Universidade do Algarve
Faculdade de Ciências e Tecnologia
TRABALHOS LABORATORIAIS
Química Geral
Engª do Ambiente
Química Inorgânica
Lic. Oceanografia
(1o Ano)
1º Semestre: 2005/2006
Índice
I. Segurança no laboratório de química ......................................................................... 2
A. Regras de segurança essenciais .............................................................................2
II. Símbolos de perigosidade .............................................................................................3
III. Material de laboratório...................................................................................................5
IV. Medições de volumes e massas. .................................................................................6
A. Medição de volumes de líquidos ...............................................................................6
B. Medição de massas .................................................................................................9
V. Tratamento de resultados ...................................................................................... … 11
A. Tipos de erros ......................................................................................................... 11
B. Precisão e exactidão.............................................................................................. 12
C. Algarismos significativos ........................................................................................ 13
1. Regras de Arredondamento........................................................................... 14
2. Manuseamento dos dados experimentais ..................................................... 17
D. Intervalos de confiança ........................................................................................... 18
E. Determinação da” melhor recta “ que passa pelos pontos experimentais ............. 21
VI. Programação e calendarização .............................................................................. …26
VII. Folha de apontamentos laboratoriais .................................................................... …27
VIII. Relatórios ............................................................................................................. …29
IX. Trabalhos práticos .................................................................................................. …31
1. Preparação e diluição de soluções .................................................................... 31
2.Gravimetria: Determinação do resíduo de uma água ......................................... 35
3. Fotometria de chama de emissão: Determinação do sódio nas águas.............. 37
4. Volumetria de Precipitação: Determinação do teor de cloretos numa água ...... 40
5. Lei dos gases: Lei de Charles e Gay-Lussac ..................................................... 42
6. Termodinâmica: Entalpia de Vaporização ......................................................... 44
7. Volumetria Áido-base: Titulação de um ácido fraco com uma base forte .......... 46
8. Volumetria Áido-base: Titulação potenciométrica .............................................. 47
9. Volumetria de Complexação: Determinação da dureza de uma água ............... 50
10. Volumetria de Redução-Oxidação: Titulação do Ferro (II) com permanganato de
potássio ............................................................................................................... 53
1
I. SEGURANÇA NO LABORATÓRIO DE QUÍMICA
A - Regras de segurança essenciais
SEMPRE
1. Familiarize-se com os procedimentos de segurança.
2. Use óculos de protecção sempre que necessário.
3. Use roupa adequada e bata.
4. Lave as mãos quando terminar a aula.
5. Prepare a experiência antes de ir para a aula, sabendo a perigosidade dos compostos
que irão ser utilizados, bem como os passos a executar durante o trabalho experimental;
6. Manuseie os químicos com cuidado.
7. Mantenha a sua área de trabalho sempre limpa.
8. Sempre que tiver alguma dúvida pergunte ao professor.
NUNCA
1. Coma ou beba no laboratório.
2. Fume no laboratório.
3. Cheire ou prove nenhum produto químico.
4. Brinque ou distraia o seu colega.
5. Corra no laboratório.
6. Trabalhe sozinho.
7. Faça trabalhos experimentais não autorizados.
Acima de tudo use BOM SENSO na forma como se comporta.
2
II. Símbolos de Perigosidade
Perigo: Por contacto, destroem o tecido vivo bem como
utensílios.
Exemplos: Bromo, ácido sulfúrico.
Cuidado: Não inalar os vapores e evitar o contacto com a pele,
olhos e vestuário.
Perigo: São substâncias que podem explodir sob determinadas
condições.
Exemplos: Permanganato de potássio, peróxido de sódio.
Cuidado:Evitar
qualquer
contacto
com
substâncias
combustíveis.
Perigo: Podem desenvolver uma acção irritante sobre a pele,
olhos e vias respiratórias.
Exemplos: Solução de amoníaco, cloreto de benzilo.
Cuidado: Não inalar os vapores e evitar o contacto com a boca
e olhos.
Perigo: A inalação, ingestão ou absorção através da pele
provoca, na maior parte das vezes lesões muito graves ou
mesmo a morte.
Exemplos: Trióxido de arsénio, cloreto de mercúrio (II).
Cuidado: Evitar qualquer contacto com o corpo e no caso de
indisposição chamar o médico.
Perigo: Quando absorvidas pelo corpo, por inalação, ingestão
ou contacto, estas substâncias provocam lesões pouco graves.
Exemplos: Piridina, tricloroetileno.
Cuidado: Evitar qualquer contacto com o corpo humano,
inclusive inalação de vapores, no caso de indisposição chamar
o médico.
3
Perigo: Fácilmente inflamáveis, sensíveis à
humidade ou água.
Exemplos: Propano, acetona, hidreto de boro
e sódio.
Cuidado: Manter afastado de fontes de calor.
4
III. Material de
Laboratório
5
IV. Medições de volumes e massas
A. Medição de volumes de líquidos
A. Material para medição de volumes:
(1) Material de Vidro
A medição de volumes é uma das acções mais frequentes num laboratório. Entre o material
volumétrico existente, distinguem-se:
Pipetas, existem dois tipos fundamentais: volumétricas e graduadas.
Volumétricas - têm uma só marca indicadora do nível a que o líquido se deve ajustar
de modo a que o valor vazado seja o valor fixo indicado na pipeta (mais rigorosas).
Graduadas – têm uma escala que permite o vazamento de quantidades variáveis de
líquido (menos rigorosas).
Balões volumétricos, o volume final deve ser ajustado, com o solvente, até ao traço.
Buretas, tubo cilíndrico graduado com uma “válvula” e com o qual é possível controlar o fluxo
e a quantidade de líquido vazado. O volume é lido na escala da bureta.
Provetas, graduadas de modo a permitir a medição de volumes variáveis e lidos até ao valor
máximo da sua escala.
6
Rigor das medições:
Pipetas volumétricas – Pipetas graduadas – Balões volumétricos – Buretas – Provetas
+ rigor
− rigor
As leituras de volume devem ser efectuadas
tendo em conta a posição do menisco,
considerando que o volume é o
correspondente à sua base, tal como
indicado na figura:
(a) Procedimento para utilização de pipetas volumétricas:
Ajustar uma "pompete" à ponta superior da pipeta, segurando sempre a pipeta pela ponta
superior (e nunca pelo meio!).
Mantendo a pipeta na posição vertical, mergulhá-la no líquido e enchê-la, por aspiração,
utilizando a "pompete", até ligeiramente acima do traço superior.
Remover quaisquer gotas de água aderentes ao exterior da pipeta, limpando-a num
movimento descendente com papel absorvente.
Deixar escorrer a água lentamente e ajustar convenientemente o menisco. Eliminar
qualquer gota em excesso que se encontre na extremidade da pipeta, encostando-a à parede
molhada dum recipiente.
Assegurar-se que não existem gotas de água aderentes ao exterior da pipeta ou às
paredes internas acima do menisco e que não há bolhas de ar nem espuma no líquido.
Deixar escoar livremente o líquido contido na pipeta para o recipiente, mantendo a pipeta
na vertical, com a extremidade encostada à parede interna do recipiente, sem a deixar
escorregar.
7
Quando terminar o escoamento visível (o menisco deve permanecer imóvel ligeiramente
acima da extremidade), manter a pipeta na mesma posição durante 3 segundos (ou, se a
pipeta tiver tempo de espera, mantê-la durante o tempo indicado).
