MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
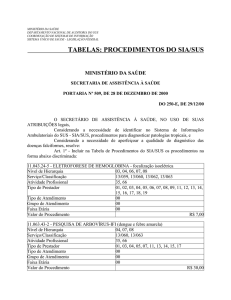
TABELAS: PROCEDIMENTOS DO SIA/SUS
MINISTÉRIO DA SAÚDE
SECRETARIA DE ASSISTÊNCIA À SAÚDE
PORTARIA Nº 347, DE 21 DE SETEMBRO DE 2000
DO 184-E, DE 22/9/00
O Secretário de Assistência à Saúde, no uso de suas atribuições legais,
Considerando a necessidade de garantir o acesso dos pacientes usuários do
Sistema Único de Saúde SUS a medicamentos considerados excepcionais, cuja relação consta
da tabela do Sistema de Informações Ambulatoriais SIA/SUS;
Considerando que os usuários destes medicamentos são pacientes crônicos
e/ou fazem seu uso por períodos prolongados e ainda o alto custo destes tratamentos;
Considerando a necessidade de estabelecer Protocolos Clínicos e Diretrizes
Terapêuticas para as diversas patologias em que estejam indicados tratamentos com os
medicamentos já citados, que contenham critérios de diagnóstico e tratamento, observando
ética e tecnicamente a prescrição médica, racionalizem a dispensação dos medicamentos
preconizados para o tratamento das doenças; regulamentem suas indicações e seus esquemas
terapêuticos e estabeleçam mecanismos de acompanhamento de uso e de avaliação de
resultados, garantindo assim a prescrição segura e eficaz;
Considerando a necessidade de que os Protocolos e Diretrizes Terapêuticas a
serem estabelecidos sejam fruto de consenso técnico e científico, que sejam formulados
dentro de rigorosos parâmetros de qualidade, precisão de indicação e posologia, que sejam
respaldados por estudos clínicos de fase 3, meta-análises de ensaios clínicos nacionais e/ou
internacionais, e
Considerando a necessidade de se promover ampla discussão destes Protocolos
e Diretrizes, possibilitando a participação efetiva da comunidade técnico científica,
sociedades médicas, profissionais de saúde e gestores do Sistema Único de Saúde na sua
formulação, resolve:
Art. 1º - Submeter à consulta pública as propostas de Protocolos Clínicos e
Diretrizes Terapêuticas contidas nos seguintes Anexos desta Portaria:
Anexo I - Esquizofrenia e Transtornos Esquizofreniformes Refratários :
Olanzapina;
Anexo II - Esquizofrenia e Transtornos Esquizofreniformes Refratários :
Risperidona;
Anexo III Esclerose Múltipla: Interfereon beta 1a e 1b e Acetato de Glatiramer;
Anexo IV- Fibrose Cística Enzimas Pancreáticas;
Anexo V Acromegalia: Ocreotida e Bromocriptina;
Anexo VI Hepatite B em Transplante Hepático: Lamivudina + HBIG.
Art. 2º - Estabelecer o prazo de 60 (sessenta) dias a contar da data da
publicação desta Portaria, para que sejam apresentadas sugestões, devidamente
fundamentadas, relativas às propostas de Protocolos Clínicos e Diretrizes Terapêuticas de que
trata o Artigo 1º.
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
§ 1º - As sugestões deverão ser encaminhadas, por escrito e em meio
magnético, para o Departamento de Sistemas e Redes Assistenciais Assessoria Farmacêutica
Ministério da Saúde, Esplanada dos Ministérios Bloco G sala 912 CEP 70.058-900 BrasíliaDF, ou para o endereço eletrônico - [email protected];
§ 2º - As sugestões enviadas deverão, obrigatoriamente, estar fundamentadas
por:
a - Estudos Clínicos de fase 3 realizados no Brasil ou exterior;
b - Meta-análises de Ensaios Clínicos.
§ 3º - As sugestões deverão ser acompanhadas por cópia dos documentos que
as fundamentem, conforme previsto no § 2º, sendo que, no caso de publicações estrangeiras,
as mesmas deverão ser enviadas na versão original, sem tradução.
Art. 3º - Determinar que o Departamento de Sistemas e Redes Assistenciais
avalie as proposições apresentadas, elaborando a versão final consolidada dos Protocolos
Clínicos e Diretrizes Terapêuticas ora submetidos à consulta pública, para que, findo o prazo
estabelecido, estes sejam publicados e entrem em vigor em todo o território nacional.
Art. 4º - Estabelecer que os medicamentos preconizados nos Protocolos ora
publicados que ainda não estejam incluídos na Tabela de Procedimentos do Sistema de
Informações Ambulatoriais do Sistema Único de Saúde, a saber Acetato de Glatiramer;
Lamivudina e HBIG, o serão após o término do período de consulta pública definido pela
presente Portaria.
Art. 5º - Esta Portaria entra em vigor na data de sua publicação.
RENILSON REHEM DE SOUZA
ANEXO I
PROTOCOLO E DIRETRIZES TERAPÊUTICAS
ESQUIZOFRENIA E TRANSTORNOS ESQUIZOFRENIFORMES REFRATÁRIOS
Medicamento: Olanzapina
1. Introdução:
Os transtornos esquizofreniformes refratários aos tratamentos convencionais foram
claramente caracterizados na portaria que preconiza seu tratamento com Clozapina e estão
citados abaixo.
Na impossibilidade de administração da Clozapina nestes pacientes, seja por efeito adverso
do tipo granulocitopenia ou convulsão, este fármaco pode ser substituído por Olanzapina. (1)
A olanzapina (10 a 20 mg/dia) foi superior à risperidona (4 to 12 mg/dia) no tratamento da
esquizofrenia. Em um estudo multicêntrico internacional, duplo-cego, prospectivo de 28
semanas com 339 pacientes a olanzapina produziu melhores respostas clínicas (mais de 40%
de diminuição nos sintomas positivos e negativos) e foi superior à risperidona no tratamento
dos sintomas negativos. (2)
2. Classificação CID 10: F20.3. Diagnóstico Clínico e Critérios de Inclusão no Protocolo de Tratamento:
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
As seguintes situações são requeridas, cumulativamente, como critérios de inclusão neste
protocolo:
3.1 - O paciente deve ter diagnóstico de Esquizofrenia pelo DSM-IV;
3.2 - A duração da doença deve ser maior do que 4 anos e o paciente não ter apresentado
nenhum funcionamento pessoal adequado há 2 anos;
3.3 - Ter apresentado falha terapêutica com Haloperidol 20-40 mg/d por 36 semanas ou com
Clorpromazina 1g/d por 36 semanas;
3.4 - Ter apresentado falha terapêutica com Thioridazina 400-800 mg/dia por 6 meses
consecutivos;
3.5 - Constar ausência de melhora dos sintomas apesar da mudança nos fatores psicossociais
estressores;
3.6 - Presença de familiar interessado, participativo, disponível e com adequado
funcionamento global;
3.7 - Paciente e familiar apresentarem-se aderentes a um serviço de atendimento psiquiátrico
ambulatorial;
3.8 - Haver adequada documentação e descrição detalhada de toda doença do paciente.
Obs. a presença de discinesia tardia como efeito colateral de neurolépticos é aceita como
critério único que permite a inclusão do paciente neste protocolo.
3.9 Ter apresentado como efeito adverso da clozapina: granulocitopenia (leucocitos totais
<3000 e/ou neutrófilos < 1500 e/ou plaquetas < 100.000/mm3)
3.10 Ter apresentado convulsão na vigência de uso de clozapina;
3.11 Paciente maior de 18 anos.
4. Critérios de Exclusão do Protocolo e Suspensão do Tratamento:
Não deverá ser incluído neste tratamento todo paciente que apresentar pelo menos uma das
situações abaixo, em que os riscos podem superar os benefícios:
4.1-São contra-indicações formais:
4.1.1- Hipersensibilidade à olanzapina;
4.1.2- Gravidez ou situação potencial de gravidez e ou lactação;
4.1.3 - História de tumor cerebral;
4.1.4 - Psicose alcoólica ou tóxica;
4.1.5 - Dependência ou abuso de drogas psicoativas;
4.1.6 - Ausência de família organizada e participante
4.1.7 - Retardo mental;
4.1.8 - Ausência de família organizada e participante;
4.1.9 - Impossibilidade de adesão e acompanhamento continuado;
4.2- São contra-indicações relativas:
4.2.1 - Doença hepática;
4.2.2 - História de tumor cerebral;
4.2.3 - Epilepsia ou condições que diminuam o limiar convulsivante;
4.2.4 - Câncer de mama ou história de;
4.2.5 - Glaucoma de ângulo estreito;
4.2.6 - História de íleo paralítico;
4.2.7 - Hiperplasia prostática significativa;
4.2.8 - Doença cardíaca ou cerebrovascular ou condições que predispõem à hipotensão;
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
4.2.9 - Paciente com risco de penumonia de aspiração;
4.2.10 - Pacientes com risco de suicídio;
4.2.11 - História de síndrome neuroléptica maligna;
5. Comitê Técnico:
Pacientes candidatos à inclusão neste protocolo deverão ser avaliados por um Comitê Técnico
composto de pelo menos 2 especialistas em psiquiatria, designados pelo gestor estadual. Os
especialistas designados deverão ser cadastrados junto ao Sistema de Farmacovigilância da
Olanzapina e deverão providenciar o preenchimento da escala BPRS de cada paciente.
6. Tratamento
Olanzapina
O tratamento deverá ser iniciado com 5 mg à noite. Pode-se aumentar a dose em 5 mg após
pelo menos 7 dias até uma dose de 10 mg/dia.
Não há evidências de que doses maiores de 10 mg/dia sejam mais eficazes. A dose máxima
permitida é de 20 mg/dia. (3)
Não é necessário ajuste de dose na insuficiência renal ou hepática.
Pacientes debilitados deverão receber no máximo 5 mg/dia.
7. Controle Clínico:
Recomendado controle de hipotensão ortostática e de parefeitos extrapiramidais.
8. Resposta Clínica :
A melhora Clínica é definida como uma diminuição de pelo menos 30% nos escores da
escala BPRS.(4) Conseguida a melhora clínica, deverá ser instituída uma redução cuidadosa
da dosagem na manutenção e acompanhamento clínico e psiquiátrico com escores trimestrais
(escala BPRS).
