTAS, M.M.; WEI, C.; FAUL,
C.; HERBERT, S.; VILLEGAS, I.; AVILA-CASADO, C.; MCGEE, M.; SUGIMOTO,
H.; BROWN, D.; KALLURI, R.; MUNDEL, P.; SMITH, P.L.; CLAPHAM, D.E. &
POLLAK, M.R. (2005). TRPC6 is a glomerular slit diaphragm-associated channel required
for normal renal function. Nature genetics, 37(7), 739–44.
RELLE, M.; CASH, H.; BROCHHAUSEN, C.; STRAND, D.; MENKE, J.; GALLE, P.R. &
SCHWARTING, A. (2011). New perspectives on the renal slit diaphragm protein podocin.
Modern pathology : an official journal of the United States and Canadian Academy of
Pathology, Inc, 24(8), 1101–10.
RISTOLA, M.; ARPIAINEN, S.; SALEEM, M.A.; MATHIESON, P.W.; WELSH, G.I.;
LEHTONEN, S. & HOLTHÖFER, H. (2009). Regulation of Neph3 gene in podocytes-key
roles of transcription factors NF-kappaB and Sp1. BMC molecular biology, 10, 83.
219
RODEWALD, R. & KARNOVSKY, M.J. (1974). Porous substructure of the glomerular slit
diaphragm in the rat and mouse. The Journal of cell biology, 60(2), 423–33.
ROELANS C. (1484). Liber de aegritudinibus infantium. Reproduced in: Sudhoff KFJ. Erstlinge
der padiatrischen Literatur. Munchen: Munchner Drucke; 1925:193–195.
ROSELLI, S.; MOUTKINE, I.; GRIBOUVAL, O.; BENMERAH, A. & ANTIGNAC, C. (2004).
Plasma membrane targeting of podocin through the classical exocytic pathway: effect of
NPHS2 mutations. Traffic, 5(1), 37–44.
RUF, R.G.; LICHTENBERGER, A.; KARLE, S.M.; HAAS, J.P.; ANACLETO, F.E.;
SCHULTHEISS, M.; ZALEWSKI, I.; IMM, A.; RUF, E.M.; MUCHA, B.; BAGGA,
A.; NEUHAUS, T.; FUCHSHUBER, A.; BAKKALOGLU, A.; HILDEBRANDT, F.
& ARBEITSGEMEINSCHAFT FÜR PÄDIATRISCHE NEPHROLOGIE STUDY
GROUP. (2004). Patients with mutations in NPHS2 (podocin) do not respond to standard
steroid treatment of nephrotic syndrome. Journal of the American Society of Nephrology :
JASN, 15(3), 722–32.
RUF, R.G.; SCHULTHEISS, M.; LICHTENBERGER, A.; KARLE, S.M.; ZALEWSKI, I.;
MUCHA, B.; EVERDING, A.S.; NEUHAUS, T. PATZER, L.; PLANK, C.; HAAS,
J.P.; OZALTIN,
F.; IMM,
A.; FUCHSHUBER,
A.;
BAKKALOGLU,
A,; HILDEBRANDT, F. & APN STUDY GROUP. (2004). Prevalence of WT1 mutations
in a large cohort of patients with steroid-resistant and steroid-sensitive nephrotic syndrome.
Kidney international, 66(2), 564–70.
RUOTSALAINEN, V.; LJUNGBERG, P.; WARTIOVAARA, J.; LENKKERI, U.; KESTILÄ,
M.; JALANKO, H.; HOLBERNG, C. & TRYGGVASON, K. (1999). Nephrin is
specifically located at the slit diaphragm of glomerular podocytes. Proceedings of the
National Academy of Sciences of the United States of America, 96(14), 7962–7.
SAKAMOTO, Y.; YOSHIDA, M.; SEMBA, K. & HUNTER, T. (1997). Inhibition of the DNAbinding and transcriptional repression activity of the Wilms’ tumor gene product, WT1, by
cAMP-dependent protein kinase-mediated phosphorylation of Ser-365 and Ser-393 in the
zinc finger domain. Oncogene, 15(17), 2001–12.