(2) Micropipetas automáticas
A necessidade de medição de pequenos volumes de líquidos, na gama do mililitro ou
microlitros, levou ao desenvolvimento de uma gama de pipetas automáticas de pontas
descartáveis. A fiabilidade destes sistemas depende em grande parte da qualidade do
instrumento, mas também de outros factores como a qualidade das pontas, o ambiente e o
operador.
Pontas descartáveis - a forma, propriedades do material e o ajuste da ponta à pipeta
influenciam o rigor da medição. É importante verificar que a ponta encaixa bem na pipeta,
testar a forma como se molha, e verificar se ficam gotas remanescentes depois de escoar o
líquido.
Condições ambientais - As fontes de erro do meio ambiente incluem a temperatura
(diferença de temperatura entre a pipeta, o fluido e a temperatura ambiente), a pressão
atmosférica e a humidade do ar. A maior contribuição para os erros ambientais é a
temperatura. É importante garantir que todos os componentes estão à mesma temperatura,
dentro de ±1ºC.
(a) Procedimento para utilização de micropipetas:
Ajustar a ponta na pipeta e ajustar o volume a medir.
Pressionar com o polegar o manípulo até à primeira paragem;
Segurando a pipeta verticalmente, introduzir a ponta cerca de 2-3 mm na amostra;
Soltar gradualmente o manípulo e observar o processo de enchimento (deve evitar-se a
turbulência no interior da ponta, para minimizar o risco de formação de aerossóis). Quando o
manípulo estiver na posição inicial, remover o polegar completamente (a ausência de pressão
melhora a precisão). Lentamente, retirar a ponta da pipeta da amostra, e limpar quaisquer
gotas de água que tenham ficado aderentes ao exterior.
8
Para escoar o volume medido, encostar a ponta da pipeta na parede do recipiente, num
ângulo de 10-45º. Colocar o polegar sobre o manípulo e pressionar de forma uniforme até à
primeira paragem. Esperar 1 segundo. Pressionar rapidamente até à segunda paragem.
Cuidados a ter ao usar micropipetas:
a pipeta e respectiva ponta devem ser escolhidas de forma a minimizar o espaço de ar
entre o pistão e o líquido;
a ponta deve ser mergulhada apenas à superfície da solução (2-3mm de profundidade);
deve molhar-se previamente a ponta com a solução a medir, para melhorar a precisão
e exactidão;
deve segurar-se a pipeta na vertical;
a aspiração deve ser feita de forma suave, e não bruscamente.
B. Medição de massas
Uma das operações mais frequentes num laboratório é a pesagem, operação pela qual
se determina a massa de uma substância. O grau de exactidão e precisão que é necessário
satisfazer numa pesagem dependem da sua finalidade.
Uma balança analítica, muito rigorosa, ±0,0001 g, tem uma capacidade que pode variar de 50
a 200 g.
Uma balança técnica é menos rigorosa, ±0,01 g, mas tem uma capacidade elevada que
pode ser de ~1000 g.
(a) Cuidados a ter durante as pesagens:
A balança deve ser mantida sempre limpa, ou seja, não se devem colocar reagentes
directamente no prato mas sim sobre uma cápsula de pesagem (ex: vidro de relógio). As
substâncias voláteis ou corrosivas devem ser pesadas em recipientes fechados.
9
A temperatura do objecto a pesar deve ser razoavelmente próxima da temperatura da
balança.
As janelas da balança devem estar fechadas durante a pesagem.
Cada passo na pesagem - taragem, colocação do objecto no prato, leitura - deve ser
feito lentamente, dando tempo suficiente à balança para atingir o equilíbrio.
O objecto a pesar deve ser cuidadosamente colocado no centro do prato da balança,
para evitar erros de excentricidade.
Terminada a pesagem, a balança deve ser limpa, se necessário, as janelas fechadas
e desligada se não for utilizada de imediato.
10
V. Tratamento de resultados
A. Tipos de erros
Todas as medições experimentais estão sujeitas a erros. O resultado de
uma análise pode ser quantitativo ou qualitativo. Quando o resultado é
quantitativo, é extremamente importante fazer uma estimativa dos erros
envolvidos na medição. Um resultado é inútil se não for acompanhado de uma
estimativa dos erros envolvidos na sua medição.
Podemos classificar os erros em três tipos:
Grosseiros (irremediáveis);
Aleatórios;
Sistemáticos
Erros grosseiros:
Não entram no padrão normal dos erros associados a uma análise.
Não devem ocorrer, e, se ocorrem e são detectados, normalmente é necessário
repetir toda a análise.
Ex:
avaria
de
um
instrumento;
distracção
do
operador;
contaminação macroscópica de um reagente, etc.
Erros Aleatórios (ou Indeterminados):
As suas fontes podem ser incerteza instrumental, do método ou do
Não são elimináveis, mas podem minimizar-se com trabalho
operador;
cuidadoso;
Reconhecem-se como uma dispersão dos valores em torno de uma
Afectam a precisão;
Podem quantificar-se pela medição da precisão (p. ex., através do
média;
desvio-padrão).
11
Erros Sistemáticos (ou Determinados):
As suas fontes podem ser erros instrumentais, do método ou do
Em princípio, são reconhecíveis e podem reduzir-se parcial ou
operador;
completamente;
Reconhecem-se pelo afastamento entre o valor verdadeiro e o valor
Afectam a exactidão;
Podem quantificar-se pela medição da diferença entre o valor
médio;
verdadeiro e valor médio.
B. Precisão e exactidão
Exactidão:
Concordância entre o valor obtido e o valor aceite como verdadeiro
Precisão:
Concordância entre os valores obtidos no mesmo ensaio repetido
várias vezes.
12
repetibilidade
precisão intermédia
Precisão
reprodutibilidade
(1) Repetibilidade
(a) Reprodutibilidade
Precisão obtida nas mesmas
condições:
mesmo laboratório
mesmo operador
mesmo equipamento
curto intervalo de tempo
Precisão obtida fazendo variar as
condições:
diferentes laboratórios
diferentes operadores
diferentes equipamentos
espaçamento no tempo
C. Algarismos significativos
O conceito de algarismos significativos permite introduzir de um modo
simples a precisão de uma medida sem explicitar a sua incerteza. Este
conceito permite também estimar a precisão de um valor que é calculado por
combinação de diferentes tipos de medida, pois a incerteza de um valor é
propagado
em
todas
as
contas
que
com
ele
forem
feitas.
13
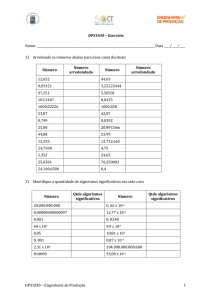
Contagem do número de algarismos significativos:
A contagem dos algarismos significativos é feita a partir do primeiro algarismo
diferente de zero, da esquerda para a direita.
Em alguns casos, se o primeiro algarismo da esquerda for maior ou igual a 5,
conta como dois algarismos significativos.
As potências de base 10 não contam como algarismos significativos.
Valor
Número de algarismos
significativos
Obs:
5,630
4
0,270
3
0,0004
1
1,0007
5
8,1 × 107
2 ou 3 ( se o primeiro
Zero à direita da vírgula
com significado
Zero
à
direita
com
significado mas o zero à
esquerda da vírgula sem
significado
Todos os zeros à esquerda
da vírgula sem significado
Todos os algarismos com
significado
Valor em notação científica.
