Metabolismo
Metabolismo de Carboidratos
Introdução:
O carboidrato, principal fonte de energia para os seres humanos, pode ser derivado
da degradação dos alimentos, das reservas corporais de glicogênio ou através da
gliconeogênese que faz a síntese endógena a partir de aminoácidos, lactato ou glicerol. São
consumidos na forma de amido, sacarose e lactose, e em menores quantidades, como
glicogênio, maltose, glicose livre e frutose livre.
Quanto à forma estrutural, podem ser classificados em:
o
Monossacarídeos: carboidratos mais simples que não sofrem hidrólise (ex: glicose,
frutose, galactose, ribose).
o
Dissacárideos: 2 moléculas de monossacarídeos com perda de uma molécula de
água. (ex: sacarose, maltose, lactose).
o
Oligossacarídeos: 3 a 10 moléculas de monossacarídeos. (ex: dextrinas).
o
Polissacarídeos:
mais
de
10
moléculas
de
monossacarídeos
com
as
correspondentes perdas de moléculas de água (ex: amido, glicogênio, celulose).
Metabolismo dos carboidratos:
Os carboidratos presentes na dieta, os quais possuem a forma estrutural de
dissacarídeos, oligossacarídeos e polissacarídeos, sofrem hidrólise para a estrutura de
monossacarídeo em diferentes segmentos no trato gastrointestinal, podendo assim ser
absorvidos.
A digestão do amido se inicia durante a mastigação pela ação da alfa-amilase
presente na saliva (ptialina), que hidrolisa as ligações glicosídicas, com a liberação de
maltose e oligossacarídeos. Contudo, a α-amilase salivar não contribui significativamente
para a hidrólise dos polissacarídeos, não apenas devido ao breve contato entre a enzima e
o substrato, mas também porque, ao atingir o estômago, a enzima é inativada pelo baixo pH
gástrico.
O amido e o glicogênio são hidrolisados no duodeno em presença da α-amilase
pancreática, que produz maltose como produto principal e oligossacarídeos chamados
dextrinas . Certa quantidade de isomaltose (dissacarídeo) também é formada.
Amido (ou glicogênio)
alfa-amilase
maltose + dextrina
A hidrólise final da maltose e dextrina é realizada pela maltase, dextrinase e
isomaltase, que se encontram na superfície das células epiteliais do intestino delgado.
Maltose + H2O
Maltase
2 D−glicose
Dextrina + H2O
Dextrinase
n D−glicose
Isomaltose + H2O
Sacarose + H2O
Lactose + H2O
Isomaltase
D-frutose + D−glicose
Sacarase
Lactase
2 D−glicose
D-galactose + D−glicose
A captação de monossacarídeos do lúmen para a célula intestinal é efetuada por
dois mecanismos:
• Transporte passivo (difusão facilitada): A glicose movimenta-se no sentido de maior para o
de menor concentração, mediada por um sistema de transporte de monossacarídeos do tipo
Na+− independente, que tem alta especificidade para D−frutose.
• Transporte ativo: Com a ajuda de co− transportador Na+−monossacarídeo (SGLT) a
glicose é captada do lúmen para a célula epitelial do intestino, sendo transportada no
sentido da menor para a maior concentração. É um processo ativo indireto envolvendo a
(Na+−K+)−ATPase (bomba de (Na+−K+), que remove o Na+ da célula, em troca de K+, com a
hidrólise concomitante de ATP, com alta especificidade por D−glicose e D−galactose.
Após a absorção, o nivel de glicose no sangue aumenta e as células β das ilhotas
pancreáticas secretam insulina (Fig 1.), um hormônio anabólico, que estimula a captação de
glicose principalmente pelo tecido adiposo e muscular, convertendo-a em lipídios
(lipogênese) ou glicogênio para armazenamento. Além disso, ainda inibe a produção de
glicose pelo fígado a partir de fontes como lactato, glicerol e aminoácidos (gliconeogênese),
estimula a síntese protéica e inibe a degradação de proteínas. O fígado, o cérebro e os
eritrócitos, não necessitam de insulina para captação de glicose por suas células (tecidos
insulino−independentes).
Outros hormônios e enzimas, além de vários mecanismos de controle, são
importantes na regulação da glicemia. Os hormônios contra-reguladores (glucagon,
epinefrina, GH e coortisol) são catabolizantes e aumentam a produção hepática de glicose,
inicialmente por aumentar a degradação do glicogênio (glicogenólise) e mais tarde pela
gliconeogênese. O hormônio do crescimento (GH) e o cortisol também aumentam a
mobilização de glicose e diminuem sua utilização.