SALVIATI, L.; SACCONI, S.; MURER, L.; ZACCHELLO, G.; FRANCESCHINI, L.;
LAVERDA, A.M.; BASSO, G.; QUINZII, C.; ANGELINI, C.; HIRANO, M.; NAINI,
A.B.; NAVAS, P.; DIMAURO, S. & MONTINI, G. (2005). Infantile encephalomyopathy
and nephropathy with CoQ10 deficiency: a CoQ10-responsive condition. Neurology, 65(4),
606–8.
SAMBROOK, J.; FRITSH, E.F. & MANIATIS, T. (1989). Molecular cloning: A laboratory
manual In: Cold Spring Harbor Laboratory Press., 2nd edition: ed. Cold Spring Harbor, NY.
220
RODRIGUEZ, S.; GAUNT, T.R. & DAY, I.N. (2009). Hardy-Weinberg Equilibrium Testing of
Biological Ascertainment for Mendelian Randomization Studies. American journal of
epidemiology.
SANTÍN, S.; GARCÍA-MASET, R.; RUÍZ, P.; GIMÉNEZ, I.; ZAMORA, I.; PEÑA, A.;
MADRID, A.; CAMACHO, J.A.; FRAGA, G.; SÁNCHEZ-MORENO, A.; COBO,
M.A.; BERNIS, C.; ORTIZ, A.; DE PABLOS, A.L.; PINTOS, G.; JUSTA,
M.L.; HIDALGO-BARQUERO, E. FERNÁNDEZ-LLAMA, P.; BALLARÍN, J.; ARS,
E.; TORRA, R. & FSGS SPANISH STUDY GROUP. (2009). Nephrin mutations cause
childhood- and adult-onset focal segmental glomerulosclerosis. Kidney international,
76(12), 1268–76.
SANTÍN, S.; TAZÓN-VEGA, B.; SILVA, I.; COBO, M.Á.; GIMÉNEZ, I.; RUÍZ, P.;
GARCÍA-MASET, R.; BALLARÍN, J.; TORRA, R.; ARS, E. & FSGS SPANISH STUDY
GROUP. (2011). Clinical value of NPHS2 analysis in early- and adult-onset steroidresistant nephrotic syndrome. Clinical journal of the American Society of Nephrology :
CJASN, 6(2), 344–54.
SAVIN, V.J.; SHARMA, R.; SHARMA, M.; MCCARTHY, E.T.; SWAN, S.K.; ELLIS, E.;
LOVELL, H.; WARADY, B.; GUNWAR, S.; CHONKO, A.M.; ARTERO, M. &
VINCENTI, F. (1996). Circulating factor associated with increased glomerular permeability
to albumin in recurrent focal segmental glomerulosclerosis. The New England journal of
medicine, 334(14), 878–83.
SCHOEB, D.S.; CHERNIN, G.; HEERINGA, S. F.; MATEJAS, V.; HELD, S.; VEGAWARNER, V.; BOCKENHAUER, D.; VLANGOS, C.N.; MOORANI, K.N.; NEUHAUS,
T.J.; KARI,
J.A.; MACDONALD,
J.; SAISAWAT,
P.; ASHRAF,
S.; OVUNC,
B.; ZENKER, M.; HILDEBRANDT, F. & GESSELSCHAFT FÜR PAEDIATRISCHE
NEPHROLOGIE (GPN) STUDY GROUP. (2010). Nineteen novel NPHS1 mutations in a
worldwide cohort of patients with congenital nephrotic syndrome (CNS). Nephrology,
dialysis, transplantation : official publication of the European Dialysis and Transplant
Association - European Renal Association, 25(9), 2970–6.
SCHULMAN, S.L.; KAISER, B.A.; POLINSKY, M.S.; SRINIVASAN, R. & BALUARTE, H.J.