Apenas se consideram os
algarismos
antes
do
expoente
algarismo da esquerda
for maior ou igual a 5,
conta
como
dois
algarismos significativos)
2 × 10-7
3,60 ×102
3600
1
3
2 ou 3 ou 4
2,36
2
Os zeros podem estar
apenas a indicar a posição
da vírgula (ex. 36,0 × 102
O número em índice indica
um valor estimado (ex. 2,36
cm medidos com uma régua
graduada em mm)
1. Regras de arredondamento
(de acordo com a norma Portuguesa NP-37/1961):
Os arredondamentos devem ser feitos de acordo com o valor do
algarismo seguinte ao qual se pretende arredondar, ou seja, quando se
14
arredondar um algarismo à casa de ordem n, deve ser ter-se em conta o
algarismo que está na casa de ordem n-1.
Se o algarismo correspondente à casa de ordem n-1 é menor que 5, o
número arredondado mantém inalterado o algarismo de ordem n (ex.: 11341
arredondado às dezenas é 11340, ou 342,53 arredondado às décimas é 342,5).
Se o algarismo correspondente à casa de ordem n-1 é maior que 5, o
número arredondado tem o aumento de uma unidade no algarismo de ordem n
(ex.: 11346 arredondado às dezenas é 11350, ou 342,57 arredondado às décimas
é 342,6)
Se o algarismo correspondente à casa de ordem n-1 é 5, e nas casas n-2,
n-3... pelo menos um algarismo é diferente de zero, o número arredondado tem
também o aumento de uma unidade no algarismo de ordem n (ex.: 11345,01
arredondado às dezenas é 11350, ou 342,552 arredondado às décimas é 342,6).
Se o algarismo correspondente à casa de ordem n-1 é 5, e nas casas n-2,
n-3... não há algarismos, ou são zeros, existem três modos de proceder ao
arredondamento:
(a) O valor a arredondar apresenta, com maior probabilidade, erro por excesso
do que por defeito (é o caso dos valores resultantes de certos métodos de
medida), neste caso o número arredondado mantém inalterado o algarismo de
ordem n.
(b) O valor a arredondar apresenta, com maior probabilidade, erro por defeito
do que por excesso (é o caso dos valores resultantes de divisões, interrompidas
quando ainda deixavam resto; e dos que resultam de certos métodos de
medida), neste caso o número arredondado tem o aumento de uma unidade no
algarismo de ordem n.
(c) Não há motivos para supor que o valor a arredondar apresenta, com maior
probabilidade, erro por excesso ou por defeito, neste caso o valor arredondado
é obtido somando uma unidade ao algarismo de ordem n se este for ímpar (ex.:
11335 arredondado à dezenas é 11340; se 342,55 arredondado às décimas é
342,6; se 43,735 arredondado às centésimas é 43,74) ou mantendo inalterado o
15
algarismo de ordem n se este for par (ex.: 11345 arredondado à dezenas é
11340; se 342,65 arredondado às décimas é 342,6; se 43,745 arredondado às
centésimas é 43,74).
16
2. Manuseamento dos dados experimentais (operações matemáticas
elementares):
Adição e subtracção: nos cálculos são utilizados todas as casas decimais,
mas o número de casa decimais significativas do resultado não pode ultrapassar o
menor número de casas significativas das parcelas. Ex.:
Multiplicação e divisão: o resultado tem o número de algarismos
significativos idêntico ao do factor com menor número de algarismos significativos
2
2
(ex.: 0,2x103,4 = 20,68 ou seja 0,2x10 ou 0,21x10 ; 0,2x140,7 = 28,14 ou seja
2
2
0,3x10 ou 0,28x10 ). Neste último caso é notário a informação dada pela
numenclatura com índice. NOTA: os números inteiros quando multiplicados por
reais não afectam o número de algarismos significativos, ou seja se um
computador custar 6.000 euros, dois computadores custam 12.000 euros e não
1x104 euros...
Logaritmos: o argumento do logaritmo e a mantissa do seu resultado
deverão ter o mesmo número de algarismos significativos (ex.: log 2,02 = 0,305)
17
D. Intervalos de confiança
É importante quantificar os erros aleatórios numa medição experimental. Isto
faz-se determinando um intervalo de confiança para o resultado final.
O intervalo de confiança representa-se como
"x ± ∆x, para um nível de confiança de α %"
e significa que há uma probabilidade α de o valor que medimos se encontrar entre
x-∆x e x+∆x.
A forma mais simples de estimar um intervalo de confiança é fazer a mesma
medição repetidas vezes. Os erros aleatórios que ocorrem em cada medição
serão diferentes. Uns serão por excesso, outros por defeito. Fazendo a média de
todos os resultados, estaremos a compensar os erros por excesso com os erros
por defeito, e, portanto, a minimizar os erros aleatórios de forma geral. Quanto
mais medições fizermos, melhor.
O valor médio de n repetições da mesma medição, xm, é uma estimativa do
valor verdadeiro da propriedade que queremos medir (chamemos a este µ ). Se
fosse possível fazer infinitas medições, conseguiríamos eliminar totalmente os
erros aleatórios. Só nesse caso é que teríamos a certeza de que o valor médio
das medições seria igual ao valor verdadeiro. Na prática, isto é impossível. Nunca
conseguimos saber o valor µ com rigor absoluto. O melhor que podemos fazer é
estimar um intervalo que tenha uma probabilidade elevada de o conter.
Sabemos que o desvio padrão é uma medida dos erros aleatórios que
ocorreram nas medições.
A maior parte dos erros aleatórios obedece a um tipo comportamento
estatístico, a que chamamos "distribuição normal" ou "distribuição de Gauss". Se
representássemos num histograma1 infinitas medições sujeitas a erros aleatórios,
este teria a forma de uma "boca de sino" designada por "curva de distribuição
1
Um histograma é um gráfico que traduz a frequência com que ocorre cada valor. No eixos das
abcissas representam-se os valores, e no eixo das ordenadas o número de vezes que cada um
ocorreu.
18
normal". Estas curvas são simétricas, e são definidas por dois parâmetros: a
média (µ) e o desvio padrão (σ)2. Na figura seguinte representam-se duas curvas
de distribuição normal com a mesma média (µ=200) e desvios padrão diferentes
(σ1=1,0 e σ2=2,5). É de salientar que:
- os valores ocorrem mais frequentemente próximo da média, e são
progressivamente menos frequentes quando nos afastamos para os extremos (o
máximo da curva está em µ);
- quanto maior o desvio padrão σ (maior é a dispersão dos valores em torno da
média µ) mais "larga" é a curva.
Uma das propriedades mais úteis das curvas de distribuição normal é que,
qualquer que seja µ e σ, cerca de 95% de todas as medições encontram-se no
intervalo µ-2σ e µ+2σ. Da mesma forma, encontra-se sempre uma percentagem
(p%) bem definida de todas as medições em qualquer intervalo µ±zσ. Isto significa
que, quando faço uma medição x, há p% de probabilidade de o valor verdadeiro,
µ, estar dentro do intervalo x±zσ. Os valores de z encontram-se tabelados em
função da probabilidade (nível de confiança). Os mais vulgarmente usados são:
2
Passaremos a designar por µ e por σ a média e o desvio padrão de uma curva de distribuição
normal, que seriam teoricamente obtidos através de infinitas medições e corresponderiam aos
valores "verdadeiros", e por xm e por s a média e desvio padrão calculados com um conjunto finito
de n pontos experimentais
19
Para calcular o intervalo de confiança, já só preciso de saber o valor de σ. Há
duas hipóteses:
- se fizer um número elevado de medições3, posso calcular o desvio padrão s e
dizer que σ ≈ s.