Pacientes que apresentarem hipersensibilidade ou intolerância deverão ser ser considerados
para inclusão no Protocolo de Risperidona.
9. Interrupção do Tratamento:
Terá indicação de interrupção de tratamento o paciente que, após12 semanas de uso de
20mg/d:
11.1 - Não apresentar melhora clínica;
11.2 - Não aderir ao tratamento e avaliações (preenchimento da escala BPRS);
11.3 - Apresentar efeitos adversos como: hipersensibilidade, hipotensão postural grave ou
doença sistêmica.
10. Consentimento Informado:
É obrigatória a orientação adequada do paciente, ou de seu responsável legal, dos potenciais
riscos e efeitos colaterais relacionados ao uso do medicamento preconizado neste protocolo, o
que deverá ser formalizado por meio da assinatura de Termo de Consentimento Informado,
de acordo com o modelo em anexo.
Bibliografia
1. Drug treatment for schizophrenia. Effective Health Care-NHS Center for Reviews and
Dissemination. 1999;5(6):1-12
2. Tran PV, Hamilton SH, Kuntz AJ et al: Double-blind comparison of olanzapine versus
risperidone in the treatment of schizophrenia and other psychotic disorders. J Clin
Psychopharmacol 1997; 17:407-418.
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
3. USP DI 2000. Information for the Health Care Professional. 20 ed. Englewood;
Micromedex Inc. 2000. Vol.1
4. Zuardi AV, Loureiro SN, Rodrigues CRC, et al. Estudo da estrutura fatorial, fidedignidade
e validade da tradução e adaptação para o português da escala de avaliação psiquiátrica breve
(BPRS) modificada. Revista ABP-PAL 1994;16(2):63-7
TERMO DE CONSENTIMENTO INFORMADO OLANZAPINA
O Paciente abaixo identificado e firmado declara para todos os efeitos legais que foi
informado de todos os benefícios, contra-indicações, potenciais efeitos colaterais, riscos e
advertências relativos ao uso do medicamento OLANZAPINA, preconizado para o
tratamento da esquizofrenia e de transtornos esquizofreniformes refratários.
Expressa, ainda, sua concordância e vontade em submeter-se ao tratamento preconizado já
referido, assumindo inteira responsabilidade e risco pelos eventuais efeitos indesejáveis que
venham a ocorrer em decorrência do mesmo.
Assim declara que:
Foi claramente informado que a medicação OLANZAPINA pode trazer os seguintes
benefícios no tratamento da esquizofrenia:
- Redução dos sintomas de sua doença e da freqüência das crises e internações hospitalares.
Foi também claramente informado a respeito das seguintes contra-indicações, potenciais
efeitos colaterais, riscos e advertências a respeito da medicação OLANZAPINA no
tratamento da esquizofrenia:
- Contra-indicação: previa hipersensibilidade à olanzapina.
- Medicações classificada na gestação como fator de risco C (significa que o risco para o bebê
não pode ser descartado mas o potencial benefício pode suplantar os riscos);
- Ajuste de dose pode ser necessário nos casos de insuficiência hepática, mas não é necessário
na insuficiência renal; A olanzapina não é removida da circulação por métodos de diálise;
- Os efeitos adversos mais comuns com a olanzapina são sonolência (26%), agitação (23%),
insônia (20%), nervosismo (16%), hostilidade (15%), tonturas (11%), ansiedade (9%),
acatisia (5%), hipertonia (4%);
- Menos freqüentemente ocorrem constipação, boca seca, rinite, and elevação de enzimas
hepáticas;
- Sintomas extrapiramidais ocorrem em menos de 10% dos pacientes tratados, uma incidência
menor do que a observada com o haloperidol;
- Aumento da prolactina também ocorre menos com a olanzapina do que com o haloperidol;
- Apetite excessivo ocorre mais com olanzapina do que com o haloperidol (24% versus
12.4%);
- Após um período médio de 16 meses de tratamento com olanzapina, os níveis de
trigliderídeos aumentam de mais ou menos 170 mg/dL para em torno de 240 mg/dL. Neste
período, os pacientes têm em média um ganho de peso de 10 kg;
- Efeitos adversos cardiovasculares: síncope (0.6%), hipotensão postural (5%), taquicardia
(4%), hipotensão (2%), dor no peito (4%), edema periférico (3%);
- Precauções: história de síndrome neuroléptica maligna; doença hepática; convulsões ou
condição que diminua o limiar convulsivante; patientes com risco de pneumonia de
aspiração; pacientes com hipertrofia prostática, glaucoma de ângulo fechado; historia de íleo
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
paralítico; gravidez; pacientes com doença cardiovascular, cerebrovascular, ou condicões que
predispoem a hipotensão postural; historia de câncer de mama; pacientes com risco de
suicídio.
- Alterações hematológicas significativas não foram descritas entretanto a experiência clínica
com a olanzapina especialmente a longo prazo premanece limitada. Há relatos de alguns
casos que apresentaram redução do número de neutrófilos com clozapina e que quando
receberam olanzapina apresentaram novamente esta redução.
O Paciente declara, ainda, estar ciente que pode suspender este tratamento a qualquer
momento, sem que este fato implique em qualquer forma de constrangimento entre ele e seu
médico, que se dispõe a continuar tratando-o em quaisquer circunstâncias.
Assim, o paciente faz sua adesão ao tratamento de forma livre, por espontânea vontade e por
decisão conjunta dele e de seu médico.
Paciente:________________________________________________
Responsável Legal (quando for o caso):______________________
Sexo do paciente: ( )Masculino ( )Feminino Idade do Paciente:___
R.G. (do paciente ou responsável legal)______________________
Endereço:_______________________________________________
Cidade: _________CEP:___________ Telefone:( )____________
__________________________
Assinatura do Paciente
_________________________
Assinatura do Responsável (quando for o caso)
Médico Responsável: ____________________CRM:_____________
Endereço do Consultório:___________________________________
Cidade:_____________ CEP: ____________ Telefone: ( )________
___________________________
Assinatura e Carimbo do Médico
_____/______/_________
Data
Obs.:
1- O preenchimento completo deste Termo e sua respectiva assinatura são imprescindíveis
para o fornecimento do medicamento
2- Este Termo ficará arquivado na farmácia responsável pela dispensação dos medicamentos
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
ANEXO II
PROTOCOLO E DIRETRIZES TERAPÊUTICAS
ESQUIZOFRENIA E TRANSTORNOS ESQUIZOFRENIFORMES REFRATÁRIOS
Medicamento: Risperidona
1.Introdução:
Os transtornos esquizofreniformes refratários aos tratamentos convencionais foram
claramente caracterizados na portaria que preconiza seu tratamento com Clozapina como
fármaco de primeira linha e estão citados abaixo.
Na impossibilidade de administração da Clozapina ou, como segunda escolha, a Olanzapina
nestes pacientes, seja por efeito adverso ou por falha terapêutica, pode ser usada a
Risperidona.(1)
A Risperidona é um aderivado benzisoxazol que bloqueia os receptores da serotonina (5HT2) e da dopamina D(2). Tem efeito clínico comparável à clozapina. Em um estudo duplocego randomizado, os pacientes foram divididos em 3 grupos recebendo 4 miligramas de
risperidona, 8 miligramas de risperidone, ou 400 miligramas de clozapina por dia durante 28
dias. Os efeitos antipsicóticos foram significativos tanto para risperidona como para a
clozapina. O pacientes do grupo de 4 miligramas de risperidona toleraram melhor o
tratamento do que aqueles com clozapina. a suspensão da clozapina forampeincipalmente
devidas a efeitos adversos, enquanto que na risperidona a maior causa de suspensão foi
devida a falta de resposta clínica. (2)
2. Classificação CID 10: F20.3. Diagnóstico Clínico e Critérios de Inclusão no Protocolo de Tratamento:
As seguintes situações são requeridas, cumulativamente, como critérios de inclusão neste
Protocolo:
3.1 - O paciente deve ter diagnóstico de Esquizofrenia pelo DSM-IV;
3.2 - A duração da doença deve ser maior do que 4 anos e o paciente não ter apresentado
nenhum funcionamento pessoal adequado há 2 anos;
3.3 - Ter apresentado falha terapêutica com Haloperidol 20-40 mg/d por 36 semanas ou com
Clorpromazina 1g/d por 36 semanas; Em caso suspeita de não adesão as soluções
intramusculares de depósito devem ser tentadas para caracterizar a ausência de resposta a
estes fármacos.
3.4 - Ter apresentado falha terapêutica com Thioridazina 400-800 mg/dia por 6 meses
consecutivos;
3.5 - Constar ausência de melhora dos sintomas apesar da mudança nos fatores psicossociais
estressores;
3.6 - Presença de familiar interessado, participativo, disponível e com adequado
funcionamento global;
3.7 - Paciente e familiar apresentarem-se aderentes a um serviço de atendimento psiquiátrico
ambulatorial;
3.8 - Haver adequada documentação e descrição detalhada de toda doença do paciente.
Obs. a presença de discinesia tardia como efeito colateral de neurolépticos é aceita como
critério único que permite a inclusão do paciente neste Protocolo.
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
3.9 Ter apresentado como efeito adverso da clozapina: granulocitopenia (leucocitos totais
<3000 e/ou neutrófilos < 1500 e/ou plaquetas < 100.000/mm3)
3.10 Ter apresentado convulsão na vigência de uso de clozapina;
3.11 Ter apresentado falha terapêutica ou efeitos adeversos que justifiquem a suspensão da
Olanzapina ou Clozapina.
3.12 Paciente maior de 18 anos.
4. Critérios de Exclusão do Protocolo e Suspensão do Tratamento:
Não deverá ser incluído neste tratamento todo paciente que apresentar pelo menos uma da
situações abaixo, em que os riscos podem superar os benefícios:
4.1- Contra-indicações formais:
4.1.1 - Hipersensibilidade à risperidona
4.1.2 - Hiperprolactinemia
4.1.3 - Crianças e adolescentes
4.1.4 - Gravidez ou situação potencial de gravidez e ou lactação;
4.1.5 - História de tumor cerebral;
4.1.6 - Psicose alcoólica ou tóxica;
4.1.7 - Dependência ou abuso de drogas psicoativas;
4.1.8 - Ausência de família organizada e participante
4.1.9 - Impossibilidade de adesão e acompanhamento continuado.