(1988). Predicting the response to cytotoxic therapy for childhood nephrotic syndrome:
superiority of response to corticosteroid therapy over histopathologic patterns. The Journal
of pediatrics, 113(6), 996–1001.
SCHULTHEISS, M.; RUF, R.G.; MUCHA, B.E.; WIGGINS, R.; FUCHSHUBER, A.;
LICHTENBERGER, A. & HILDEBRANDT, F. (2004). No evidence for
genotype/phenotype correlation in NPHS1 and NPHS2 mutations. Pediatric nephrology
(Berlin, Germany), 19(12), 1340–8.
SCHUMACHER, V.; SCHÄRER, K.; WÜHL, E.; ALTROGGE, H.; BONZEL, K.E.;
GUSCHMANN, M.; NEUHAUS, T.J.; POLLASTRO, R.M.; KUWERTZ-BRÖKING,
221
E.; BULLA, M.; TONDERA, A.M.; MUNDEL, P.; HELMCHEN, U.; WALDHERR,
R.; WEIRICH, A. & ROYER-POKORA, B. (1998). Spectrum of early onset nephrotic
syndrome associated with WT1 missense mutations. Kidney international, 53(6), 1594–600.
SCHWARZ, K.; SIMONS, M.; REISER, J.; SALEEM, M.A.; FAUL, C.; KRIZ, W.; SHAW,
A.S.; HOLZMAN, L.B. & MUNDEL, P. (2001). Podocin, a raft-associated component of
the glomerular slit diaphragm, interacts with CD2AP and nephrin. The Journal of clinical
investigation, 108(11), 1621–9.
SELLIN, L.; HUBER, T.B.; GERKE, P.; QUACK, I.; PAVENSTÄDT, H. & WALZ, G. (2003).
NEPH1 defines a novel family of podocin interacting proteins. FASEB journal : official
publication of the Federation of American Societies for Experimental Biology, 17(1), 115–
7.
SHARPLES, P.M.; POULTON, J. & WHITE, R.H. (1985). Steroid responsive nephrotic
syndrome is more common in Asians. Archives of disease in childhood, 60(11), 1014–7.
SHIH, N.Y.; LI, J.; COTRAN, R.; MUNDEL, P.; MINER, J.H. & SHAW, A.S. (2001). CD2AP
localizes to the slit diaphragm and binds to nephrin via a novel C-terminal domain. The
American journal of pathology, 159(6), 2303–8.
SIMONS, K. & IKONEN, E. (1997). Functional rafts in cell membranes. Nature, 387(6633),
569–72.
SIMONS, M.; SCHWARZ, K.; KRIZ, W.; MIETTINEN, A.; REISER, J.; MUNDEL, P. &
HOLTHÖFER, H. (2001). Involvement of lipid rafts in nephrin phosphorylation and
organization of the glomerular slit diaphragm. The American journal of pathology, 159(3),
1069–77.
SMART, E.J.; GRAF, G.A.; NIVEN, M.A.; SESSA, W.C.; ENGELMAN, J.A.; SCHERER, P.
E.; OKAMOTO T. & LISANTI, M. P. (1999). Caveolins , Liquid-Ordered Domains , and
Signal Transduction, 19(11), 7289–7304.
SMOYER, W.E. & MUNDEL, P. (1998). Regulation of podocyte structure during the
development of nephrotic syndrome. Journal of molecular medicine, 76(3-4), 172–83.
SRIVASTAVA, T.; SIMON, S.D. & ALON, U.S. (1999). High incidence of focal segmental
glomerulosclerosis in nephrotic syndrome of childhood. Pediatric nephrology, 13(1), 13–8.
STOLL, R.; LEE, B.M.; DEBLER, E.W.; LAITY, J.H.; WILSON, I.A.; DYSON, H.J. &
WRIGHT, P.E. (2007). Structure of the Wilms tumor suppressor protein zinc finger domain
bound to DNA. Journal of molecular biology, 372(5), 1227–45.
