EXCREÇÃO DE COMPOSTOS NITROGENADOS
NAS DIFERENTES ESPÉCIES*
Introdução
As proteínas compõem cerca de 18% do peso corporal dos animais e podem ter diferentes
funções como: reguladoras do metabolismo (enzimas e hormônios), elementos estruturais
(membranas, músculos e tecido conjuntivo), substâncias de transporte (O2 pela hemoglobina e
elétrons pelo citrocromo c) osmorreguladores (albumina), componentes de ácido nucleico
(nucleoproteínas) e defensores do organismo (imunoglobulinas e os interferons).
As proteínas vindas da dieta são hidrolisadas na luz intestinal pela ação das muitas enzimas
proteases e peptidases, resultando na produção de aminoácidos livres, que em sua maioria são
transportados para o fígado via sangue portal. Além da proteína da dieta outra fonte de
aminoácidos é o catabolismo das proteínas tissulares que devem ser constantemente repostas.
O reservatório sanguíneo de aminoácidos é essencial para a síntese proteica, e ainda para a
formação de muitos outros compostos nitrogenados essenciais para a função tissular adequada
como as purinas, pirimidinas, neurotransmissores e heme. O catabolismo dos aminoácidos
envolve a remoção do grupo amino e utilização dos α-cetoácidos resultantes para a oxidação em
CO2 e formação de ATP e síntese de glicose e lipídios.
Os aminoácidos podem sofrer degradação oxidativa com liberação de grupos amino,
basicamente em três situações: durante o turnover proteico com uma parte dos aminoácidos
liberados na quebra das proteínas não sendo utilizados; em dietas ricas em proteína onde os
aminoácidos são ingeridos em excesso o excedente não pode ser armazenado e é catabolizado; e
durante o jejum severo ou no diabetes melito quando os carboidratos não estão acessíveis ou
não são utilizados adequadamente ocorre hidrolise das proteínas corporais para a produção de
energia.
A amônia liberada nesses processos precisa ser convertida em algum composto passível de
ser excretado. O acúmulo de amônia no organismo leva a intoxicação caracterizada por vômitos,
recusa de alimentos ricos em proteína, ataxia intermitente, irritabilidade, letargia e atraso
mental. Sendo assim, a seguir veremos as principais formas de remoção dos grupos amino dos
aminoácidos e a forma na qual são convertidos e excretados em cada espécie animal.
____________________________
*Seminário apresentado pela aluna RAQUEL MELCHIOR na disciplina BIOQUÍMICA DO TECIDO
ANIMAL, no Programa de Pós-Graduação em Ciências Veterinárias da Universidade Federal do Rio
Grande do Sul, no primeiro semestre de 2013. Professor responsável pela disciplina: Félix H. D.
González.
Liberação de grupos amino
As proteínas consumidas na dieta podem servir de fonte de energia dependendo da espécie,
quase 90% da energia no caso dos carnívoros e uma pequena fração nos herbívoros, ou podem
ser oxidadas para a produção de energia em caso de dietas com excesso de proteína. A oxidação
dos aminoácidos derivados das proteínas dos alimentos é a fonte da maioria dos grupos amino e
a proteólise intracelular que ocorre normalmente em todos os tecidos contribui com uma
pequena parte dos grupos amino, em situações normais. A maior parte dos aminoácidos é
metabolizada no fígado, e a amônia gerada em excesso nos outros tecidos é transportada até ele
para ser convertida na forma apropriada de excreção.
A degradação oxidativa dos aminoácidos ocorre por rotas catabólicas diferentes para cada
um dos 20 aminoácidos proteicos embora todas terminem em um dos seguintes metabolitos:
piruvato, acetil-CoA, ou outros compostos intermediários do ciclo de Krebs. Com exceção dos
que terminam em acetil-CoA, todos os aminoácidos constituem substratos precursores da
gliconeogênese.