4.2- Contra-indicações relativas:
4.2.1 - Síndrome neuroléptica maligna
4.2.2 - Discinesia tardia
4.2.3 - Prolongamento do intervalo QT do eletrocardiograma
4.2.4 - Doença cardiovascular ou cerebrovascular que predispõe a hipotenão ortostática
4.2.5 - Hipothermia ou hipertermia;
4.2.6 - Diagnóstico prévio de câncer de mama ou tumor dependente de prolactina;
4.2.7 - Insuficiência renal;
4.2.8 - Insuficiência hepatic;
4.2.9 - Doença de Parkinson;
4.2.10 - História de convulsão ou epilepsia;
4.2.11 - Alterações patológicas nas contagens sanguíneas;
4.2.12 - Retardo mental;
5. Comitê Técnico:
Pacientes candidatos à inclusão neste Protocolo deverão ser avaliados por um Comitê
Técnico composto de pelo menos 2 especialistas em psiquiatria, designados pelo gestor
estadual. Os especialistas designados deverão ser cadastrados junto ao Sistema de
Farmacovigilância da Olanzapina e deverão providenciar o preenchimento da escala BPRS de
cada paciente.
6. Tratamento
Risperidona
Iniciar com 1 miligrama duas vezes ao dia para evitar efeito de primeira dose (bloquei alfaadrenérgico). A dose pode ser aumentada em 1 miligrama duas vezes ao dia até uma dose
alvo de 6 miligramas por dia (3 mg duas vezes ao dia) seja alcançada no terceiro dia.
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
As doses recomendadas são de 4 to 8 mg/dia. A segurança de doses acima de 16mg/dia não
foram avaliadas suficientemente em ensaios clínicos.
A dose máxima permitida é de 16 mg/dia. (3) Doses acima de 6 mg/dia em geral não são
recomendadas pelo laboratório fabricante e estão mais associadas a efeitos adversos
extrapiramidais e sedativos.
Se descontinuada a Risperdona deve ser reiniciada conforme primeira dose (acima). Pacientes
com insuficiência renal, hepática ou debilitados a dose máxima recomendada é de 3 mg/dia.
A administração simultânea com alimentos não interfere na biodisponibilidade da
risperidona.
7. Controle Clínico:
Sedação é o efeito adverso mais prevalente com Risperidona. Outros efeitos adversos
incluem cefaléia, boca seca, constipação, visão borrada e retenção urinária. Palpitações,
nervosismo e lombalgia podem ocorrer.
8. Resposta Clínica :
A melhora Clínica é definida como uma diminuição de pelo menos 30% nos escores da
escala BPRS. (4)
Conseguida a melhora clínica, deverá ser instituída a menor dose eficaz no tratamento de
manutenção. O acompanhamento clínico e psiquiátrico deve envolver o preenchimento da
escala BPRS a cada 3 meses.
9. Interrupção do Tratamento:
Terá indicação de interrupção de tratamento o paciente que, após12 semanas de uso de 16
mg/d:
7.1 - Não apresentar melhora clínica;
7.2 - Não aderir ao tratamento e avaliações (preenchimento da escala BPRS);
7.3 -Apresentar efeitos adversos como: hipersensibilidade, hipotensão postural grave ou
doença sistêmica.
10. Consentimento Informado:
É obrigatória a orientação adequada do paciente, ou de seu responsável legal, dos potenciais
riscos e efeitos colaterais relacionados ao uso do medicamento preconizado neste Protocolo,
o que deverá ser formalizado por meio da assinatura de Termo de Consentimento Informado,
de acordo com o modelo em anexo.
Bibliografia
1. Drug treatment for schizophrenia. Effective Health Care-NHS Center for Reviews and
Dissemination. 1999;5(6):1-12
2. Heinrich K, Klieser E, Lehmann E et al: Risperidone versus clozapine in the treatment of
schizophrenic patients with acute symptoms: a double blind, randomized trial. Prog NeuroPsychopharmacol Biol Psychiat 1994; 18:129-137.
3. USP DI 2000. Information for the Health Care Professional. 20 ed. Englewood;
Micromedex Inc. 2000. Vol.1
4. Zuardi AV, Loureiro SN, Rodrigues CRC, et al. Estudo da estrutura fatorial, fidedignidade
e validade da tradução e adaptação para o português da escala de avaliação psiquiátrica breve
(BPRS) modificada. Revista ABP-PAL 1994;16(2):63-7
TERMO DE CONSENTIMENTO INFORMADO RISPERIDONA
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
O Paciente abaixo identificado e firmado declara para todos os efeitos legais que foi
informado de todos os benefícios, contra-indicações, potenciais efeitos colaterais, riscos e
advertências relativos ao uso do medicamento RISPERINONA, preconizado para o
tratamento dos transtornos esquizofrênicos refratários.
Expressa, ainda, sua concordância e vontade em submeter-se ao tratamento preconizado já
referido, assumindo inteira responsabilidade e risco pelos eventuais efeitos indesejáveis que
venham a ocorrer em decorrência do mesmo.
Assim declara que:
- Foi claramente informado que a medicação RISPERINONA pode trazer os seguintes
benefícios no tratamento: redução dos sintomas de sua doença e da freqüência das crises e
internações hospitalares.
Foi também claramente informado a respeito das seguintes contra-indicações, potenciais
efeitos colaterais, riscos e advertências a respeito da medicação RISPERINONA no
tratamento dos transtornos esquizofrênicos refratários.
- Medicação classificada na gestação como fator de risco C (significa que o risco para o bebê
não pode ser descartado mas o potencial benefício pode suplantar os riscos);
- Contra-indicado em casos de hipersensibilidade (alergia) componentes da formulação;
- Efeitos adversos relatados (entre 1-10%) são hipotensão (especialmente ortostática),
taquicardia, arritmias, anormalidades eletrocardiográficas (na repolarização ventricular),
síncope, sedação, cefaléia, tonturas, ansiedade, reações extrapiramidais, reações distônicas,
parkinsonismos, discinesia tardia, síndrome neuroléptica maligna, alteração na regulação
central de temperatura, fotosensibulidade, amenorréia, galactorréia, ginecomastia, disfunção
sexual, constipação, íleo adinâmico, xerostomia, náuseas e vômitos, ganho de peso, retenção
urinária, incontinência urinária priaprismo, agranulocitose, leucopenia, icterícia colestática,
borramento da visão, pigmentação da retina, diminuição da acuidade visual;
- Convulsões podem raramente (<1%) ocorrer;
- O risco de ocorrer superdosagem é pequeno e, quando ocorre, é bem tolerado;
O Paciente declara, ainda, estar ciente que pode suspender este tratamento a qualquer
momento, sem que este fato implique em qualquer forma de constrangimento entre ele e seu
médico, que se dispõe a continuar tratando-o em quaisquer circunstâncias.
Assim o paciente faz sua adesão ao tratamento de forma livre, por espontânea vontade e por
decisão conjunta dele e de seu médico.
Paciente:______________________________________________
Responsável Legal (quando for o caso):______________________
Sexo do paciente: ( )Masculino ( )Feminino Idade do Paciente:__
R.G. (do paciente ou responsável legal)_______________________
Endereço:_______________________________________________
Cidade: __________CEP:___________ Telefone:( )____________
__________________________ Assinatura do Paciente
_________________________ Assinatura do Responsável (quando for o
caso)
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
Médico Responsável: ____________________CRM:_____________
Endereço do Consultório:___________________________________
Cidade:______________ CEP: ___________ Telefone: ( )________
___________________________
Assinatura e Carimbo do Médico
_____/______/_________
Data
Obs.:
1- O preenchimento completo deste Termo e sua respectiva assinatura são imprescindíveis
para o fornecimento do medicamento
2- Este Termo ficará arquivado na farmácia responsável pela dispensação dos medicamentos
ANEXO III
PROTOCOLO E DIRETRIZES TERAPÊUTICAS
ESCLEROSE MÚLTIPLA
Medicamentos: Intereferon Beta 1a ou 1b ou Acetato de Glatiramer (Copolímero-1)
1- Introdução:
A esclerose múltipla (EM) é uma doença de caráter geralmente progressivo na qual a
inflamação e desmielinização da substância branca do sistema nervoso central resulta em
vários sinais e sintomas neurológicos. Após 10 anos do início dos sintomas 50% dos
pacientes estarão inaptos para fazer atividades caseiras e de trabalho. (1)
Apesar de ser considerada como uma doença autoimune, a resposta clínica aos
imunossupressores tem sido desapontadora.(2) A introdução recente de imunomoduladores
como o interferon beta produziu diminuição da frequência e severidade das recidivas e talvez
da progressão da doença. (3)Em pacientes ambulatoriais portadores da forma "surto-remissão"
o interferon beta 1a ou 1b diminui a frequência dos surtos.(4,5)
Apesar de 10% dos pacientes apresentarem uma evolução favorável, recomenda-se iniciar
tratamento o mais precoce possível a todos os casos. Entretanto, na forma progressiva da
doença, as evidências da eficácia deste tratamento são apenas de pequenas séries de casos
(nível C).(6)
O advento de Acetato de Glatiramer representou um avanço importante, sendo recomendado
como fármaco de escolha para casos de falha ao interferon, seja por ausência de resposta
clínica, seja por efeitos adversos. Ensaios clínicos apresentam um perfil de tolerância muito
bom, sem alterações laboratoriais ou efeitos adversos endocrinológicos descritos.(7)
2- Classificação CID 10:
3- Diagnóstico e Critérios de Inclusão:
As seguintes situações são requeridas, cumulativamente, como critérios de inclusão neste
Protocolo:
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
3.1 - Portador de Esclerose Múltipla Definitiva caracterizada clinica ou laboratorialmente
pelos critérios de Poser e colaboradores; (8)
3.2 - Idade entre 18 a 50 anos;
3.3 - Portadores de esclerose múltipla forma definida clinicamente como "surto-remissão";
3.4 - Doença caracterizada como ativa: pela história clínica e ou por neuroimagem com
ressonância magnética;
3.5 - Apresentar pontuação na escala EDSS de 0 a no máximo 6,5; (9)
3.6 - Ter capacidade de deambular com ou sem ajuda;
3.7 - Ter apresentado pelo menos 2 surtos antes do início do tratamento;
3.8 - Paciente ou familiar capaz de assegurar que a adesão ao tratamento será mantida e que a
monitorização dos efeitos adeversos será adequadamente realizada tanto pela família como
pelo médico neurologista prescritor;
3.9 - Paciente foi avaliado por neurologista em Centro Especializado em Esclerose Múltipla
credenciado pelo Comitê Brasileiro de Tratamento e Pesquisa em Esclerose Múltipla e pelo
gestor estadual;
3.10 - Foram excluídas outras doenças que possam se assemelhar a Esclerose Múltipla.