TAVTIGIAN, S.V.; DEFFENBAUGH, A.M.; YIN, L.; JUDKINS, T.; SCHOLL, T.;
SAMOLLOW, P.B.; DE SILVA, D.; ZHARKIKH, A. & THOMAS, A. (2006).
222
Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of
eight recurrent substitutions as neutral. Journal of medical genetics, 43(4), 295–305.
THOMPSON, J.D.; HIGGINS, D.G. & GIBSON, T.J. (1994). CLUSTAL W: improving the
sensitivity of progressive multiple sequence alignment through sequence weighting,
position-specific gap penalties and weight matrix choice. Nucleic acids research, 22(22),
4673–80.
TORY, K.; MENYHÁRD, D.K.; NEVO, F.; O GRIBOUVAL, A.; KERTI, P.; STRÁNER, C.;
ARRONDEL, T. TULASSAY, G.; MOLLET, A. & PERCZEL, C.A. (2013). Mutationdependent recessive inheritance in NPHS2-associated steroid-resistant nephrotic syndrome.
Beyond Mendel’s laws. European Journal of Human Genetics, 21, Supple(C19.4), 50.
TRYGGVASON, K.; PATRAKKA, J. & WARTIOVAARA, J. (2006). Hereditary proteinuria
syndromes and mechanisms of proteinuria. The New England journal of medicine, 354(13),
1387–401.
TSUDA, M.; OWADA, M.; TSUCHIYA, M.; MURAKAMI, M. & SAKIYAMA, T. (1999).
WT1 nephropathy in a girl with normal karyotype (46,XX). Clinical nephrology, 51(1), 62–
3.
TSUKAGUCHI, H.; SUDHAKAR, A.; LE, T.C.; NGUYEN, T.; YAO, J.; SCHWIMMER, J.A.;
SCHACHTER,
A.D.; POCH,
E.; ABREU,
P.F.; APPEL,
G.B.; PEREIRA,
A.B.; KALLURI, R. & POLLAK, M.R. (2002). NPHS2 mutations in late-onset focal
segmental glomerulosclerosis: R229Q is a common disease-associated allele. The Journal
of clinical investigation, 110(11), 1659–66.
VERMA, R.; WHARRAM, B.; KOVARI, I.; KUNKEL, R.; NIHALANI, D.; WARY,
K.K.; WIGGINS, R.C.; KILLEN, P. & HOLZMAN, L.B. (2003). Fyn binds to and
phosphorylates the kidney slit diaphragm component Nephrin. The Journal of biological
chemistry, 278(23), 20716–23.
VINEY, R. L.; MORRISON, A.A.; VAN DEN HEUVEL, L.P.; NI, L.; MATHIESON, P.W.;
SALEEM, M.A. & LADOMERY, M.R. (2007). A proteomic investigation of glomerular
podocytes from a Denys-Drash syndrome patient with a mutation in the Wilms tumour
suppressor gene WT1. Proteomics, 7(5), 804–15.
VÖLKER, L.A.; SCHUREK, E.M.; RINSCHEN, M.M.; TAX, J.; SCHUTTE, B.A.;
LAMKEMEYER, T.; UNGRUE, D.; SCHERMER, B.; BENZING, T. & HÖHNE, M.
(2013). Characterization of a short isoform of the kidney protein podocin in human kidney.
BMC nephrology, 14, 102.
WAGNER, N.; WAGNER, K.D.; SCHOLZ, H.; KIRSCHNER, K.M., & SCHEDL, A. (2006).
Intermediate filament protein nestin is expressed in developing kidney and heart and might
223
be regulated by the Wilms’ tumor suppressor Wt1. American journal of physiology.
Regulatory, integrative and comparative physiology, 291(3), R779–87.
WANG, Z.Y.; QIU, Q.Q. & DEUEL, T.F. (1993). The Wilms’ tumor gene product WT1
activates or suppresses transcription through separate functional domains. The Journal of
biological chemistry, 268(13), 9172–5.