O catabolismo dos aminoácidos ocorre principalmente no fígado e uma pequena parte no
rim. A primeira etapa da degradação oxidativa dos aminoácidos é a remoção do grupo amino
que ocorre em duas rotas integradas: a transaminação e a desaminação oxidativa
Transaminação
Este é o método mais comum da remoção dos grupos amino dos aminoácidos. Quando os Laminoácidos chegam ao fígado, o primeiro passo no seu catabolismo é a remoção dos grupos αamino promovida por enzimas chamadas aminotransferases ou transaminases. Nessas reações
de transaminação, o grupo α-amino é transferido para o átomo de carbono α do α-cetoglutarato,
produzindo o respectivo α-cetoácido análogo do aminoácido e glutamato. Esta é uma reação
reversível e há ampla distribuição das transaminases pelos tecidos, algumas são mitocondriais
outras são citossólicas e outras estão em ambos os compartimentos celulares. O glutamato
conduz os grupos amino para serem utilizados por vias biossintéticas ou, então, para uma
sequencia final de reações pelas quais são formados produtos nitrogenados degradados que, a
seguir, são excretados.
As aminotransferases requerem como coenzima o piridoxal-fosfato (forma enzimática da
vitamina B6), o qual se encontra como grupo prostético das transaminases. Estas enzimas
catalisam as reações conhecidas como reações de “pig-pong”, nas quais um primeiro
aminoácido se liga ao sitio ativo da enzima e perde seu grupo amino, produzindo um αcetoácido. Esse grupo amino se incorpora a piridoxal-fosfato que se transforma em
piridoxamina-fosfato. Em seguida o α-cetoglutarato se liga ao sitio ativo da piridoxamina e
aceita seu grupo amino transformando-se em glutamato, como mostra a Figura 1.
2
Figura 1. Reações de transaminação. Fonte: Lehningher, 2002.
Desaminação oxidativa
É nesta rota que o glutamato que recolhe os grupos amino de vários aminoácidos nas
reações de transaminação é oxidado e desaminado por ação da glutamato desidrogenase nas
mitocôndrias das células hepáticas.
Figura 2. Reação de desaminação oxidativa. Fonte: Champe, 2006.
No caso de alguns tecidos a amônia livre é combinada ao glutamato para formar glutamina e
ser transportada até o fígado, pela ação da glutamina sintetase em uma reação que requer ATP
para ativar o glutamato. Quando chega a mitocôndria hepática a amônia é liberada da glutamina
por ação da glutaminase e retorna a glutamato. A glutamina é um importante transportador de
grupos amino no sangue, pois não possui cargas elétricas o que lhe confere capacidade de
3
atravessar com facilidade as membranas. A formação de glutamina é essencial para o transporte
de grupos amino para muitos processos biológicos, além é claro, de conduzi-los até o ciclo da
ureia para excreção, quando estão em excesso.
Figura 3. Transporte de grupos amino via glutamina. Fonte: Lehningher, 2002.
Os grupos amino liberados nos músculos se ligam ao glutamato e este pode ser convertido à
glutamina (receber amônia) ou transferir seu grupo α-amino para o piruvato pela ação da
alanina-aminotransferase formando alanina, para serem levados até o fígado. Essa reação pode
ser revertida em ambos os tecidos e a alanina também não possui cargas elétricas o que lhe
permite passar facilmente pelas membranas. A alanina também pode remover piruvato do
músculo e leva-lo até o fígado para servir de precursor da glicose na via de gliconeogênese e a
glicose produzida pode voltar para o músculo para servir de energia no ciclo conhecido como
4
glicose-alanina. Ocorre que no citosol hepático a alanina aminotransferase transfere o grupo
amino da alanina para o α-cetoglutarato formando piruvato e glutamato. O glutamato entra na
mitocôndria para liberar a amônia e o piruvato será convertido em glicose.
Figura 4. Transporte de grupos amino via alanina. Fonte: Lehningher, 2002.
Excreção
Uma parte da amônia oriunda da desaminação dos aminoácidos é usada para a formação de
compostos nitrogenados biologicamente úteis e o restante precisa ser convertido a uma forma
menos tóxica pra ser excretado.
Em geral a amônia é convertida em ureia ou acido úrico no fígado e só uma pequena parte
pode ser coletada pelos rins. Mas quando o organismo está em acidose metabólica, os rins
aumentam a capitação de glutamina que está transportando amônia dos tecidos para o fígado,
nestes casos a amônia liberada ai pela ação da glutaminase e glutamato desidrogenase não é
levada até o fígado ou convertido em ureia, e sim é diretamente excretada na urina.