4- Critérios de Exclusão:
Serão excluídos deste protocolo de tratamento todos os pacientes que apresentarem pelo
menos uma das situações abaixo:
4.1 - Esclerose Múltipla forma progressiva;
4.2 - Esclerose Múltipla forma surto-remissão caracterizada como doença muito avançada:
com pontuação na escala EDSS maior do que 6,5;
4.3 - Pacientes do sexo feminino onde a possibilidade de concepção não pode ser
adequadamente controlada;
5- Centros de Referência:
Recomenda-se a criação de Centros de Referência a serem cadastrados pelo gestor estadual
para avaliação diagnóstica e quantificação da disfunção dos pacientes com Esclerose
Multipla. Este trabalho será realizado por neurologistas clínicos cuja aferição será
considerada como indispensável para a dispensação dos medicamentos.
6- Benefícios esperados com o tratamento:
- Melhora sintomática;
- Diminuição da frequência e severidade das recidivas;
- Redução do número de internações hospitalares.
7- Apresentação:
Existem 3 diferentes produtos disponíveis contendo interferon beta recombinante:
- Interferon Beta 1b administração subcutânea
- Interferon Beta 1a administração intramuscular
- Interferon Beta 1a administração subcutânea
O IFN beta-1a é a forma glicosilada e mais semelhante ao IFN humano e mais hidrosolúvel.
Estudo recente demonstrou a superioridade do IFN beta-1b sobre a resposta de marcadores
plasmáticos da atividade do interferon.(10) Entretanto não há evidência de que estes achados
se traduzam em superioridade clínica.(11) Assim adotamos a assertiva da eficácia clínica
semelhante das diferentes formas comerciais de interferon beta no tratamento da esclerose
múltipla.
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
O copolímero-1 ou glatiramer acetato ou copaxone 20 mg existe na apresentação de ampolas
para injeção sub-cutânea, sendo esta a única via de administração. Não pode ser usada
intramuscular ou intravenosamente.
8- Tratamento:
Esquema de administração: os 3 esquemas terapêuticos utilizando interferon beta abaixo
serão aceitos.
Produto
IFN beta-1b
IFN beta-1a
IFN beta-1a
Dose
8 MUI
12 MUI
6 MUI
Via
Subcutânea
Subcutânea
Intramuscular
intervalo
cada 2 dias
3 vezes por semana
uma vez por semana
Conteúdo de IFNbeta
250mg
44mg
30mg
Nos casos preconizados no presente Protocolo, poderá ser utilizado o Acetato de Glatiramer, da
seguinte forma:
Produto
Acetato de Glatiramer
Dose
20 mg
Via
Subcutânea
Intervalo
diariamente
9- Interrupção do tratamento:
Não há critérios definitivos na literatura para decidir sobre a suspensão do tratamento exceto
na vigência de efeitos adversos graves ao interferon.
10- Monitorização:
Recomenda-se hemograma e provas de função hepática avaliados mensalmente nos primeiros
6 meses e após a critério clínico. O uso deve ser interrompido em caso de aparecimento de
efeitos colaterais graves.(12)
Não há necessidade de exame do líquor ou ressonância magnética para o acompanhamento
clínico destes pacientes.
Depressão, hepatite e hipersensibilidade são parefeitos potenciais.
11- Tempo de tratamento: indeterminado.(12)
12- Consentimento Informado
É obrigatória a orientação adequada do paciente, ou de seu responsável legal, dos potenciais
riscos e efeitos colaterais relacionados ao uso do medicamento preconizado neste Protocolo,
o que deverá ser formalizado por meio da assinatura de Termo de Consentimento Informado,
de acordo com os modelos em anexo.
Referências:
1. Rudick RA. Disease Modifying drugs for relapsing-remitting multiple scleorsis and future
directions for multiple sclerosis therapeutics. Arch Neurol 1999;56(9):1079-84
2. Giovannoni G, Miller DH. Multiple Sclerosis and its treatment. J R Coll Physicians Lond
1999;33(4):315-22
3. PRISMS (Prevention of Relapses and Disability by Interferon b-1a Subcutaneouly in
Multiple Sclerosis). Randomized double-blind placebo-controlled study of interferon b-1a in
relapsing/remitting multiple sclerosis. Lancet 1998;352:1498-504
4. van Oosten BW, Truyen L, Barkhof F, et al. Choosing drug therapy form multiple
sclerosis. An update. Drugs. 1988;56(4):555-69
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
5. Rice GP, Oger J, Duquette P, et al. Treatment with interferon beta-1b improves quality of
life in multiple sclerosis. Can J Neurol Sci 1999;26(4):276-82
6. Patti F, L'Episcopo MR, Cataldi ML, et al. Natural interferon-beta treatment of relapsingremitting and secondary-progressive multiple sclerosis patients. A two year study. Acta
Neurol Scand 1999;100(5):283-9
7. Johnson KP. Therapy of relapsing forms. In: Burks JS, Johnson KP. Multiple Sclerosis.
Demos Med.Plubish.Inc. New York. 2000
8. Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis:
guidelines for research protocols. Ann Neurol 1983;13:227-31
9. Kurtzke JF. Rating neurological impairment in multiple sclerosis: na expanded disability
status scale (EDSS). Neurology 1983;33:1444-52
10. Deisenhammer F, Mayringer A, Dilitz E, et al. A comparative study of the relative
bioavailability of different interferon beta preparations. Neurology 2000;54:2055-60
11. RederAT, Antel JP. Injecting rationale into interferon-B therapy. Neurology
2000;54:2034-5
12. Brazilian Committee for the Treatment and Research in Multiple Sclerosis. O Consenso
do BCTRIMS. BCTRIMS News. 2000;1(maio):1
TERMO DE CONSENTIMENTO INFORMADO INTERFERON BETA 1 a
O Paciente abaixo identificado e firmado declara para todos os efeitos legais que foi
informado de todos os benefícios, contra-indicações, potenciais efeitos colaterais, riscos e
advertências relativos ao uso do medicamento INTERFERON BETA 1A, preconizado para o
tratamento da esclerose múltipla.
Expressa, ainda, sua concordância e vontade em submeter-se ao tratamento preconizado já
referido, assumindo inteira responsabilidade e risco pelos eventuais efeitos indesejáveis que
venham a ocorrer em decorrência do mesmo.
Assim, declara que:
Foi claramente informado que a medicação INTERFERON BETA 1A pode trazer os
seguintes benefícios no tratamento esclerose múltipla:
- Melhora sintomática, diminuição da frequência e severidade das recidivas. Redução do
número de internações hospitalares.
Foi também claramente informado a respeito das seguintes contra-indicações, potenciais
efeitos colaterais, riscos e advertências a respeito da medicação INTERFERON BETA 1A no
tratamento esclerose múltipla.
- Medicação classificada na gestação como fator de risco C (significa que risco para o bebê
não pode ser descartado, mas um benefício potencial pode ser maior que os riscos);
- O interferon beta 1A deve ser evitado durante a lactação, uma vez que não existem trabalhos
avaliando a sua segurança nesta situação;
- Contra-indicado em casos de hipersensibilidade (alergia) a interferon beta natural ou
recombinante, albumina humana ou outro componente da formulação;
- Interferon beta 1A deve ser utilizado com cautela em pacientes com história de depressão,
convulsões, supressão de medula óssea ou doenças cardíacas;
- Efeitos adversos comuns (1-10%) são taquicardia, síncope, insuficiência cardíaca (raro),
cefaléia, letargia, convulsões, labilidade emocional, ansiedade, ideação suicida, sonolência,
agitação e confusão; também pode ocorrer alopécia, náuseas, vômitos, diarréia, perda de
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
peso, leucopenia, trombocitopenia, anemia, elevação de transaminases hepáticas (leve e
transitória), dor no local de administração (até 80%), fraqueza, alterações das provas de
função renal, sintomas tipo gripais (febre, náuseas, dores musculares);
- Dispnéia e sinusites já foram descritos com o uso do INF-beta 1A;
- O risco de ocorrer efeitos adversos aumenta com a superdosagem;
Paciente declara, ainda, estar ciente que pode suspender este tratamento a qualquer momento,
sem que este fato implique em qualquer forma de constrangimento entre ele e seu médico,
que se dispõe a continuar tratando-o em quaisquer circunstâncias.
Assim o paciente faz sua adesão ao tratamento de forma livre, por espontânea vontade e por
decisão conjunta dele e de seu médico.
Paciente:_____________________________________________
Responsável Legal (quando for o caso):____________________
Sexo do paciente: ( )Masculino ( )Feminino Idade do Paciente:___
R.G. (do paciente ou responsável legal)______________
Endereço:_______________________________________________
Cidade: __________CEP:___________ Telefone:( )___________
__________________________
Assinatura do Paciente
_________________________
Assinatura do Responsável (quando for o caso)
Médico Responsável: ____________________CRM:_____________
Endereço do Consultório:____________________________________
Cidade:______________ CEP: ____________ Telefone: ( )_______
___________________________
Assinatura e Carimbo do Médico
_____/______/_________ Data
Obs.:
1 - O preenchimento completo deste Termo e sua respectiva assinatura são imprescindíveis
para o fornecimento do medicamento
2 - Este Termo ficará arquivado na farmácia responsável pela dispensação dos medicamentos
TERMO DE CONSENTIMENTO INFORMADO
INTERFERON BETA 1 b
O Paciente abaixo identificado e firmado declara para todos os efeitos legais que foi
informado de todos os benefícios, contra-indicações, potenciais efeitos colaterais, riscos e
advertências relativos ao uso do medicamento INTERFERON BETA 1B, preconizado para o
tratamento esclerose múltipla.
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
Expressa, ainda, sua concordância e vontade em submeter-se ao tratamento preconizado já
referido, assumindo inteira responsabilidade e risco pelos eventuais efeitos indesejáveis que
venham a ocorrer em decorrência do mesmo.