Fig 1.Produção da Insulina
O glucagon é um peptídeo secretado pelas células alfa das ilhotas de Langerhans do
pâncreas que age no fígado estimulando a glicogenólise e a gliconeogênese. A secreção de
glucagon é regulada principalmente pelas concentrações de glicose no plasma, sendo níveis
baixos estimuladores e níveis altos inibidores. A insulina antagoniza seus efeitos e inibe sua
liberação pelo pâncreas.
A epinefrina (adrenalina) é uma catecolamina secretada pela medula adrenal que
estimula a produção de glicose, diminui sua utilização e aumenta o nível de glicose no
sangue ao estimular a secreção de glucagon e inibir a secreção de insulina pelo pâncreas. A
epinefrina parece ter um papel importante na contra-regulação da glicose, quando a
secreção de glucagon está prejudicada (exemplo: nos casos de pancreatite crônica,
hemocromatose e pancreatectomia). A agressão física ou emocional aumenta a produção
de epinefrina, liberando glicose para fins energéticos. Tumores da medula adrenal
(feocromocitomas)
secretam
epinefrina
e
norepinefrina
em
excesso,
produzindo
hiperglicemia moderada, enquanto os estoques de glicogênio estiverem disponíveis no
fígado (fig2).
O hormônio do crescimento é um peptídeo secretado pela hipófise anterior (adeno-
hipófise) sob a regulação do hipotálamo e que tem a função de estimular a gliconeogênese,
aumentando a lipólise e antagonizando a captação de glicose estimulada pela insulina. É
inibido pela somatostatina.
O cortisol, secretado pelo córtex da adrenal em resposta ao hormônio
adrenocorticotrófico (ACTH), estimula a gliconeogênese e aumenta a degradação de
proteínas e lipídeos. Pacientes com síndrome de Cushing tem cortisol aumentado devido a
um tumor ou à hiperplasia do córtex adrenal, podendo desenvolver hiperglicemia. Ao
contrário, indivíduos com doença de Addison apresentam insuficiência adrenocortical devido
à destruição ou à atrofia do córtex adrenal, podendo levar a hipoglicemia.
Tiroxina ou tetraiodotironina (T4), uma amina secretada pela tireóide, não está
envolvida na homeostasia da glicose, mas estimula a glicogenólise e aumenta a velocidade
de esvaziamento gástrico e de absorção intestinal de glicose. Tais fatores podem produzir
intolerância à glicose em indivíduos com tireotoxicose, mas geralmente a glicemia de jejum
é normal.
A somatostatina é um hormônio protéico produzido pelas células delta do pâncreas e
em diversos locais do organismo, que embora não pareça ter um efeito direto sobre o
metabolismo glicídico, inibe a liberação do hormônio do crescimento pela hipófise, a
secreção de glucagon e de insulina. Sua secreção é regulada pelos altos níveis de glicose e
aminoácidos.
Pâncreas
Cel Delta
Fígado
Glucagon
Cel alfa
Cel Beta
Glicose
Glicogenólise e
Gliconeogênese
Epinefrina
Somatostatina
Insulina
Cortisol, GH
Tecido adiposo
Captação de Glicose Lipogênese
Músculo
Captação de
glicose - Glicólise
Fig 2. Regulação glicêmica e os hormônios relacionados. Setas cheias
significam estimulação e as setas tracejadas significam inibição (3).
Vias metabólicas:
o
Glicolítica: Essa via refere-se à degradação de glicose desde sua forma inicial até a
formação de piruvato e pode ocorrer na presença ou ausência de oxigênio. A enzima
marca-passo desta via é a fosfofrutoquinase (FFK).
- Glicólise aeróbica: ocorre na presença de O2, e ativa o ciclo de krebs e a cadeia
respiratória para a produção de energia.
- Glicólise anaeróbica: ocorre na ausência de O2, não ativa o ciclo de krebs e a
cadeia respiratória, tendo como produto final o ácido Lático.
o
Glicogênese: transforma a glicose em glicogênio para o seu armazenamento, ou
seja, o armazenamento de energia.
o
Glicogenólise: transforma a glicogênio em glicose, para usar a energia armazenada.
o
Via das pentoses: transformação da glicose em pentose.