- se não for possível fazer um número suficientemente grande de medições,
calculo o desvio padrão, s, e em vez de multiplicar por z multiplico por outro factor,
o t de student.
O valor t de student encontra-se tabelado em função do nível de risco, (100-p),
e do número de graus de liberdade, gl. Este é dado por gl = n - 1 quando estamos
a fazer uma média de n medições.
Na Tabela 1 encontram-se alguns valores deste parâmetro4.
3
O que é um "número elevado de medições" varia, conforme os casos. Em geral, considera-se
n>30 suficientemente elevado.
4
Também pode calcular-se t numa folha de cálculo excel (versão inglesa) com a função
TINV(risco, gl).
20
O intervalo de confiança obtido para uma única medição x será então
No entanto, geralmente fazem-se n medições (são necessárias para
determinar s), e o valor médio dessas medições, xm, é uma aproximação melhor
ao valor verdadeiro do que as medições individuais. Demonstra-se que o desviopadrão da média, sm é igual ao desvio-padrão dos valores individuais, s, dividido
pela raiz quadrada do número de valores usados na média. O melhor intervalo de
confiança que conseguimos assim obter com n medições será:
E. Determinação da “melhor recta” que passa pelos pontos experimentais
Frequentemente fazem-se medições de uma propriedade que varia linearmente
com outra (por exemplo, a absorvência de uma solução pode variar linearmente
21
com a sua concentração, segundo a lei de Lambert-Beer). No entanto, as
medições estão sempre sujeitas a erros aleatórios, pelo que, em geral, os pontos
experimentais não coincidem com uma recta. Nestes casos, é necessário
determinar a equação (y=mx+b) da recta que melhor se ajusta ao conjunto do
dados experimentais. A este tipo de cálculo chama-se "regressão linear".
Um dos métodos mais usados para fazer regressão linear é o método dos
mínimos quadrados. Neste método, procura-se minimizar a distância "vertical" de
cada ponto experimental x a uma recta teórica, mx+b (ver figura seguinte)
O método parte de dois pressupostos muito importantes:
1. os erros aleatórios ocorrem apenas nas ordenadas (y), e não nas abcissas (x)
2. a ordem de grandeza dos erros aleatórios não varia ao longo da recta. Com
estes pressupostos, o método calcula os "residuais", que são a distância, na
vertical, de cada ponto experimental, yi, à recta: i y iy ) − (onde ýi representa o
valor esperado de y, valor que yi teria se não tivesse erro, ou seja, se tivesse
"caído" sobre a recta). A função U é a soma dos quadrados dos residuais, e é uma
medida do afastamento de todos os pontos experimentais a uma recta teórica de
declive m e ordenada na origem b:
22
Na função U, as incógnitas são m (o declive da recta) e b (a ordenada na
origem). Pode calcular-se o mínimo desta função derivando e igualando a zero. O
resultado deste cálculo dá as seguintes fórmulas para m e b:
onde N é o número de pontos experimentais (xi, yi). Os parâmetros Sxx, Syy e Sxy
podem calcular-se por:
Da regressão linear retira-se outro parâmetro muito importante, o desvio
padrão dos residuais, sy:
O desvio padrão dos residuais é uma quantificação dos erros aleatórios que
afastam os pontos da recta. Pode usar-se para determinar o desvio padrão do
declive, sm e da ordenada na origem, sb:
23
Assim, podemos determinar a equação da recta que melhor passa pelos
pontos experimentais, com intervalo de confiança para o declive e ordenada na
origem:
(Note-se que, neste caso, o número de graus de liberdade para o t de student é n2, e não n-1) Um parâmetro que traduz de forma simples se o ajuste da recta é
bom ou não é o coeficiente de correlação. O cálculo deste pode ser feito utilizando
a expressão:
O coeficiente de correlação pode tomar valores entre +1 e –1, quando |r|=1
então existe uma relação linear entre x e y (os resultados experimentais podem
ser descritos por uma recta), se r=0 existe uma independência completa entre os
valores de x e y (os resultados não apresentam qualquer relação linear).
Em métodos instrumentais de análise a regressão linear é frequentemente
usada para construir com soluções padrão uma recta de calibração, que
posteriormente é usada para determinar a concentração de uma amostra. Nestes
casos, o desvio padrão sc associado à concentração C determinada a partir da
recta é:
24
onde L é o número de réplicas da amostra que foram lidas,
c
é a média das L
leituras da amostra, e é a média das leituras das N soluções padrão que foram
usadas para construir a recta.
O intervalo para a concentração da amostra será então C ± 2 s C, para 95% de
confiança.
25
VI. Programação e calendarização das aulas práticas
Aulas
Data
1ª aula
22 de Setembro
Introdução
2ª aula
29 de Setembro
Preparação de soluções e Diluição de soluções
3ª aula
6 de Outubro
4ª aula
13 de Outubro
Gravimetria
Fotometria de chama de emissão: determinação de
Na+
Volumetria de precipitação: Titulação de Cl-
5ª aula
20 de Outubro
Lei de Charles
6ª aula
27 de Outubro
Segurança em laboratórios
7ª aula
3 de Novembro
Programa
Determinação de ∆ H vap (Clausius – Clapeyron)
8ª aula
10 de Novembro
Volumetria Ácido- base
9ª aula
17 de Novembro
Potenciometria
10ªaula
24 de Novembro
Volumetria de Complexação: Dureza da água
11ª aula
12 de Dezembro
Teste prático
26
Data: ______________
Nome:_________________________________________________No:_________
_______
Curso:_____________________________Disciplina:_____________________
APONTAMENTOS LABORATORIAIS
Título do trabalho:
1. Objectivo do trabalho:
2. Introdução:
3. Parte experimental:
3.1
Material utilizado
3.2
Reagentes utilizados
27
4. Resultados experimentais e cálculos:
5. Conclusões:
28
VIII. Relatórios
Cada trabalho prático deverá ser:
bem preparado,
bem executado e
bem discutido para ser avaliado como BOM.
Preparação da aula:
O estudante deverá preparar o trabalho prático.
O estudante deverá relacionar o objectivo do trabalho prático com os
aspectos teóricos da disciplina.
Quais aspectos teóricos são abordados / demonstrados no trabalho
prático?
O estudante deverá utilizar o protocolo prático, os apontamentos das aulas,
a bibliografia recomendada e outras fontes de informação a sua disposição afim
de elaborar as secções 1-5:
1. Título do trabalho: breve e conciso, nome completo do estudante,
número de inscrição. Data.
2. Objectivo: o objectivo pretendido.
3. Introdução e fundamento teórico: descrição da natureza do
problema, o âmbito e método. Discussão da metodologia.
geral da investigação (max A4).
4. Material: listas de material corrente de laboratório podem ser colocadas
no anexo. A preparação das soluções e reagentes utilizados assim com
as suas concentrações e volumes utilizados e aparelhos utilizados
devem constar nesta secção.
5. Procedimento: deve ser escrita de modo completo e explícito de forma
a que um investigador competente possa repetir as experiências.
Este trabalho será apresentado no início da aula laboratorial. Pode ser
apresentado como “documento de trabalho” ou seja 1ª versão. A versão
definitiva será entregue em conjunto com o relatório final.