Assim, declara que:
Foi claramente informado que a medicação INTERFERON BETA 1B pode trazer os
seguintes benefícios no tratamento esclerose múltipla.
- Melhora sintomática, diminuição da frequência e severidade das recidivas. Redução do
número de internações hospitalares.
Foi também claramente informado a respeito das seguintes contra-indicações, potenciais
efeitos colaterais, riscos e advertências a respeito da medicação INTERFERON BETA 1B no
tratamento esclerose múltipla.
- Medicação classificada na gestação como fator de risco C (significa que risco para o bebê
não pode ser descartado, mas um benefício potencial pode ser maior que os riscos);
- O interferon beta 1B deve ser evitado durante a lactação, uma vez que não existem trabalhos
avaliando a sua segurança nesta situação;
- Precaução deve ser tomada em pacientes com depressão e/ou ideação suicida, com
supressão de medula óssea, com doenças cardiovasculares e pulmonares, pacientes diabéticos
propensos a cetoacidose diabética, alterações neuropsiquiátricas ou convulsões, doença renal
ou hepática e infecções por herpes zoster;
- Contra-indicado em casos de hipersensibilidade (alergia) a interferon beta natural ou
recombinante, albumina humana ou outro componente da formulação e a produtos derivados
da E. coli;
- Efeitos adversos relatados são reações locais no sítio de administração, sintomas tipo
gripais, desordens menstruais, depressão (inclusive com ideação suicida), ansiedade, fadiga,
tonturas, letargia, insônia, sonolência, palpitações, dor torácica, desordens vasculares
periféricas, hipertensão, insuficiência cardíaca, leucopenia, anemia e trombocitopenia,
discrasias sangüíneas, dispnéia, laringite, cistite, queixas gastrointestinais, convulsões,
cefaléia e alterações de transaminases hepáticas
- O risco de ocorrer efeitos adversos aumenta com a superdosagem;
Paciente declara, ainda, estar ciente que pode suspender este tratamento a qualquer momento,
sem que este fato implique em qualquer forma de constrangimento entre ele e seu médico,
que se dispõe a continuar tratando-o em quaisquer circunstâncias.
Assim o paciente faz sua adesão ao tratamento de forma livre, por espontânea vontade e por
decisão conjunta dele e de seu médico.
Paciente:________________________________________
Responsável Legal (quando for o caso):___________________
Sexo do paciente: ( )Masculino ( )Feminino Idade do Paciente:___
R.G. (do paciente ou responsável legal)______________________
Endereço:_______________________________________________
Cidade: __________CEP:__________ Telefone:( )____________
__________________________
_________________________
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
Assinatura do Paciente
Assinatura do Responsável (quando for o caso)
Médico Responsável: ____________________CRM:_____________
Endereço do Consultório:__________________________
Cidade:______________ CEP: ____________ Telefone: ( )_______
___________________________
Assinatura e Carimbo do Médico
_____/______/_________
Data
Obs.:
1- O preenchimento completo deste Termo e sua respectiva assinatura são imprescindíveis
para o fornecimento do medicamento
2- Este Termo ficará arquivado na farmácia responsável pela dispensação dos medicamentos
TERMO DE CONSENTIMENTO INFORMADO
ACETATO DE GLATIRAMER (COPOLÍMERO-1)
O Paciente abaixo identificado e firmado declara para todos os efeitos legais que foi
informado de todos os benefícios, contra-indicações, potenciais efeitos colaterais, riscos e
advertências relativos ao uso do medicamento ACETATO DE GLATIRAMER
(COPOLÍMERO-1), preconizado para o tratamento da esclerose múltipla.
Expressa, ainda, sua concordância e vontade em submeter-se ao tratamento preconizado já
referido, assumindo inteira responsabilidade e risco pelos eventuais efeitos indesejáveis que
venham a ocorrer em decorrência do mesmo.
Assim declara que:
Foi claramente informado que a medicação ACETATO DE GLATIRAMER
(COPOLÍMERO-1) pode trazer os seguintes benefícios no tratamento esclerose múltipla:
- Melhora sintomática, diminuição da frequência e severidade das recidivas. Redução do
número de internações hospitalares.
Foi também claramente informado a respeito das seguintes contra-indicações, potenciais
efeitos colaterais, riscos e advertências a respeito da medicação ACETATO DE
GLATIRAMER (COPOLÍMERO-1) no tratamento da esclerose múltipla:
- Medicação classificada na gestação como fator de risco B (significa que risco para o bebê é
pouco provável e os benefícios potenciais provavelmente sejam maiores que os riscos);
- Contra-indicado em casos de hipersensibilidade (alergia) componentes da formulação;
- Efeitos adversos comuns (10%) são dor e irritação no local da injeção, dor no peito (até
26%) e dores difusas;
- São também relatados (entre 1 e 10%) taquicardia, vasodilatação, flushing, ansiedade,
depressão, tonturas, eritema, urticária, eosinofilia, tremores, dispnéia, diaforese;
- O risco de ocorrer superdosagem é pequeno e, quando ocorre, é bem tolerado;
O paciente declara, ainda, estar ciente que pode suspender este tratamento a qualquer
momento, sem que este fato implique em qualquer forma de constrangimento entre ele e seu
médico, que se dispõe a continuar me tratando em quaisquer circunstâncias.
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
Assim, o paciente faz sua adesão ao tratamento de forma livre, por espontânea vontade e por
decisão conjunta dele e seu médico.
Paciente:_____________________________________________
Responsável Legal (quando for o caso):_________________
Sexo do paciente: ( )Masculino ( )Feminino
Idade do Paciente:_____
R.G. (do paciente ou responsável legal)_____________________
Endereço:_______________________________________________
Cidade: __________CEP:___________ Telefone:( )___________
Assinatura do Paciente
_________________________
Assinatura do Responsável (quando for o caso)
Médico Responsável: ____________________CRM:_____________
Endereço do Consultório:__________________________
Cidade:______________ CEP: ____________ Telefone: ( )_______
___________________________
Assinatura e Carimbo do Médico
_____/______/_________
Data
Obs.:
1- O preenchimento completo deste Termo e sua respectiva assinatura são imprescindíveis
para o fornecimento do medicamento
2- Este Termo ficará arquivado na farmácia responsável pela dispensação dos medicamentos
Anexo III Protocolo Esclerose Múltipla - Intef
ANEXO IV
PROTOCOLO E DIRETRIZES TERAPÊUTICAS
FIBROSE CÍSTICA
Medicamento: Enzimas Pancreáticas
1. Introdução:
Desordem genética (autossômica recessiva) caracterizada por infecções crônicas das vias
aéreas, que leva ao desenvolvimento de bronquiectasias, insuficiência pancreática exócrina e
disfunções intestinais, anormalidades das glândulas sudoríparas e disfunção urogenital(1). A
incidência estimada é de 1:3000 nascidos vivos entre caucasianos caindo para 1:17000 entre
afro-americanos e 1:90000 entre asiáticos.
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
Apresenta uma morbi-mortalidade muito elevada com apenas 34% dos pacientes chegando a
idade adulta e menos de 10% ultrapassando os 30 anos de idade sobrevida média é de 28
anos(1).
2. Classificação CID 10: E84.3. Diagnóstico Clínico:
A maioria dos pacientes apresenta-se sintomática nos primeiros anos de vida; 15% dos
pacientes, já nas primeiras 24 horas de vida, apresentam o íleo meconial. Outras
manifestações comuns nos primeiros dois anos de vida são sintomas respiratórios
predominantemente tosse e infiltrados pulmonares recorrentes e retardo no desenvolvimento.
Até 4% dos pacientes são diagnosticados somente na fase adulta.
O curso clínico da doença se caracteriza por períodos de remissão e exacerbação, com
aumento da freqüência e severidade das exacerbações com o passar do tempo. Pneumotórax é
comum ( 10%). Pequenas quantidades de sangue no escarro costumam estar associados a
infecções em pacientes com doença grave. Insuficiência respiratória e cor pulmonale são
eventos finais. Do aspecto do trato digestório, a síndrome do íleo meconial se apresenta com
distensão abdominal, impossibilidade de evacuação e vômitos. Adultos também podem
apresentar quadro semelhante ao íleo meconial (com dor em quadrante ínfero-direito, perda
do apetite, vômitos e ocasionalmente uma massa palpável). No trato genito-urinário observase puberdade tardia, azoospermia em até 95% dos homens e infertilidade em 20% das
mulheres(1).
4. Diagnóstico Laboratorial:
A primeira anormalidade funcional observada no pulmão de uma criança com fibrose cística
é o aumento na relação volume residual-capacidade pulmonar total. Mudanças posteriores
incluem diminuição na capacidade residual total e no volume expiratório forçado no primeiro
segundo (VEF-1). Alterações radiológicas iniciam por hiper-expansão, evoluindo para sinais
de impactação de muco culminando com bronquiestasias. A análise do teor de cloro no suor
(juntamente com a clínica compatível) faz o diagnóstico. Apenas 1-2% dos pacientes vão
apresentar níveis normais de cloro(1).
5. Exames Subsidiários Exigidos:
Dosagem de Cl- em suor.
6. Critérios de Inclusão:
As seguintes situações são requeridas, cumulativamente, como critérios de inclusão neste
Protocolo:
- Critérios clínicos compatíveis com o diagnóstico de Fibrose Cística;
- Dosagem de Cl- em suor, compatível com Fibrose Cística.
7. Critérios de Exclusão:
Não deverá ser incluído neste tratamento todo o paciente que apresentar pelo menos uma das
situações abaixo:
- Hipersensibilidade a proteínas de suínos:
- Pancreatite aguda ou crônica agudizada.
8. Casos Especiais:
Pacientes com colonopatia fibrosante devem utilizar doses baixas de lipase (500 a
2500U/kg/refeição)
9. Tratamento:
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
9.1. Fármaco: enzimas pancreáticas - pancreatolipase
9.2. Dose: deve-se iniciar com 1.000 U/kg de lipase por refeição em menores de 4 anos e 500
U/kg em maiores de 4 anos; usualmente metade de dose é utilizada para lanches. A dose total
diária deve refletir aproximadamente 3 efeições e 2-3 lanches. Se sinais e sintomas de malabsorção persistem, incrementos nas doses podem ser realizados. Não se sabe a segurança de
doses entre 2.500 e 6.000 U/kg por refeição; acima desta dose o risco de colonopatia
fibrosante tem sido descrito(2,3,4). No caso do desenvolvimento de colonopatia, a dose deve
ser reduzida para 500 a 2.500U/kg(2,5). O uso de antiácidos aumenta a biodisponibilidade por
diminuir a inativação pelo pH baixo(4).