Glicólise (do grego, glykos, doce e lysis, romper) é a principal via de degradação
catabólica da glicose, sendo composta de uma série de reações enzimáticas que ocorrem
no citosol de todas as células humanas. O produto final é a formação de duas moléculas de
Piruvato, sendo que a energia liberada durante o processo é conservada na forma de duas
moléculas de adenosina Trifosfato (ATP) e duas de nicotinamida adenina dinucleotídeo
(NADH). Tal processo é resumido na equação química abaixo:
Glicose + 2 ADP + 2 Pi + 2 NAD+ → 2 piruvato + 2 NADH + 2 H+ + 2 ATP + 2 H2O
Na situação de baixo suprimento de oxigênio (hipóxia) ou em células sem mitocôndrias,
o piruvato produzido pela glicólise é transformado em lactato, processo denominado glicólise
anaeróbica.
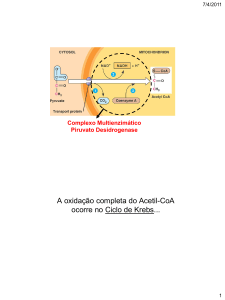
Quando o suprimento de oxigênio é adequado, o piruvato é transformado em acetil−CoA
nas mitocôndrias e o grupo acetil da acetil−CoA é totalmente oxidado no ciclo do ácido
cítrico (seqüência de reações enzimáticas que ocorrem durante o processo de respiração
celular, no interior da mitocôndria) com a formação de duas moléculas de CO2. Essa
conversão é um processo de oxidação irreversível, no qual o grupo carboxílico do piruvato
(composto por 3 carbonos) é removido na forma de CO2 e os dois carbonos remanescentes
tornam-se o grupo Acetil da acetil-CoA. Outros aminoácidos e ácidos graxos sem a
formação intermediária de piruvato também produzem acetil-CoA, sendo este o ponto de
convergência do metabolismo degradativo de carboidratos, aminoácidos e ácidos graxos.
A partir de Acetil-CoA, a via glicolítica passa a apresentar dois papéis:
Gerar ATP.
Fornecer componentes para a síntese de ácidos graxos e outras substâncias
lipídicas
e
seus
derivados
(Triglicérides,
Fosfolípides,
Pigmentos
Carotenóides, Colesterol e seus ésteres, Ácidos Biliares, Vitamina K e
Hormônios Esteróides).
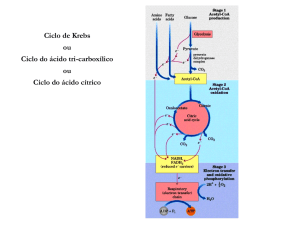
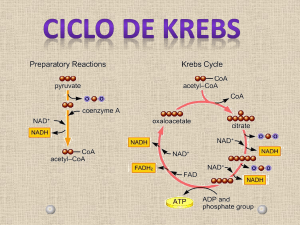
Na primeira reação do Ciclo de Krebs, a Acetil-CoA doa seu grupo Acetil a um
composto de 4 carbonos, denominado Oxalacetato, para formar um composto de 6
carbonos, o Citrato. O Citrato é então transformado em Isocitrato, também uma molécula de
6 carbonos, o qual é desidrogenado com perda de CO2 para produzir um composto de 5
carbonos, o a-Cetoglutarato. Este último submete-se a perda de uma molécula de CO2 e
adição de uma molécula de CoA (coenzima A) para produzir um composto de 4 carbonos o
Succinil-CoA. . Este é então convertido em Succinato a partir da liberação da CoA e
produção de GTP. O Succinato formado sofre outro processo de desidrogenação com
formação de FADH, como receptor dos prótons, formando o Fumarato. O Fumarato é
hidratado sendo convertido em Malato que, por sua vez, será desidrogenado, com formação
de NADH e Oxalacetato, reiniciando-se o Ciclo.
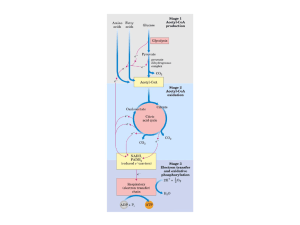
A energia liberada no Ciclo de Krebs é conservada em carreadores de elétrons
reduzidos denominados NADH (Nicotinamina-Adenina-Dinucleotídeo Hidrogenada) e FADH
(Flavina-Adenina-Dinucleotídeo).