Todos os animais excretam uma pequena porção dos compostos nitrogenados na forma de
amônia. No entanto, a excreção nesse caso se da na forma de amônio (NH4+), ou seja, a
molécula de amônia (NH3) precisa ganhar mais um hidrogênio e passar a amônio para poder ser
5
excretada. Esse mecanismo, se utilizado para excretar toda a amônia produzida pelo organismo,
levaria a um processo de alcalose metabólica.
O destino da amônia produzida pelas diferentes reações de desaminação varia com o tipo de
animal e seu habitat. Como a amônia é altamente tóxica, os tecidos animais são equipados com
diferentes mecanismos para converter amônia em substâncias atóxicas ou para posterior
utilização no anabolismo pelo animal ou excreção. Os meios mais importantes para a excreção
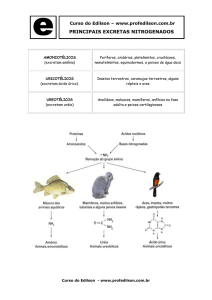
do excesso de nitrogênio são: formação e excreção de ureia em vertebrados terrestres
(ureotélicos); síntese de acido úrico em aves e repteis que vivem no solo (uricotélicos) e
eliminação direta em animais aquáticos (amonotélicos).
Formação de ureia
O ciclo da ureia foi descoberto em 1932 por Krebs e Henseleit, antes mesmo da descoberta
do ciclo do ácido cítrico. Este processo ocorre no fígado e é nele que a através de uma série de
reações dois grupos amino são incorporados a uma molécula de CO2 para formar uma molécula
de ureia.
A desaminação dos aminoácidos ocorre principalmente no fígado e os íons amônio
resultantes podem ser transformados em ureia no fígado na maioria dos vertebrados terrestres.
Os íons amônio e o dióxido de carbono (originário basicamente do ciclo de Krebs) interagem
com ATP, formando fosfato de carbamoil nas mitocôndrias. Os geradores primários de íons
amônio mitocondriais são a glutamato-desidrogenase e a glutaminase. O fosfato de carbamoilsintetase I requer N-acetilglutamato para sua atividade. O N-acetilglutamato é um composto
produzido a partir de acetil-CoA e glutamato e é sintetizado em quantidades maiores quando
estão presentes quantidades maiores de aminoácidos, fornecendo assim um sinal para aumentar
a síntese de ureia durante o excesso de aminoácidos.
O grupo carbamoil é transferido do fosfato de carbamoil para a ornitina formando citrulina,
uma reação catalisada pela ornitina-transcarbamoilase das mitocôndrias. Após o transporte da
citrulina ao citossol, a arginino-succinato-sintetase catalisa então a condensação de aspartato
com citrulina para produzir arginino-succinato. Esta síntese é regida por clivagem do ATP em
AMP e pirofosfato inorgânico (PPi) e pela subsequente hidrolise de PPi em dois Pi. A argininosuccinase rompe então o arginino-succinato em fumarato e arginina. Esta ultima é rompida
hidroliticamente pela arginase para formar ureia e ornitina, completando assim o ciclo. A
arginase é encontrada em grandes quantidades apenas no fígado em animais que excretam ureia.
O fumarato gerado na reação de arginino-succinase é prontamente hidratado em malato e
reoxidado em oxalacetato para aceitar um grupo amino, em geral proveniente do glutamato, a
fim de regenerar aspartato.
6
Figura 5. Ciclo da ureia. Fonte: Schmidt-Nielsen, 1996.
Em resumo o ciclo da ureia ocorre em 5 passos:
1. Grupo amino + CO2 → carbamil fosfato (catalisada pela carbamil fosfato sintetase I e
consome 2 moléculas de ATP).
2. Ornitina + grupo carbamil → citrulina + Pi (catalisada pela ornitina transcarbamilase).
3. Aspartato + citrulina → argininossuccinato (catalisada pela argininossuccinato com
consumo de 1 molécula de ATP).
4. Argininossuccinato → arginina + fumarato (hidrolise pela argininossuccinato liase).
5. Arginina → ornitina + ureia (hidrolise pela arginase).
O fumarato produzido é um intermediário do ciclo do acido cítrico, então os ciclos são
interconectados em um processo dito “bicicleta de Krebs”. Cada ciclo pode funcionar de
maneira independente dependendo do transporte de intermediários entre a mitocôndria e o
citosol para estabelecer comunicação.