As diferentes apresentações das enzimas apresentam variabilidade muito grande de efeito,
sendo fortemente recomendada a manutenção da mesma preparação durante todo o
tratamento; caso seja necessário a troca, deve haver acompanhamento médico(4).
Apresentações com elevadas concentrações de lipase parecem apresentar a mesma eficácia
que apresentações de baixa concentração(4,6), porém não são recomendadas para crianças com
menos de 15 anos(7). Dose máxima diária não deve ultrapassar 10.000 U / kg de lipase(7), caso
não se consiga controle adequado dos sintomas, doses maiores poderão ser utilizadas após avaliação por comitê de especialistas nomeados
pelo Estado.
9.3. Tempo de tratamento: indefinidamente
9.4. Monitorização: o ajuste da dose será realizado através da dosagem de nitrogênio e do
conteúdo de gordura das fezes(4).
Efeitos adversos são hiperuricemia, náuseas e vômitos, colonopatia fibrosante
(principalmente com doses elevadas), hiperuricosúria e cristalúria (cristais de ácido úrico),
hipersensibilidade pulmonar (devido à inalação do pó); anafilaxia é raro.
10. Consentimento Informado:
É obrigatória a cientificação do paciente, ou de seu responsável legal, dos potenciais riscos e
efeitos colaterais relacionados ao uso dos medicamentos preconizados neste Protocolo, o que
deverá ser formalizado por meio da assinatura de Termo(s) de Consentimento Informado, de
acordo com o modelo em anexo.
Referências:
1. Boucher RC. Cystic Fibrosis. In Fauci AS, et al. Harrison's Principles of Internal Medicine.
14th ed. 1998
2. Borowitz DS, et al. Use of pancreatic enzyme supplements for patients with cystic fibrosis
in the context of fibrosing colonopathy. J Pediatr 1995 Nov;127(5):681-4
3. FitzSimmons SC, et al. High dose pancreatic-enzime supplements and fibrosing
colonopathy in chindren with cystic fibrosis. NEJM 1997:336:1283-89.
4. MICROMEDEX
5. Stevens JC, et al. Pancreatic enzime supplementation is cystic fibrosis patients before and
after fibrosing colonophaty. JPGN 1998;26:80-84.
6. Gan KH, et al. Comparison of a high lipase pancreatic enzyme extract with a regular
pancreatin preparation in adult cystic fibrosis patients. Alimentary Pharmacology &
Therapeutics 1994; 8(6): 603-7
7. Powell CJ. Colonic toxicity from pancreatins: a contemporary safety issue. Lancet
1999;353:911-15.
TERMO DE CONSENTIMENTO INFORMADO
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
ENZIMAS PANCREÁTICAS
O Paciente abaixo identificado e firmado declara para todos os efeitos legais que foi
informado de todos os benefícios, contra-indicações, potenciais efeitos colaterais, riscos e
advertências relativos ao uso do medicamento PANCRELIPASE (enzima pancreática),
preconizado para o tratamento da fibrose cística.
Expressa, ainda, sua concordância e vontade em submeter-se ao tratamento preconizado já
referido, assumindo inteira responsabilidade e risco pelos eventuais efeitos indesejáveis que
venham a ocorrer em decorrência do mesmo.
Assim declara que:
Foi claramente informado que a medicação PANCRELIPASE (enzima pancreática) pode
trazer os seguintes benefícios no tratamento da fibrose cística:
- Diminuição do volume de fezes e da quantidade de gordura delas;
- Diminuição do apetite, das cólicas abdominais e da flatulência;
- Aumento de peso e melhor crescimento e desenvolvimento.
Foi também claramente informado a respeito das seguintes contra-indicações, potenciais
efeitos colaterais, riscos e advertências a respeito da medicação PANCRELIPASE (enzima
pancreática) no tratamento da fibrose cística:
- Medicação classificada na gestação como fator de risco C (significa que risco para o bebê
não pode ser descartado, mas um benefício potencial pode ser maior que os riscos);
- Contra-indicado em casos de hipersensibilidade (alergia) a produtos derivados de porco e
em casos de pancreatite aguda ou crônica agudizada;
- Os comprimidos não devem ser mastigados ou triturados;
- Efeitos adversos raros são reação alérgica, irritação da boca, encurtamento da respiração,
obstrução nasal, dificuldade para respirar, sibilância (chiado no peito), diarréia, náuseas,
vômitos, dor abdominal, hiperuricemia, hiperuricosúria;
- Efeito adverso raro e dose dependente é a colonopatia fibrosante, relacionada
principalmente com doses superiores a 10.000 U/dia;
O paciente declara, ainda, estar ciente que pode suspender este tratamento a qualquer
momento, sem que este fato implique em qualquer forma de constrangimento entre ele e seu
médico, que se dispõe a continuar tratando-o em quaisquer circunstâncias.
Assim, o paciente faz sua adesão ao tratamento de forma livre, por espontânea vontade e por
decisão conjunta dele e seu médico.
Paciente:________________________________________________
Responsável Legal (quando for o caso):___________________
Sexo do paciente: ( )Masculino ( )Feminino
Idade do Paciente:_____
R.G. (do paciente ou responsável legal)____________________
Endereço:_______________________________________________
Cidade: __________CEP:___________ Telefone:( )___________
__________________________
Assinatura do Paciente
_________________________
Assinatura do Responsável (quando for o caso)
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
Médico Responsável: ____________________CRM:_____________
Endereço do Consultório:____________________________
Cidade:______________ CEP: ____________ Telefone: ( )_______
___________________________
Assinatura e Carimbo do Médico
_____/______/_________
Data
Obs.:
1- O preenchimento completo deste Termo e sua respectiva assinatura são imprescindíveis
para o fornecimento do medicamento
2- Este Termo ficará arquivado na farmácia responsável pela dispensação dos medicamentos
ANEXO V
PROTOCOLO E DIRETRIZES TERAPÊUTICAS
ACROMEGALIA
Medicamento: Acromegalia - Ocreotida
1. Introdução:
Desordem debilitante resultante da exposição, a longo prazo, a elevados níveis de hormônio
do crescimento (GH). A incidência anual é de 3-4 casos/milhão com prevalência de 40-90
casos/milhão. O risco relativo de mortalidade, em relação a população normal, é 2-3, sendo as
principais causas de mortalidade complicações cardiovasculares e respiratórias. Tal risco é
revertido com o tratamento. A principal causa da hipersecreção de GH é a presença de
adenoma benigno de pituitária; e os efeitos do GH são mediados por fator de crescimento tipo
insulina I (IGF-I)(1,2).
2. Classificação CID 10: E22.0
3. Diagnóstico Clínico:
Devido à apresentação clínica ser insidiosa, o diagnóstico comumente é retardado de 4 a 10
anos desde o início da doença(3,4). A característica clínica marcante na acromegalia é a
desfiguração cosmética; manifestações articulares, neuropatia e cardiopatia estão entre as
principais manifestações sistêmicas. Os pacientes podem apresentar macrognatia,
crescimento exagerado de mãos e pés, hipertrofia de tecidos moles. Outras características
incluem cefaléia, hiperidrose, bócio, osteoartrite, síndrome do túnel do carpo, fadiga,
distúrbios visuais, aumento do número de sinais cutâneos, hirsutismo, polipose colônica,
macroglossia, apnéia do sono e sonolência, desordens reprodutivas (amenorréia, impotência,
perda do libido), hipogonadismo, e doenças cardiovasculares (insuficiência cardíaca,
arritmias, hipertensão)(1-5). Sintomas relacionados à massa tumoral também podem ocorrer,
uma vez que a maioria dos pacientes apresenta volumosos adenomas hipofisários.
4. Diagnóstico Laboratorial:
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
A dosagem normal de IGF-I descarta o diagnóstico; estando esta elevada, deve ser realizado a
dosagem de GH após sobrecarga de glicose (50-100g). Se houver supressão dos níveis de GH
(< 2ng/ml) descarta o diagnóstico, caso contrário o confirma. Após a confirmação deste,
deve-se verificar a origem da secreção aumentada de GH: tomografia computadorizada ou
ressonância magnética da pituitária deve ser realizada, e se for visualizada massa selar,
confirma-se o diagnóstico de adenoma pituitária. Raramente pode-se observar sela vazia. Se
não forem observadas alterações selares, deve ser realizado exame de imagem do tórax e/ou
abdome em busca de fonte ectópica para a produção exagerada de GH (acromegalia extrapituitária)(1,3-5).
5. Exames Subsidiários Exigidos:
Dosagem de GH após sobrecarga de glicose e/ou dosagem de IGF-1, exame de imagem
confirmando adenoma hipofisário.
6. Critérios de Inclusão:
Confirmação diagnóstica, ter sido refratário ao tratamento cirúrgico ou ter contra-indicação
para tal.
7. Critérios de Exclusão:
Colelitíase sintomática(6) ou hipersensibilidade conhecida (3,7)
8. Casos Especiais:
Pacientes muito sintomáticos (cefaléia ou apnéia do sono, por exemplo) podem ser incluídos
no protocolo enquanto aguardam a cirurgia.
9. Tratamento:
9.1. Fármaco:
9.1.1. Octreotide:. Dose: 100-250mg, SC, 3x ao dia, até um máximo de 1500mg/dia(2,3,6-8). Na
preparação de depósito ( Octreotide-LAR ) iniciar com a dose de 20 mg IM a cada 28 dias,
aumentando a dosagem até o máximo de 40 mg/28 dias, dependendo da avaliação de
GH/IGF-1. Tal dose pode ser reduzida para 10mg a cada 28 dias de acordo com a resposta
clínico-laboratorial. A administração da preparação de depósito deve ser realizada por um
profissional da saúde. As doses devem ser ajustadas para a função renal.