Trabalho em laboratório
Os estudantes deverão apresentar-se com batas, pontualmente no
laboratório às horas indicadas no horário, e iniciar o trabalho prático preparado,
respeitando sempre as normas de segurança e de utilização dos laboratórios.
29
Relatórios (cont.)
A versão definitiva das secções 1-5 será entregue em conjunto com o
relatório final.
O estudante deverá utilizar o protocolo prático, os apontamentos das aulas,
a bibliografia recomendada e outras fontes de informação a sua disposição afim
de elaborar as secções 6-11:
6. Resultados:
dados apresentados em tabela,
cálculos utilizados,
tratamento estatístico dos dados,
gráficos,
breve descrição dos pontos notáveis.
7. Resultados:
tentar explicar os resultados obtidos,
relacionar os resultados com o objectivo,
discutir os problemas,
comparar os resultados com os de outros grupos práticos, e/ou
publicados na literatura.
8. Conclusão: resumo dos itens mais importantes da discussão.
9. Bibliografia: normalizada, citando todas as fontes de informação
utilizadas na preparação da aula e do relatório. É imprescindível que toda a
bibliografia citada no texto conste nas referências bibliográficas e vice-versa.
10. Anexos: listas etc
11. Resumo: uma condensação muito breve do trabalho, o que foi feito, os
processos gerais usados, os principais resultados e conclusões. (Max. 300
palavras).
12. Assinatura.
Este relatório deverá ser entregue no início da aula laboratorial seguinte,
dactilografado (tamanho 12 ou 14) a espaço e meio em papel A4, folhas soltas,
numa só face e com margens de aproximadamente 3cm de cada lado.
Os gráficos deverão ser apresentados em papel milimétrico.
30
IX. Trabalhos práticos
1. Preparação e Diluição de Soluções
Objectivo: Familiarizar os alunos com o manuseamento de material
volumétrico, balança analítica e preparação de
soluções
Introdução:
Uma
solução
é
uma
mistura
homogénea de dois ou mais componentes, o
facto de ser homogénea significa que a sua
composição é a mesma em todos os pontos.
Chama-se
solvente
ao
componente
predominante na mistura, e solutos aos
componentes existentes em menor quantidade.
Concentração de uma solução é a a quantidade de soluto presente numa
dada quantidade de solução. A concentração pode exprimir-se em várias
unidades, sendo as mais comuns:
Molaridade
Molalidade
moles de soluto
M, mol/L ou mol.dm-3
volume de solução
moles de soluto
massa de solvente
moles de soluto
massa de solvente
Percentage m em massa do soluto
m ou mol/kg
Molalidade
massa de soluto
volume de solvente
Massa de soluto
100%
Massa do soluto massa de solvente
mg/L (ou ppm), g/L, g.dm-3
31
Em geral, para preparar uma solução de concentração conhecida,
dissolve-se uma quantidade pré-determinada de soluto num volume rigoroso de
solvente. Se o soluto for um padrão primário e as medições de massa e volume
forem rigorosas, a concentração da solução será rigorosamente conhecida.
Podemos preparar soluções diluídas por diluição de soluções mais
concentradas. Neste caso, para obter uma solução de concentração C2 é
necessário pipetar um volume V1 da solução mais concentrada (de concentração
C1) para um balão de volume V2, e completa-se o volume deste até ao traço com
o solvente. Sempre que se fazem diluições é válida a relação:
C1.V1 = C2.V2
Neste trabalho, pretende-se preparar uma solução de concentração de
NaCl por pesagem e dissolução, e soluções menos concentradas por diluição
desta.
Observações:
1)
A quantidade de soluto a ser pesada deve ser calculada de acordo com a
concentração pretendida.
2)
Verter para um recipiente pequeno e limpo (copo), uma quantidade suficiente
de composto, será desse copo que serão retiradas as quantidades previstas.
NOTA: Não deve introduzir qualquer espátula ou similar dentro do frasco que
contém o composto.
3)
O NaCl e o KCl são padrões primários. Devem ser previamente secos na
estufa, para remover quaisquer moléculas de água de hidratação.
4)
Para pesar quantidades rigorosas deve usar-se uma balança analítica de
precisão ±0,0001 g. (Nota: Para pesagens menos rigorosas pode usar-se
uma balança técnica de precisão ±0,01 g).
5)
Na pesagem deve ser utilizado um copo especial ou papel de filtro.
32
Procedimento experimental:
a) Soluções de NaCl:
1) Num copo de 100 mL, pese rigorosamente a quantidade de NaCl necessária
para preparar 250 mL de uma solução aquosa 0,250 M.
2) Com a ajuda de uma vareta de vidro, dissolva todo o NaCl usando a menor
quantidade de água destilada possível.
3) Quando todo o sólido estiver dissolvido, transfira o líquido para um balão
volumétrico de 250 mL. Lave cuidadosamente o copo várias vezes com um
esguicho de água destilada, deitando as águas de lavagem no balão, de modo
a transferir quantitativamente todo o NaCl. Tenha o cuidado de não deixar nível
do líquido ultrapassar o traço do balão volumétrico.
4) Tape o balão e agite para homogeneizar a solução.
5) Adicione cuidadosamente água destilada com o esguicho, até ajustar
exactamente o nível de líquido à marca do balão. Tenha atenção para não
cometer erros de paralaxe.
6) Homogeneíze novamente a solução.
7) Identifique o balão.
Preparação de soluções diluídas:
Por diluição da solução anterior, prepare soluções de concentração:
2x10-3 M
2x10-2 M
5x10-2 M
0,1 M
0,2 M.
Calcule os volumes mais adequados a cada diluição, sabendo que existem
no laboratório pipetas volumétricas de:
1, 2, 3, 5, 10, 20, 25 e 50 mL;
e ainda balões volumétricos de:
10, 20, 25, 50, 100, 200, 250, 500 e 1000 mL.
Sugestão: para cada solução, escolha primeiro o balão que pretende usar, e
calcule depois o volume que é necessário pipetar.
33
Cálculos:
Calcule rigorosamente a concentração das soluções que preparou, em
molaridade e em mg/L.
Bibliografia:
[1] R. Chang, Química, 1994, 5ª Edição, Alfragide-Portugal, p 7-34; 525-568.
[2] J. E. Brady e J. R. Holum, Chemistry - The study of matter and its changes, 1993, 1ª
Edição, New York, p. 404-457.
34
2. Gravimetria
“Determinação do resíduo de uma água”
Material:
placa de Petri
balança
pipeta volumétrica de 25 ml
Solução Padrão de NaCl preparada na aula anterior
banho Maria ou estufa
exsicador
Procedimento experimental:
1.
Pesar metade de uma placa de Petri.
2.
Pipetar 25 ml da solução de NaCl que preparou na aula anterior.
3.
Aquecer até evaporação total.
4.
Deixar arrefecer em exsicador.
5.
Pesar.
6.
Repetir 3-5 até obter dois pesos consecutivos iguais.
7.
Determinar o peso do resíduo.
Peso da placa (g)
Peso placa + resíduo
Peso resíduo
Concentração da sol. padrão
g/l
Concentração determinada
g/l
Concentração da sol. padrão
M
Concentração determinada
M
35
8. Calcular a partir dos dados da experiência a concentração da solução inicial
em g/l e M.