O fumarato pode ser convertido em malato e este, em oxalacetato no citosol, esses
intermediários podem sofrer metabolização no próprio citosol ou ser transportado para o interior
da mitocôndria para uso no ciclo de Krebs. Também o aspartato formado na mitocôndria pode
ser transportado no citosol e servir como doador de nitrogênio na reação do ciclo da ureia.
Nos animais ruminantes os níveis de ureia são mais elevados (15-40 mg/dL) devido a
absorção de amônia pelo rúmen. Além disso, nos ruminantes a amônia é metabolizada no fígado
a ureia e esta deve voltar ao rúmen via sangue ou via saliva. Isto significa uma poupança de
energia para a produção de ureia e água. Quando a amônia alcança o rúmen ela é rapidamente
desdobrada em amônia e CO2 pela enzima uréase, produzida pelas bactérias. Os micro7
organismos ureolíticos e proteolíticos utilizam esta amônia para a síntese da sua própria
proteína. À medida que o bolo alimentar avança no processo de digestão estes microorganismos são arrastados junto. Quando chegam ao abomaso (estômago químico) em função
do baixo pH e ação das enzimas as proteínas inclusive a dos micro-organismos é degradada e os
aminoácidos vão para a corrente sanguínea via sistema portal para serem usados na síntese
proteica.
É raro, mas podem ocorrer distúrbios metabólicos da síntese da ureia se qualquer uma das
cinco enzimas do ciclo for deficiente. A toxicidade da amônia faz com que as primeiras reações
do ciclo ocorram dentro da mitocôndria para que os íons amônia não caiam na corrente
sanguínea. A toxicidade da amônia se deve ao fato de sua capacidade de ligação com o αcetoglutarato que é um composto intermediário do ciclo de Krebs. Em caso de excesso de
amônia pode ocorrer uma grande utilização do α-cetoglutarato, diminuindo sua disponibilidade
para o ciclo de Krebs e eventualmente este e a cadeia de transporte de elétrons podem parar.
Além disso, a ureia é uma base forte e pode provocar alcalose. A sintomatologia clinica da
intoxicação por amônia são: vômitos, irritabilidade, letargia, ataxia intermitente e retardamento
mental. As dietas pobres em proteínas e pequenas refeições frequentes resultam em
concentrações mais baixas de amônia sanguínea, e, portanto, em menos sintomas.
Formação e excreção do acido úrico
A excreção de ureia obriga a excreção conjunta de água. No caso dos repteis que vivem em
solo e as aves que possuem seu desenvolvimento embrionário em ovos com cascas
impermeáveis a água não é possível à excreção de ureia, pois não há excreção de água. Além
disso, a reserva de água no interior desse ovo é limitada e a excreção precisou ser adaptada para
ocorrer na forma de produtos insolúveis (acido úrico precipitado).
A via do ácido úrico é uma importante rota para a eliminação dos compostos nitrogenados
em aves e repteis. Nesses animais a via biossintética das purinas foi adaptada para fazer a
excreção de N na forma de ácido úrico. Uma vez que o acido úrico também é o principal
subproduto da degradação das purinas (adenina e guanina) nos primatas, pássaros e repteis.
O acido úrico nas aves é formado no fígado a partir da amônia. A hepatotectomia provoca
acumulo de amônia e aminoácidos no sangue das aves, exatamente como ocorre nos mamíferos.
Acredita-se também que o rim, assim como o fígado, possa ser um local de síntese de acido
úrico nas aves.
Para a formação de acido úrico há considerável gasto energético uma vez que inicialmente
são formadas adenina e guanina a partir do nitrogênio de alguns aminoácidos e carbono oriundo
de moléculas doadoras. Posteriormente adenina e guanina são transformadas em acido úrico por
uma via em que o grupo fosfato é perdido pela ação da 5’-nucleotidase. O adenilato libera a
8
adenosina, que é então desaminada para inosina pela adenosina desaminase. A inosina é
hidrolisada para liberar a D-ribose e a base purínica hipoxantina. A hipoxantina é oxidada
sucessivamente para xantina e para ácido úrico pela xantina oxidase, uma flavoenzima que
contém um átomo de molibdênio e quatro centros ferro-enxofre em seu grupo prostético, como
se pode ver na Figura 6. O mesmo ocorre com a guanina, esta sofre remoção hidrolítica do seu
grupo amino para liberar xantina que também é convertida em ácido úrico pela xantina oxidase.