9.1.2. Bromocriptina: alternativa com eficácia inferior ao octreotide, pode ser utilizada
conjuntamente com o primeiro em casos de resistência(9). Dose: inicia-se com 2,5mg/dia com
aumentos gradativos a cada 3-7 dias (dose usual de 20-30mg/dia) (3,4)
9.2. Tempo de tratamento: até obtenção da cura da moléstia.
9.3. Monitorização: O benefício esperado com o tratamento é a normalização dos níveis de
GH e de IGF-I, com conseqüente melhora dos sinais e sintomas. Teste supressão do GH e
dosagem IGF-I devem ser realizados em 1 mês, não havendo resposta o tratamento deve ser
suspenso(3,4,6,10). Após deve ser realizado acompanhamento a cada 4 meses com avaliação
clínica e laboratorial.
10. Referências:
1. Melmed S. Clinical manifestations of acromegaly. CD-ROM UpToDate 6.3.
2. Newman CB. Medical Therapy for Acromegaly. Endocrinol Metabol North Am
1999;28(1):171-190.
3. Melmed S, et al. Current Treatment Guidelines for Acromegaly. J Clin Endocrinol Metab
1998;83:2646-52.
4. Melmed S. Diagnosis of acromegaly. CD-ROM UpToDate 6.3.
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
5. Shimon I, Melmed S. Management of pituitary tumors. Ann Intern Med 1998;129:472483.
6. Ezzat S, et al. Octreotide Treatment of Acromegaly. A Randomized, Multicenter Study.
1992;117:711-18.
7. Melmed S. Treatment of acromegaly. CD-ROM UpToDate 6.3.
8. Lamberts S, et al. Octreotide. NEJM 1996;334:246-54.
9. Lamberts SWJ, et al. A comparison among the growth hormone-lowering effect in
acromegaly of the somatostatin analog SMS 201-995, bromocriptine, and the combination of
both drugs. J Clin Endocrinol Metab 1986;63:16-19.
10. Chanson P. Predicting the efrfects of long-term medical treatment in acromegaly. At what
cost? For waht benefits? Eur J Endocrinol 1997;136:359-61.
TERMO DE CONSENTIMENTO INFORMADO
ACROMEGALIA - OCREOTIDA
O Paciente abaixo identificado e firmado declara para todos os efeitos legais que foi
informado de todos os benefícios, contra-indicações, potenciais efeitos colaterais, riscos e
advertências relativos ao uso do medicamento OCTREOTIDA, preconizado para o
tratamento da acromegalia.
Expressa, ainda, sua concordância e vontade em submeter-se ao tratamento preconizado já
referido, assumindo inteira responsabilidade e risco pelos eventuais efeitos indesejáveis que
venham a ocorrer em decorrência do mesmo.
Assim declara que:
Foi claramente informado que a medicação OCTREOTIDA pode trazer os seguintes
benefícios no tratamento da acromegalia:
- Melhora da sintomatologia;
- Redução das complicações.
Foi também claramente informado a respeito das seguintes contra-indicações, potenciais
efeitos colaterais, riscos e advertências a respeito da medicação OCTREOTIDA no
tratamento da acromegalia:
- Medicação classificada na gestação como fator de risco B (significa que risco para o bebê é
muito improvável);
- Contra-indicado em casos de colelitíase e de hipersensibilidade conhecida (alergia grave);
- Efeitos como náuseas, dores abdominais, diarréia, má absorção de gorduras e flatulência
podem ocorrer e são dependentes da dose; tais efeitos costuma desaparecer espontaneamente
em 10 a 14 dias de uso;
- São comuns edema, fadiga, cefaléia, tonturas, vertigens, anorexia (perda do apetite),
depressão, hipotireoidismo, galactorréia, constipação, icterícia (amarelão), hepatite, dor no
local de administração;
- Efeitos raros são dor torácica, reação hipertensiva, ansiedade, febre, alopécia (queda de
cabelo), eritema (vermelhidão), tromboflebite, paralisia facial, tremor, ardência ocular,
desconforto na garganta, rinorréia e encurtamento da respiração;
- A secreção de insulina é também diminuída, podendo ocorrer hiperglicemia;
- Incidência elevada (20-30%) de colelitíase por pedras de colesterol, embora a maioria seja
assintomática;
- O risco de ocorrer efeitos adversos aumenta com a superdosagem;
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
O Paciente declara, ainda, estar ciente que pode suspender este tratamento a qualquer
momento, sem que este fato implique em qualquer forma de constrangimento entre ele e seu
médico, que se dispõe a continuar tratando-o em quaisquer circunstâncias.
Assim o paciente faz sua adesão ao tratamento de forma livre, por espontânea vontade e por
decisão conjunta dele e de seu médico.
Paciente:________________________________________________
Responsável Legal (quando for o caso):____________________
Sexo do paciente: ( )Masculino ( )Feminino
Idade do Paciente:_____
R.G. (do paciente ou responsável legal)____________________
Endereço:_______________________________________________
Cidade: __________CEP:___________ Telefone:( )___________
__________________________
Assinatura do Paciente
_________________________
Assinatura do Responsável (quando for o caso)
Médico Responsável: ____________________CRM:_____________
Endereço do Consultório:____________________________________
Cidade:______________ CEP: ____________ Telefone: ( )_______
___________________________
Assinatura e Carimbo do Médico
_____/______/_________
Data
Obs.:
1- O preenchimento completo deste Termo e sua respectiva assinatura são imprescindíveis
para o fornecimento do medicamento
2- Este Termo ficará arquivado na farmácia responsável pela dispensação dos medicamentos.
ANEXO VI
PROTOCOLO E DIRETRIZES TERAPÊUTICAS
HEPATITE B EM CANDIDATOS A TRANSPLANTE HEPÁTICO
Medicamento: Lamivudina associada a Gama Globulina Hiperimune (HBIG)
1- Introdução:
A recorrência de infecção do fígado pelo vírus da hepatite B (VHB) é das principais causas
de morbi-mortalidade após o transplante hepático em pacientes submetidos ao procedimento
por doenças hepáticas causadas por esse vírus.(1) Nos pacientes que apresentam replicação
viral ativa pré-transplante, definida pela positividade do DNA do VHB, a taxa de recorrência
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
é significativamente maior, quando comparados a pacientes DNA do VHB negativos (96%
versus 29% em 2 anos, respectivamente). (2) A administração intravenosa de altas doses de
gamaglobulina hiperimune (HBIG) no período pós-operatório diminui a taxa de recorrência
do VHB, porém seu efeito é menor em pacientes com altas taxas de replicação viral. (3) Desta
maneira, em alguns centros, pacientes DNA do VHB positivos (avaliados pelo teste
quantitativo de hibridização molecular ou pelo "branched chain-DNA") foram excluídos da
lista de espera. (4) Mais recentemente, foi evidenciado que a administração de lamivudina
para pacientes com hepatopatias crônicas DNA do VHB- positivos suprime a replicação viral
praticamente em todos casos. (5-10) Um ensaio multicêntrico norte-americano mostrou que a
administração de lamivudina, na dose de 100 mg/dia, antes e depois do transplante, sem a
administração concomitante de HBIG, era capaz de impedir a infecção do enxerto. (11)
Entretanto, estudos com maior seguimento demostraram recorrência de infecção do enxerto
por vírus mutantes, sugerindo que a monoterapia provavelmente não seja suficiente. (12-13) Por
essa razão, a utilização combinada de HBIG e lamivudina parece ser a estratégia mais
adequada para evitar a infecção do enxerto em pacientes transplantados por doenças causadas
pelo VHB. A administração associada de lamivudina e HBIG (altas doses intravenosas)
demonstrou ausência de recorrência da infecção pelo VHB em um seguimento médio de 346
dias. (14)
Entretanto, a utilização das doses convencionais de HBIG aumenta marcadamente os custos
do transplante em pacientes VHB positivos. Ademais, sua produção é limitada, a droga deve
ser administrada via intravenosa e seu uso tem sido associado com reações imunes e com
intoxicação mercurial.
Mais recentemente surgiram protocolos que utilizam a associação de lamivudina e doses de
HBIG menores e/ou a via intramuscular (15-16).
Em 1999, McCaughan e colaboradores demonstraram em estudo multicêntrico utilizando a
associação lamivudina e baixas doses HBIG previniu reinfecção do enxerto em todos os
pacientes.(17) Mais recentemente, o mesmo grupo australiano publicou sua experiência usando
esquema idêntico em um grupo de 37 pacientes AgHBs positivos (19 HBeAg positivos),
submetidos a transplante hepático. Em nenhum paciente registrou-se evidências de infecção
pelo VHB. (18) Assim neste protocolo sugere-se adotar a experiência do grupo Australiano que
combina lamivudina com dose baixa de HBIG intramuscular.
2 - Diagnóstico e Critérios de Inclusão no Protocolo de Tratamento:
- Candidato a transplante hepático - doença hepática terminal por virus B: HBSAg (+),
HBeAg (+ ou -).
- Todos deverão realizar quantificação do HBV-DNA pelo método de hibridização molecular.
Se o teste HBV-DNA for positivo, devem ser tratados com lamivudina por 4 a 6 meses e
reavaliados, se persistirem com HBV-DNA (+) não se indica transplante.
3- Critérios de Exclusão:
Não deverá ser incluído neste tratamento todo o paciente que apresentar pelo menos uma das
situações abaixo:
- candidato portador de cirrose por vírus B cujo HBV-DNA não negativar com 4 a 6 meses de
lamivudina;
- candidato a transplante por cirrose por vírus C.
4- Tratamento e Esquema de administração:
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
Lamivudina:
- 100 mg via oral por dia administrada por não mais de 6 meses antes do transplante
(idealmente 4 semanas);
- No dia do transplante recebe 200 mg;
- No pós-operatório imediato, assim que tiver via oral, recomeça com 100 mg/dia mantendo
para o resto da vida.
HBIG:
Recomenda-se a administração intramuscular de HBIG 400 UI no pós-operatório imediato
seguida de 400 UI intramuscular por dia durante 7 dias, seguida de uma administração por
semana até a alta e então uma administração por mês.
5- Acompanhamento:
- Dosar HBsAg e Anti-HBs e ALT+AST mensalmente nos primeiros 6 meses e após de 6 em
6 meses.
- Se Anti-HBs 100 UI: manter a dose de HBIG. Se menor do que 100UI: aumentar a dosagem
de HBIG para 800 UI - IM /mês.
6- Tempo de tratamento:
Durante toda a vida.
7- Apresentação:
Lamivudina: comprimidos de 100 mg.