9. Compare os valores obtidos na experiência aos valores da aula anterior.
36
3. Fotometria de Chama de Emissão
“Determinação do Sódio em Águas”
Objectivo:
Utilização da técnica de fotometria de chama de emissão para determinar
os teores em sódio (Na) em águas.
Introdução:
O modelo actualmente aceite para a estrutura do átomo indica que este é
constituido por um núcleo (de protões e neutrões) e por electrões. Os electrões
distribuem-se em torno do núcleo, ocupando cada electrão um nível bem definido
de energia.
Os electrões podem ser excitados (passando para níveis de energia
superiores) por absorção de um quanta de energia, emitido por uma chama ou por
uma descarga eléctrica. Quando os mesmos electrões passam de novo para o
estado de energia mais baixo (estado fundamental) podem, dependendo do tipo
de decaimento, emitir também um quanta de energia sob a forma de um ou vários
fotões [1].
Eh (estado excitado)
e
Eh
fotão
e
E1 (estado fundamental)
(Absorção de energia)
E1
(Emissão de energia)
A energia do fotão (Efotão) é igual à diferença entre os dois níveis de
energia, e inversamente proporcional ao comprimento de onda do fotão (), de
acordo com a seguinte equação:
E = Eh – E1 = Efotão = hc/ = h
em que c é a velocidade da luz, 3.00x108 m/s, h é a constante de Planck,
6.63x1034 J.s/fotão e a frequência do fotão ( = c/).
37
A emissão de mais de um fotão por cada quanta de energia absorvido é
devido ao facto de para cada electrão serem possíveis vários níveis de energia
excitados, deste modo pode ser obtido um espectro de riscas com diferentes de
emissão.
Efotão (1)
Efotão (2)
Efotão (3)
Cada elemento tem um espectro de riscas característico.
Esta propriedade pode ser usada para determinar a concentração de sódio
e potássio na água do mar, usando um fotómetro de emissão de chama. Sendo a
intensidade da radiação emitida a um dado proporcional à concentração do
elemento na solução.
Este trabalho tem como objectivo a determinação dos teores em sódio (Na)
e potássio (K) na água do mar, utilizando como técnica de análise a fotometria de
chama de emissão.
A existência destes elementos em quantidades elevadas em águas
subterrâneas pode ser indicativa de poluição por actividades humanas ou animais,
em especial tratando-se de zonas onde não existem minerais contendo sódio (sal
gema). Estações de tratamento de esgotos e a actividade agrícola contribuem
normalmente para a existência de sódio nas águas superficiais. Por outro lado,
próximo da costa, a proliferação dos furos subterrâneos pode baixar o nível
freático abaixo do nível da água do mar, causando a contaminação dos poços e
furos com água salina e o aumento do nível de sódio nestas águas [2], como
acontece na costa algarvia.
38
Procedimento experimental:
Utilize as soluções de NaCl preparadas na
última aula. Estas soluções servirão de padrão para
calibrar o fotómetro de chama.
a) Soluções de amostra
Prepare a amostra diluindo a amostra de água
Fotómetro de Chama
1:200.
b) Determinação dos teores em sódio em água
Calibração do fotómetro de emissão de chama com filtro de sódio:
1. Utilizando água destilada pura (branco) calibra-se o aparelho para ler
o valor 0 (zero) e com a solução de NaCl mais concentrada o valor 100.
2. Efectuar as medidas para as soluções de NaCl diluídas e para a
amostra.
Cálculos:
1) Calcule rigorosamente a concentração das soluções de NaCl que preparou, em
molaridade e em mg/L.
2) Traçar as rectas log I/I0 versus Concentração para as soluções de sódio.
3) Calcular os teores em sódio e da água analisada.
Bibliografia:
[1] J.A. Bevan, Laboratory Manual for principles of General Chemistry, 5th Ed.,
John Wiley & Sons, 1994.
[2] S. E. Kegley, J. Andrews, The Chemistry of Water, University Science Books,
1998.
[3] R. Chang, Química, 1994, 5th Ed., Alfragide-Portugal, p 7-34; 525-568.
[4] J. E. Brady, J. R. Holum, Chemistry - The study of matter and its changes, 1th
Ed., 1993,New York, p. 404-457.
39
4.Volumetria de Precipitação
“Determinação do teor em cloretos de uma água”
Introdução:
O teor em cloretos de uma solução pode ser determinado, por volumetria
de precipitação, sendo a detecção do ponto de equivalência feita pela formação de
um precipitado vermelho de cromato de prata (método de Mohr).
Neste método é feita a titulação dos iões cloreto com o nitrato de prata, de
que resulta a formação de um precipitado branco de cloreto de prata (Kps = 1,8 x
10-10, a 25 ºC).
Ag+ (aq) + Cl - (aq)
AgCl (s)
Esta titulação é feita em presença de ião cromato, que actua como
indicador e confere à solução uma coloração amarela.
Quando o ião cloreto tiver sido completamente consumido, a adição de um
excesso de titulante terá como consequência a reacção dos iões prata com o ião
cromato presente na solução, formando um precipitado vermelho de cromato de
prata (Kps = 1,1 x 10-12, a 25 ºC). A precipitação deste sal, dada a sua maior
solubilidade, só ocorre após o consumo do ião cloreto, o que permite a sua
utilização como indicador, mas só se verifica quando a sua solubilidade for
atingida, o que acarreta a necessidade de realização de um ensaio em branco.
2 Ag+ (aq) + CrO4 2- (aq)
Ag2CrO4 (s)
Neste processo é importante que o pH da solução se mantenha entre 6,5 e
10, a fim de evitar reacções secundárias. O acerto do pH faz-se mediante a adição
de carbonato de cálcio ou de ácido nítrico.
Aplicação: A maioria das águas contém iões cloreto dissolvidos
provenientes de contaminação por água do mar ou da adição de cloro como
desinfectante. A determinação destes iões faz-se, segundo a NP-423, pelo método
descrito neste trabalho.
40
Parte experimental:
1. Determinação da concentração da solução de nitrato de prata:
- Deitar num gobelé 10 cm3 da solução-padrão de NaCl e verificar o seu pH, com
papel indicador.
- Se a solução apresentar pH inferior a 6,5 , adicionar CaCO3 (s) puro.
- Se a solução apresentar pH superior a 10, adicionar HNO 3, até pH inferior a 6,5
e em seguida ajustar como no caso anterior.
- Adicionar 1 cm3 de K2CrO4 a 5% e preparar uma bureta com a solução de
AgNO3 a titular.
- Titular a solução de NaCl com a solução contida na bureta, até obtenção de uma
coloração vermelha, persistente à agitação.
- Fazer mais dois ensaios iguais ao anterior; nos cálculos, usar a média dos
volumes obtidos.
- Efectuar um ensaio a branco, usando como titulado 10 cm3 de água destilada.
- Determinar a concentração da solução de nitrato de prata, usando o valor
corrigido de volume de titulante gasto.
2. Determinação do teor em cloretos de uma água:
- Medir para um gobelé 100 cm3 da água em análise e verificar o seu pH,
ajustando-o se for caso disso.
- Titular com a solução-padrão de AgNO3, na presença de 1 cm3 de K2CrO4 a
5%, até obtenção de coloração vermelha persistente à agitação.
- Fazer mais dois ensaios iguais ao anterior; nos cálculos, usar a média dos
volumes obtidos.
- Efectuar um ensaio a branco, usando como titulado 100 cm3 de água destilada.
- Calcular o teor em cloretos da água analisada expresso em mg/dm 3 de Cl-.