WEBER, S.; GRIBOUVAL, O.; ESQUIVEL, E.L.; MORINIÈRE, V.; TÊTE, M.J.;
LEGENDRE, C.; NIAUDET, P. & ANTIGNAC, C. (2004). NPHS2 mutation analysis
shows genetic heterogeneity of steroid-resistant nephrotic syndrome and low post-transplant
recurrence. Kidney international, 66(2), 571–9.
WEI, C.; MÖLLER, C.C.; ALTINTAS, M.M.; LI, J.; SCHWARZ, K.; ZACCHIGNA, S.; XIE,
L.; HENGER, A.; SCHMID, H.; RASTALDI, M.P.; COWAN, P.; KRETZLER, M,;
PARRILA, R.; BENDAYAN, M.; GUPTA, V.; NIKOLIC, B.; KALLURI, R.;
CARMELIET, P.; MUNDEL, P & REISER, J. (2008). Modification of kidney barrier
function by the urokinase receptor. Nature medicine, 14 (1): 55-63.
WITTOP KONING, T.H. & SCHÜMPERLI, D. (1994). RNAs and ribonucleoproteins in
recognition and catalysis. European journal of biochemistry / FEBS, 219(1-2), 25–42.
YAN, K.; KHOSHNOODI, J.; RUOTSALAINEN, V. & TRYGGVASON, K. (2002). N-linked
glycosylation is critical for the plasma membrane localization of nephrin. Journal of the
American Society of Nephrology : JASN, 13(5), 1385–9.
YASUNAMI, M.; SUZUKI, K.; HOUTANI, T.; SUGIMOTO, T. & OHKUBO, H. (1995).
Molecular characterization of cDNA encoding a novel protein related to transcriptional
enhancer factor-1 from neural precursor cells. The Journal of biological chemistry, 270(31),
18649–54.
ZENKER, M.; AIGNER, T.; WENDLER, O.; TRALAU, T.; MÜNTEFERING, H.; FENSKI, R.;
PITZ,
S.; SCHUMACHER,
V.; ROYER-POKORA,
B.; WÜHL,
E.; COCHAT,
P.; BOUVIER, R.; KRAUS, C.; MARK, K.; MADLON, H.; DÖTSCH, J.; RASCHER,
W.; MARUNIAK-CHUDEK, I.; LENNERT, T.; NEUMANN, L.M. & REIS, A. (2004).
Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and
distinct eye abnormalities. Human molecular genetics, 13(21), 2625–32.
ZHAO, C. & MENG, A. (2005). Sp1-like transcription factors are regulators of embryonic
development in vertebrates. Development, growth & differentiation, 47(4), 201–11.
ZHOU, W. & HILDEBRANDT, F. (2012). Inducible podocyte injury and proteinuria in
transgenic zebrafish. Journal of the American Society of Nephrology : JASN, 23(6), 1039–
47.
224
ZHU, L.; YU, L.; WANG, C.D.; LV, J.C.; LI, G.S.; ZHANG, H. & WANG, H.Y. (2009).
Genetic effect of the NPHS2 gene variants on proteinuria in minimal change disease and
immunoglobulin A nephropathy. Nephrology, 14(8), 728–34.
ZWINGER T. (1722). Anasarca puerorum. In T. J. Basel E & Eds, Paedioatreia practica
curationem puerorumque morborum puerilium etc. (p. Cap 119). In: Dock W, ed.
Proteinuria: the story of 250 years of trials, errors and rectifications. Bull N Y Acad Med.
1974;5:659-666.
225
226
ANEXOS
227
ANEXO 1
228
ANEXO 2
Tabela 1: Material fixado em parafina
Material
Concentração
(ng/µl)
M1 A101/93
19,31
M2 38136
0, 2817
M3 55909
35
M4 75369
7, 263
M5 111141/95
14,49
M6 B04679/06
11,98
M7 A197/91
864,4
229