Esses co-fatores reduzidos são oxidados produzindo
prótons (H+) e elétrons. Os elétrons são transferidos para o O2 ao longo de uma cadeia de
moléculas carreadoras de elétrons, conhecida como Cadeia Respiratória, onde haverá a
formação de H2O.
Durante esse processo de transporte de elétrons, energia é liberada e conservada na
forma de ATP, em um processo chamado de Fosforilação Oxidativa .
Metabolismo de Proteínas
Os principais constituintes das proteínas são os aminoácidos, compostos, por
definição, tanto um grupo amina quanto uma carboxila. Após serem absorvidos do jejuno
para o sangue, atingindo uma concentração normal de 35 a 65mg/dl, são transportados para
todas as células do organismo, sobretudo para o fígado, através de transporte ativo ou
facilitado.
Quando as células chegam no seu potencial máximo de armazenamento, inicia-se o
processo de degradação dos aminoácidos, que consiste na conversão e excreção do grupo
amino em uréia e oxidação da cadeia carbônica em piruvato, acetilCoA e intermediários do
ciclo de Krebs.
O processo de retirada do grupo amino dos aminoacidos é chamado de
desaminação e ocorre principalmente pela transaminação no fígado, durante a qual existe a
formação de amônia. Esta, por sua vez, é retirada do sangue através de sua conversão em
uréia, que é excretada na urina.
Após a remoção do grupo amino do aminoácido, o que resta é uma cadeia carbônica
na forma de α-cetoácido. Esta cadeia poderá ter como destinos finais, dependendo do tecido
e do estado fisiológico considerados, os seguintes caminhos:
ciclo de Krebs, fornecendo energia
utilização pela via gliconeogênese, para produção de glicose
conversão a triacilgliceróis e armazenamento
A maior parte dos aminoácidos produz piruvato ou intermediários do ciclo de Krebs
(oxaloacetato, α-cetoglutarato, succinil-CoA e fumarato), sendo chamados de glicogênicos,
pois são precursores da gliconeogênese. Apenas a leucina produz exclusivamente corpos
cetônicos, sendo cetogênico. Existem aminoácidos tanto glicogênicos quanto cetogênicos,
sendo então chamados de glicocetogênicos.
A degradação de cada aminoácido (fig. 3 e 4) e seu composto referente podem ser
agrupados de acordo com o produto formado, sendo:
A-Alanina, cisteína, glicina, serina, treonina, triptofano convertidos a piruvato
B-Asparagina, aspartato convertidos a oxaloacetato
C-Fenilalanina, tirosina, aspartato convertidos a fumarato
D-Isoleucina, valina, metionina, treonina convertidos a succinil-CoA
E-Glutamato, glutamina, prolina, arginina, histidina convertidos a α-cetoglutarato
F-Leucina, lisina, triptofano, fenilalanina, isoleucina, treonina, tirosina acetil-CoA
Aminoácido
s
Cadeia
Carbônica
Grupo Amino
Uréia
Piruvato
AcetilCoA
Intermediários do ciclo de Krebs.
Fig. 3 : Esquema geral da degradação de aminoácidos.
A
Piruvato
Acetil-CoA
F
α -Cetoglutarato
E
Succinil-CoA
D
B
Oxaloacetato
Fumarato
C
Fig. 4 : Esquema representando a degradação dos aminoácidos e seus produtos
formados.
Metabolismo de Lipídios
Os lipídios representam a maior reserva energética do organismo, fornecendo um
maior rendimento energético por grama de lipídeo metabolizado que os carboidratos. São
adquiridos a partir da dieta e endogenamente, sendo distribuídos por todo o organismo
através das lipoproteínas.
Os triacilgliceróis são os lipídios mais abundantes, sendo armazenados nas células
adiposas. Sua mobilização, quando necessário, ocorre pela ação da lipase dos adipócitos,
com liberação de glicerol e ácidos graxos. Tal reação química é demonstrada abaixo:
Triacilglicerol
Lipase
Glicerol
+
Ácidos Graxos
O glicerol, no fígado e tecidos, se transformará em um intermediário da glicólise ou
da gliconeogênese. Os ácidos graxos serão utilizados como fonte de energia nos tecidos, a
partir do processo de β-oxidação nas mitocôndrias, tendo como produto acetil-CoA. Durante
o jejum prolongado, acorre um aumento excessivo da mobilização de ácidos graxos,
havendo uma produção significativa de acetil-CoA e corpos cetônicos (ácido acetoacético, βhidroxibutírico e acetona).