41
5. Leis dos gases: Lei de Charles e Gay-Lussac
Objectivo: Determinar o efeito da temperatura no volume de uma dada massa de
gás a pressão constante.
Introdução:
Um dos factos notáveis no comportamento dos gases é que, apesar das
diferenças nas suas propriedades químicas, todos obdecem ao mesmo conjunto
de leis físicas: Leis dos gases.
As grandezas mensuráveis importantes de um gás são: volume (V),
pressão (P), temperatura (T) e massa (m). As relações entre estas quatro
grandezas foi determinda numa série de experiências sucessivas das quais será
feita referência nesta breve introdução apenas a duas. Robert Boyle (1627-1691)
determinou experimentalmente que que o volume de um gás varia na razão
inversa da pressão (de notar que o volume de um gás é sempre igual ao do seu
contentor, uma vez que os gases se expandem para ocupar todo o espaço que
está disponível). Assim,
P1V1=P2V2
A relação entre a pressão e o volume de um gás com uma determinada
massa, mantendo a temperatura constante é expressa como a lei de Boyle:
PV=constante.
A relação entre o volume de uma amostra de gás, a pressão constante, e a
sua temperatura absoluta pode ser expressa através da Lei de Charles. Segundo
esta lei os gases aproximam-se do comportamento da lei de Charles quando a um
aumento de temperatura, a pressão constante, corresponde a um aumento do seu
volume.
V1/V2=T1/T2
42
Procedimento experimental:
a) Introduzir cerca de 750 mL de água desionizada num copo de precipitação de
1L
b) Colocar cerca de 6 mL de azeite numa proveta graduada de 10 mL.
c) Introduzir a proveta de boca para baixo no copo.
d) Colocar o termómetro dentro do copo. Aguardar alguns minutos e verificar qual
a temperatura da água e registar o volume de ar existente na proveta (levantar
o menisco do azeite até à superfície da água para efectuar a leitura).
e) Colocar a barra de agitação no copo.
f) Colocar o sistema sobre uma placa de aquecimento com agitação, já quente.
g) Ligar o sistema de agitação.
h) Registar o volume ocupado pelo ar na proveta, para cada incremento de 5 ºC,
até a temperatura atingir 70 ºC.
Cálculos e discussão de resultados:
1) Registar num gráfico o volume ocupado pelo ar em função da temperatura.
2) Traçar uma recta prolongando-a até cortar o eixo dos xx.
3) Interpretar os resultados obtidos.
Bibliografia:
[1] R. Chang, Química, 1994, 5th Ed., Alfragide-Portugal, p 7-34; 525-568.
43
6. Termodinâmica: Entalpia de vaporização
Objectivo: Aplicação das leis da termodinânica para determinar a entalpia de
vaporização da água.
Introdução:
As moléculas de um líquído não estão numa rede rigída, estando portanto
em movimento constante. A cada temperatura existe sempre energia suficiente
para que uma determinada quantidade de moléculas passe do estado líquído ao
gasoso (processo de vaporização). Assim, a pressão de vapor de um líquido (ou
de um sólido) pode ser representada como função da temperatura, num diagrama
que permite identificar os equilíbrios que se estabelecem entre as diferentes fases.
Na figura seguinte está representado esquematicamente o diagrama de fases para
a água pura:
subli
P
maç
gasoso
ão
sólido
fus
ão
vaporiza
ção
líquido
T
Figura 1 – Diagrama de fases para a água pura
A entalpia de vaporização (Hvap) é uma medida da força com que as
moléculas estão ligadas num liquído (força intrermolecular) e pode ser definida
como a energia necessária para vaporizar uma mole de um liquído (entalpia de
vaporização molar). A grandeza Hvap pode ser determinada experimentalmente
utilizando a equação de Clausius-Clapeyron, que relaciona a pressão de vapor de
um líquido (P) com a temperatura absoluta (T):
H v ap 1
C
ln P
R T
44
Esta equação tem a forma da equação linear (y=mx+b) considerando: y=lnP, m=(-Hvap/R), x=(1/T)
e b=C.
Procedimento experimental:
Para calcular a entalpia de vaporização da água:
1) Registar a pressão atmosférica.
2) Encher uma proveta de 10 mL até ~2/3 do seu volume com água destilada.
Tapar com o dedo e inverter a proveta para um copo de 500 mL,
previamente cheio com água destilada (Nota: uma amostra de ~4 a 5 mL
de água destilada deve ficar retida na proveta).
3) Aquecer até 80 ºC, com agitação o copo contendo a proveta. Registar o
volume de ar na proveta.
4) Deixar arrefecer a água, com agitação constante, registando em intervalos
de 5 ºC a temperatura da água e o volume correspondente de ar, até 40 ºC.
5) Arrefecer rapidamente até ~1 a 5 ºC, com gelo. Registar o volume
correspondente.
Cálculos:
a) Elaborar um gráfico lnPvap versus 1/T.
b) Calcular o número de moles de ar na proveta (PV=nRT, R=0.0821 L.atm.K 1.mol-1)
c) Calcular a entalpia de vaporização (R=8,31 J.K-1.mol-1)
Bibliografia:
[1] R. Chang, Química, 1994, 5ª Edição, Alfragide-Portugal, p 223-265.
[2] J. E. Brady e J. R. Holum, Chemistry- The study of matter and its changes,
1993, 1ª Edição, New York, p. 136-169.
45
7. Volumetria ácido-base
“Titulação de um ácido fraco com uma base forte”
Encher a bureta com uma solução de NaOH de concentração conhecida.
Ensaio preliminar:
Pipetar para um erlenmeyer um pequeno volume (1-5ml) do ácido a analisar.
Adicionar 1 gota de fenolftaleina (indicador).
Titular até virar o indicador (rosa pálido).
Ensaio definitivo:
A partir do resultado do ensaio preliminar, determinar o volume a pipetar para
se utilisar quase uma bureta inteira de solução titulante.
Pipetar o volume adequado de ácido a analisar para um erlenmeyer.
Adicionar 1 gota de fenolftaleina (indicador).
Titular até virar o indicador (rosa pálido).
Repetir a experiência.
Calcular a concentração do ácido.
46
8. Volumetria ácido-base
“Titulação potenciométrica”
Objectivo: Estudo da curva de variação do pH
Introdução:
Uma medida da concentração de iões H+ numa solução é o pH. Define-se
como
pH = -log (H+)
em que (H+) é a actividade dos iões H+ em solução.
O pH de uma solução pode ser medido usando um medidor de pH. Este é
constituído por um eléctrodo combinado de vidro e um potenciómetro, e mede
uma diferença de potencial que é directamente proporcional ao pH.
O ponto de equivalência de uma titulação ácido-base pode ser detectado
seguindo a evolução do pH à medida que o titulante é adicionado - titulação
potenciométrica.
Numa titulação de HCl com NaOH, o pH inicial da solução de HCl é baixo.
À medida que se adiciona NaOH, todo o OH- reage com o ácido, e o pH começa a
subir gradualmente. Quando o número de moles de base adicionados iguala o
número de moles de ácido inicialmente presentes, pequenas adições de base
causam grandes variações de pH. Perto do ponto de equivalência, a variação de
pH com a adição de base é muito brusca e a curva de titulação torna-se
praticamente vertical. Depois de todo o ácido ser consumido, a adição de mais
base sobe o pH cada vez mais lentamente, até que o pH da solução seja
semelhante ao da base (Figura 1a).