Imunoglobulina Hiperimune Anti-HBs - frascos ampolas com 2 ml (100 UI) e 10 ml (500UI).
8- Desfechos esperados com o tratamento:
- Redução da taxa de recorrência do vírus B em pacientes transplantados;
- Aumento da taxa de sobrevida pós-transplante em portadores de cirrose por vírus B.
9 -Riscos:
Os efeitos adversos da Lamivudina incluem diarréia e outros distúrbios gastrointestinais,
cefaléia, fadiga, insônia, artralgias, mialgias, enzimas hepátidas elevadas, rash cutâneo,
infecções de vias aéreas superiores. Neutropenia ocorre infrequentemente com as doses
recomendadas de lamivudina. Acidose lática e hepatomegalia grave com esteatose, incluindo
casos fatais foram relatados.
A administração de HBIG, por ser produto de origem humana está associada a reações
alérgicas e risco de choque anafilático.
10- Logística:
Os fármacos preconizados deverão ser fornecidos diretamente aos centros transplantadores
devidamente cadastrados no SNT e sob coordenação regional do gestor estadual.
O uso ambulatorial será coordenado pelo gestor estadual com registros trimestrais atualizados
dos dados de todos os pacientes.
11- Consentimento Informado:
É obrigatória a cientificação do paciente, ou de seu responsável legal, dos potenciais riscos e
efeitos colaterais relacionados ao uso dos medicamentos preconizados neste Protocolo, o que
deverá ser formalizado por meio da assinatura de Termo(s) de Consentimento Informado, de
acordo com o modelo em anexo.
Referências:
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
1. Wiesner RH. Who and when to list patients for liver transplantation. In: Treatment of Liver
Diseases. Arroyo V, Bosch J, Bruguera M, Rodes J, Tapias JMS. Eds. Masson. Barcelona.
1999. 159-75p
2. Samuel D, Bismuth A, Mathieu D, et al. Passive immunoprophylaxis after liver
transplantation in HbsAg-positive patients. Lancet 1991;337:813-15
3. Samuel D, Müller R, Alexander G, Fassati I, Ducot B, Benhamou J-P et al. Liver
transplantation in European patients with hepatitis B surface antigen. N Engl J Med 193; 329:
1842-47.
4. Muller R, Samuel D, Fassati LR, et al. EUROHEP consensus on the management of liver
transplantation for hepatitis B virus infection. J Hepatol 1994;21:1140-43
5. Dienstag JL, Perrillo RP, Schiff ER, et al. A preliminary trial of lamivudine for chronic
hepatitis B. N Engl J Med 1995;333:1657-61
6. Lai CL, Chein RN, Leung NW, et al. One year trial of lamivudine for chronic hepatitis B.
Asia Hepatitis Lamivudine Study. N Engl J Med 1998;339:61-7
7. Dienstag JL, Schiff ER, Wright TL, et al. Lamivudine as initial treatment for chronic
hepatitis B in United States. N Engl J Med 1999;341:1256-63
8. Tassopoulos NC, Volpes R, Patore G, et al. Efficacy of lamivudine im patients with
hepatitis B e antigen-negative/hepatitis B virus DNA-positive (precore mutant) chronic
hepatitis B. Lamivudine Precore Mutant Study Group. Hepatology 1999;29(3):889-96
9. Honkoop P, de Man RA, Niesters HG et al. Quantitiative hepatitis B virus DNA
assessment by limiting dilution polymerase chain reaction in chronic hepatitis B patients:
evidence of continuing viral suppression with longer diration and higher dose of lamivudine
therapy. J Viral Hepat 1998; 5(5):307-12
10. Villeneuve J-P, Condreay LD, Willems B, Pomier-Layrargues G, Fenyes D, Bilodeau M
et al. Lamivudine treatment for decompensated cirrhosis resulting from chronic hepatitis B.
Hepatology 2000; 31: 207-210
11. Grellier R Mutimer D, Ahmed M, et al. Lamivudine prophylaxis against reinfection in
liver transplantation for hepatitis B cirrosis. Lancet 1996;348:1212-15
12. Ling R, Mutimer D, Ahmed M, et al. Selection of mutations in the hepatitis B virus
polymerase during therapy of transplant recipiens with lamivudine. Hepatology 1996;24:71113
13. Mutimer D, Pillay D, Dragon E, et al. High pre-treatment serum hepatitis B virus titre
predicts failure of lamivudine prophylaxis and graft re-infection after liver transplantation. J
Hepatology 1999;30:715-21
14. Markowitz JS, Martin P, Conrad AJ, et al. Prophylaxis against hepatitis B recurrence
following liver transplantation using combination lamivudine and hepatitis B immune
globulin. Hepatology 1998;28:585-89
15. Yoshida EM, Siegfried RE, Partovi N, et al. Liver transplantation for chronic hepatitis B
infection with the use of combination lamivudine and low-dose hepatitis B immune globulin.
Liver Transpl Surg 1999;5:520-25
16. Yao FY, Osorio RW, Roberts JP, et al. Intramuscular hepatitis B immune globulin
combined with lamivudine for prophylaxis aginst hepatitis B recurrence after liver
transplantation. Liver Transplant Surg 1999;5:491-96
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
17. McCaughan GW, Spencer J, Koorey D, et al. Lamivudine therapy in patients undergoing
liver tranplantation for hepatitis B virus precore mutant-associated infection: high resistance
rate in treatment of recurrence but universal prevention of used as prophylaxis with very-low
dose hepatitis B immune globulin. Liver Transpl Surg 1999;5:512-19.
18. Angus PW, McCaughan GW, Gane EJ, Crawford DHG, Harley H and the Australasin
Liver Transplant Study Group. Liver Transplantation 2000; 6: 429-33.
TERMO DE CONSENTIMENTO INFORMADO
Lamivudina e Gama Globulina Hiperimune (HBIG)
O Paciente abaixo identificado e firmado declara para todos os efeitos legais que foi
informado de todos os benefícios, contra-indicações, potenciais efeitos colaterais, riscos e
advertências relativos ao uso dos medicamentos Lamivudina e Gama Globulina Hiperimune
(HBIG), preconizados, em associação, para o tratamento da hepatite por vírus B em candidato
a transplante hepático.
Expressa, ainda, sua concordância e vontade em submeter-se ao tratamento preconizado já
referido, assumindo inteira responsabilidade e risco pelos eventuais efeitos indesejáveis que
venham a ocorrer em decorrência do mesmo.
Assim declara que:
Foi claramente informado que a associação de Lamivudina e Gama Globulina Hiperimune
(HBIG) podem trazer os seguintes benefícios no tratamento da hepatite por vírus B em
candidato a transplante hepático:
- Redução da taxa de recorrência do vírus B em pacientes transplantados;
- Aumento da taxa de sobrevida pós-transplante em portadores de cirrose por vírus B.
Foi também claramente informado a respeito das seguintes contra-indicações, potenciais
efeitos colaterais, riscos e advertências a respeito da associação de Lamivudina e Gama
Globulina Hiperimune (HBIG) no tratamento da hepatite por vírus B em candidato a
transplante hepático:
- Medicação classificadas na gestação como fator de risco C (significa que risco para o bebê
não pode ser descartado, mas um benefício potencial pode ser maior que os riscos);
- Contra-indicado para casos de hipersensibilidade a qualquer componente (lamivudina e
HBIG) e se trombocitopenia e deficiência de Imunoglobulina A (HBIG);
- Deve ser feito ajuste da dose de acordo com a função renal (lamivudina);
- Efeitos adversos comuns com a lamivudina incluem: cefaléia (dor de cabeça), insônia,
fadiga, dores pelo corpo, náuseas, vômitos, diarréia, neuropatia periférica, parestesias
(formigamentos), sintomas nasais, tosse; efeitos comuns do HBIG são dores musculares,
sensibilidade e rigidez muscular no local de acesso;
- Também podem ocorrer (1-10%) tonturas, depressão, febre, calafrios, rash cutâneo
(vermelhidão da pele), anorexia (diminuição de apetite); dor abdominal, dispepsia (azia),
elevação de amilase, neutropenia (baixa das células brancas do sangue), anemia (baixa das
células vermelhas do sangue), elevação de transaminases (provas de lesão hepática), dores
musculares e articulares com a lamivudina; com o HBIG pode ocorrer rubor, calafrios e
náuseas;
MINISTÉRIO DA SAÚDE
DEPARTAMENTO NACIONAL DE AUDITORIA DO SUS
COORDENAÇÃO DE SISTEMAS DE INFORMAÇÃO
SISTEMA ÚNICO DE SAÚDE - LEGISLAÇÃO FEDERAL
TABELAS: PROCEDIMENTOS DO SIA/SUS
- Raramente (<1%) pode ocorrer, com lamivudina, pancreatite, trombocitopenia (baixa da
quantidade de plaquetas do sangue) e hiperbilirrubinemia (amarelão); efeitos raros do HBIG
são febre, letargia, uriticária, angioedema, eritema, vômitos, dores musculares e reações de
hipersensibilidade (alergia);
O Paciente declara, ainda, estar ciente que pode suspender este tratamento a qualquer
momento, sem que este fato implique em qualquer forma de constrangimento entre ele e seu
médico, que se dispõe a continuar tratando-o em quaisquer circunstâncias.
Assim, o paciente faz sua adesão ao tratamento de forma livre, por espontânea vontade e por
decisão conjunta dele e de seu médico.
Paciente:__________________________________________
Responsável Legal (quando for o caso):_____________________
Sexo do paciente: ( )Masculino ( )Feminino Idade do Paciente:____
R.G. (do paciente ou responsável legal)______________________
Endereço:_______________________________________________
Cidade: ______________CEP:___________Telefone:( )_________
________________________
Assinatura do Paciente
____________________________
Assinatura do Responsável (quando for o caso)
Médico Responsável: _______________________CRM:__________
Endereço do Consultório:____________________________
Cidade:______________ CEP:___________ Telefone: ( )________
__________________________
Assinatura e Carimbo do Médico
_______/______/_________
Data
Obs.:
1- O preenchimento completo deste Termo e sua respectiva assinatura são imprescindíveis para o
fornecimento do medicamento
2- Este Termo ficará arquivado na farmácia responsável pela dispensação dos medicamentos
Anexo VI Protocolo Hep B em Trnspl. Hep
|