O colesterol pode ser encontrado em todas as células do organismo, sendo obtido
através da dieta e endogenamente pela síntese no fígado, tendo como precursora a acetilCoA. Por volta de 80% do colesterol é transformado em ácido cólico, o qual participará da
formação de sais biliares. O restante é utilizado para a produção de hormônios, vitamina D e
estruturação de membranas.
Os lipídios, devido sua característica hidrofóbica, necessitam de lipoproteínas para
serem transportados aos tecidos e órgãos e poderem exercer suas funções metabólicas. As
lipoproteínas são divididas em cinco grupos:
Quilomícrons
Lipoproteína de muito baixa densidade (VLDL)
Lipoproteína de densidade intermediária (IDL)
Lipoproteína de baixa densidade (LDL)
Lipoproteína de alta densidade (HDL)
Os
quilomícrons
são
constituídos
principalmente
de
triglicerídeos
e
são
responsáveis pelo transporte exógeno a partir do tubo digestivo. Após sua absorção, sofrem
ação das lipoproteínas lípases, sendo hidrolisados a monoacilglicerol, glicerol e ácidos
graxos, que posteriormente serão utilizados para fornecimento de energia.
A VLDL também é rica em triglicerídeos, contendo cerca de 10% de colesterol,
sendo sintetizada pelo fígado.
A IDL é composta por quantidades proporcionais de colesterol e triglicerídeos, sendo
formada após hidrólise da VLDL, tendo vida curta.
A LDL é sintetizada no fígado e em algumas células específicas no organismo, sendo
fonte importante para síntese de hormônios esteróides e possuindo maior concentração de
colesterol. A remoção de cerca de 40 a 60% de LDL do plasma é feita através de sua
ligação com receptores na membrana celular; o restante é feito através do sistema celular
de limpeza. Em uma situação em que exista aumento dos níveis de LDL, as células
removedoras captam maior quantidade de lipoproteína. Devido a este mecanismo, as
células do músculo liso e os macrófagos da parede arterial se transformam em células
espumosas por conterem excesso de ésteres de colesterol, levando ao início da placa
aterosclerótica.
A HDL é composta por proteínas, fosfolipídeos e colesterol. Acredita-se que a
ingestão de grandes quantidades de colesterol estimule sua produção. A HDL exerce papel
importante na retirada de colesterol dos tecidos e retorno deste da periferia para o fígado.
Conclusão
Portanto, a partir dos temas discutidos, observamos a grande importância a respeito do
conhecimento e entendimento dos processos fisiológicos, para que também possamos
entender os estados patológicos e a atuação de medicamentos nestes.
Referências:
1. I Diretriz brasileira de diagnóstico e tratamento da síndrome metabólica. Arquivos
Brasileiros de Cardiologia, v. 84, supl 1, p. 28-01, 2005
2. Atualização brasileira sobre diabetes / Sociedade Brasileira de Diabetes. Rio de Janeiro :
Diagraphic, p 42-23, 2005
3. Burtis Carl A, Ashwood,Edward R. Tietz Fundamentos de Bioquímica Clínica 4º ed. Rio de
Janeiro: Guanabara-Koogan, p 363-234, 1998
4. ECKEL R, Grundy S, Zimmet P: The Metabolic Syndrome. Lancet 365: 1428-1415, 2005
5. Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults.
Executive summary of the third report of the national cholesterol education program (NCEP).
JAMA, 285: 2486-97, 2001
6. HALPERN A, Corrêa Mancini M: Obesidade. In: Antonio Carlos Lopes. Tratado de Clínica
Médica. São Paulo : Roca, p 3570-3555, 2006
7.Júnior, Armando Miguel. Diabetes no idoso – controle dietético e energético. 2007.
Disponível em http://www.medicinageriatrica.com.br/2007/07/25/ . Acesso em 18/04/2008.
8. KAHN R, Buse J, Ferrannini E, Stern M: The Metabolic Syndrome: Time for a Critical
Appraisal. Diabetes Care 28: 2304-2289, 2005
9.NELSON, D. L., COX, M. M. Lehninger: Princípios de bioquímica. 3 ed. São Paulo:
Sarvier, 2002. p. 269-96.
10.STRYER, L. Bioquímica. 4 ed. Rio de Janeiro: Guanabara-Koogan, 1996. p. 419-36.
11.VOET, D., VOET, J.G., PRATT, C.W. Fundamentos de bioquímica. Porto Alegre: Artmed,
2000. p. 353-81.