O ponto de equivalência pode ser determinado por métodos gráficos. Um
método é o método da primeira derivada. O ponto de equivalência é o ponto de
inflexão da curva de titulação, e ocorre quando o seu declive é máximo. Portanto,
podemos determinar o ponto de equivalência calculando o declive em cada ponto
da titulação próximo do ponto de equivalência. Para calcular o declive em cada
47
ponto faz-se a diferença de pH entre duas adições sucessivas de base e divide-se
pH
pela diferença de volumes correspondente (Figura 1b).
12
10
8
6
4
18
19
20
21
22
V (ml)
Veq=20,6 ml
Figura 1 - Exemplo de uma curva de titulação ácido-base (a) e curva da primeira derivada (b).
Procedimento experimental:
1) Pipetar para um copo de 250 mL, 20 mL de ácido clorídrico.
2) Pôr no copo um agitador magnético
3) Encher uma bureta com NaOH 0,1M e colocar num suporte sobre o copo.
4) Introduzir os eléctrodos do aparelho de pH no copo (adicionar água à
solução de modo a que os eléctrodos fiquem mergulhados)
5) Ligar o aparelho e agitador e registar o pH inicial.
6) Adicionar pequenos volumes de base e registar o pH. Quando as variações
de pH se mostrarem mais acentuadas, passar a adicionar volumes de base
mais pequenos (cerca de 0,1 mL). Quando as variações de pH novamente
se mostrarem menos pronunciadas passar a adicionar volumes de cerca de
0,5 mL. Dar por terminadas as variações quando por adição de base não se
verificar variação sensível do pH.
48
Cálculos:
a) Fazer o gráfico pH versus volume de base adicionada.
b) Determinar o volume de base gasto até ao ponto de equivalência, pelo
método da primeira derivada.
c) Calcular a concentração de ácido.
Bibliografia:
[1] R. Chang, Química, 1994, 5ª Edição, Alfragide-Portugal, p 671-737-755.
[2] J. E. Brady e J. R. Holum, Chemistry- The study of matter and its changes,
1993, 1ª Edição, New York, p. 680-736.
49
9. Volumetria de Complexação
“Determinação da dureza de uma água”
Introdução: O teor de uma solução em iões cálcio pode ser determinado
por volumetria de complexação com EDTA (ácido etilenodiaminotetracético) ou
com o seu sal dissódico (1), por este ser mais solúvel em meio aquoso, na
presença de indicadores metalocrómicos (Negro de Eriocrómio T (2) - Ério T - ou
Murexide, consoante a determinação).
OH
N N
Na+ - OOC
Na+ - OOC
OH
H2C
N CH2
H2C
CH2 COOH
CH2 N
CH2 COOH
HO3S
(1)
NO2
(2)
azul a 6,3 < pH < 11,5
O sal dissódico do EDTA forma com os iões cálcio complexos estáveis, de
acordo com a equação:
Ca 2+ + H2(edta) 2-
Ca(edta) 2-
+ 2H+
Quanto ao indicador, também ele complexa os iões cálcio, formando
compostos de cor distinta da que apresenta quando livre em solução. No entanto,
estes são menos estáveis do que os complexos de EDTA, pelo que a adição deste
reagente à solução provoca a sua destruição, libertando o indicador. O final da
titulação é assim indicado pelo aparecimento de uma coloração azul, reveladora
de que o indicador se encontra livre, isto é, de que todos os iões cálcio existentes
na solução se encontram complexados com o EDTA.
50
Aplicação: A dureza total de uma água é devida à presença de catiões
cálcio (Ca2+) e magnésio (Mg2+), bem como de quantidades vestigiais de catiões
Fe2+, Al3+, Cu2+, sob a forma de carbonatos, hidrogenocarbonatos, sulfatos,
cloretos e nitratos. Estes iões não são particularmente tóxicos, sendo os VMA
(valores máximos admitidos por lei) relativamente elevados; no entanto, a sua
presença numa água pode torná-la imprópria para certos usos industriais já que as
águas duras apresentam grande dificuldade em fazer espuma com detergentes,
impossibilitando a sua utilização para lavagens, e grande tendência em formar
incrustações nas tubagens, canalizações e recipientes com que contacta. Por
vezes, interessa conhecer a dureza cálcica de uma água, apenas devida aos iões
cálcio, o que se consegue precipitando os iões magnésio, sob a forma de
Mg(OH)2, por adição de NaOH:
Mg 2+ (aq) +
2
OH - (aq)
Mg(OH)2 (s)
Ambas as durezas se exprimem em mg.dm -3 ou em ppm de CaCO3, por se
considerar que a dureza total da água se deve essencialmente à presença deste.
Considera-se dureza temporária de uma água, a dureza associada à presença
de iões HCO3-, que se eliminam por ebulição:
2 HCO3- (aq)
CO32- (aq) + CO2 (g) + H2O (l)
CO32- (aq) + Ca2+ (aq)
CaCO3 (s)
A dureza permanente de uma água será, portanto, a dureza devida aos sais de
cálcio e magnésio não eliminados por ebulição (sulfatos, cloretos, etc.) e
corresponderá à diferença entre a dureza total e a dureza temporária:
Dureza total = Dureza permanente + Dureza temporária
As normas vigentes na determinação da dureza de águas são as NP-506 e
NP-507 e pressupõem que a mesma seja feita pelo método aqui descrito.
51
Parte experimental:
1. Aferição da solução de EDTA:
- Deitar, num erlenmeyer, 25 cm3 de solução-padrão de Ca2+ 0,01 M e diluir com
25 cm3 de água desionizada
- Adicionar 2 cm3 da solução tampão de NH4+/NH3 e 3 gotas da solução de Ério
T.
- Preparar uma bureta com a solução de EDTA e titular a solução contida no
erlenmeyer até obtenção de uma cor azul, persistente à agitação.
- Fazer mais dois ensaios iguais ao anterior.
2. Determinação da dureza de uma água:
- Medir, para um erlenmeyer, 100 cm3 da água em estudo e adicionar-lhe 4 cm3
da solução tampão de NH4+/NH3 e 6 gotas da solução de Ério T.
- Preparar uma bureta com a solução padrão de EDTA e titular a solução contida
no erlenmeyer até obtenção de uma cor azul, persistente à agitação.
- Fazer mais dois ensaios iguais ao anterior.
- Calcular a dureza da água expressa em mg/dm3 e em ppm de CaCO3.
52
10. Volumetria de Redução-oxidação
“Titulação do Ferro (II) com Permanganato de Potássio”
Introdução:
O Fe(II) duma solução será oxidado a Fe(III) pelo MnO4- em meio ácido de
acordo com a reacção:
5Fe2+ + MnO4- + 8H+
5Fe3+ + Mn2+ +4H2O
Procedimento experimental:
1. Pipetar para um Erlenmeyer 10 mL da solução a titular.
2. Adicionar um volume duplo de H2SO4 0.2 M (20 mL) e 1 mL de ácido
fosfórico concentrado (esta solução deve ser titulada de imediato).
3. Titular com a solução padrão de permanganato de potássio até que a
solução adquira uma tonalidade rosa pálido.
4. Repetir o ensaio três vezes.
Cálculos:
a) Calcular a concentração de Fe(II) em solução.
b) Calcular a média e o desvio padrão.
Bibliografia:
[1]. R. Chang, Química, 1994, 5ª Edição, Alfragide-Portugal
53
Alice Newton
28/05/2017
54