Figura 6. Síntese de ácido úrico (principais reações). Enzimas: (1) ribose 5-fosfato pyrophosphokinase,
(2) glutamina fosforribosil pirofosfato amidotransferase, (3) fosforribosil sintetase glicinamida, (4)
amido-ligase, (5) sintetase específica, (6) xantina oxidase. Fonte: D’Mello, 2003.
O acido úrico é filtrado livremente no glomérulo e é também secretado pelos túbulos. A
secreção tubular é o processo dominante e é responsável por 90% da excreção total. A presença
do sistema porta renal fornece uma grande fonte de sangue para os túbulos para ser depurado em
vez de serem fornecidas pelas arteríolas eferentes, e assim maiores quantidades podem ser
secretadas para os túbulos. As grandes quantidades de acido úrico nos túbulos superam a sua
9
solubilidade, e a precipitação ocorre. O acido úrico continua através dos túbulos na forma
precipitada e aparece na urina como um coágulo esbranquiçado. Como o acido úrico não
permanece na solução, ele não contribui para a pressão osmótica efetiva do fluido tubular, e a
perda obrigatória de água é evitada.
Segundo Sonne et al. (1946), citados por Waldroup et al. (2005), o aumento do requerimento
da glicina em aves jovens ocorre em função da maior excreção de nitrogênio, pois cada
molécula de ácido úrico excretada representa a perda de uma molécula de glicina pelas aves.
O metabolismo das purinas e purimidinas, bases nitrogenadas, possuem uma independência
celular. Isto é, já no intestino delgado aquelas que não foram utilizadas no metabolismo normal
são convertidas em ácido úrico que é excretado ou quando em excesso pode acumular-se nas
articulações no processo conhecido como Gota. No entanto, esta não é a principal via de
excreção, uma vez que grande parte das purinas é absorvida para o fígado e, este sim, encarregase de convertê-las em ácido úrico e excretá-lo por via urinária.
A doença conhecida como gota, que nada mais é do que um processo inflamatório doloroso
resultante do acúmulo de ácido úrico nas articulações, pode ser resultado de uma hiperatividade
da enzima PRPP-sintetase ou por redução da atividade da enzima HGPRTase. Mas ambos são
problemas hereditários, reversíveis mediante a diminuição de alimentação rica em material
celular (carnes vermelhas, principalmente) e uso de medicamentos bloqueadores da síntese de
ácido úrico, como por exemplo, a droga conhecida como alopurinol um inibidor da xantina
oxidase responsável pela conversão das purinas em ácido úrico.
Figura 7. Formação de ácido úrico no intestino e no fígado. Fonte:Vieira, 2003.
10
Excreção direta
O amônio urinário origina-se nos túbulos distais dos rins de mamíferos, em grande parte da
hidrolise de glutamina por glutaminase e em segundo lugar da desaminação de outros
aminoácidos. Os cetoácidos resultantes são usados principalmente para gliconeogênese. A
amônia formada em células tubulares renais reage com íons hidrogênio, produzindo íons
amônio durante a acidose metabólica. A excreção de íons amônio é negligível durante a
alcalose. A excreção de íons amônio ajuda a conservar íons sódio, que são os principais cátions
na regulação da concentração eletrolítica e do equilíbrio acidobásico no sangue. Tal reposição
de íons sódio com íons amônio verifica-se em maior proporção durante a cetose, quando estão
sendo eliminados corpos cetônicos pela urina. Os animais aquáticos que excretam excesso de
nitrogênio como íons amônio produzem a maior parte da amônia excretora pela ação da
glutaminase sobre a glutamina.
Nos animais amoniotélicos (excretores de amônia) o catabolismo dos aminoácidos inicia
com uma transaminação envolvendo o α-cetoglutarato formando-se glutamato. Glutamato
igualmente pode ser formado pela desidrogenase glutâmica utilizando-se de amônia livre. No
entanto, o glutamato não pode atravessar as membranas celulares devido as suas cargas
negativas em pH fisiológico sendo necessário a ligação com mais amônia, processo catalisado
pela glutamina sintetase. A molécula de glutamina formada é neutra, não toxica e capaz de se
translocar e transportar N na forma amídica para o fígado ou para as guelras no caso dos peixes.
Nas guelras a glutamina sofre ação da glutaminase aí presente e libera o N na forma de amônia
que é excretada para o meio exterior.
Existem 4 mecanismos conhecidos para a excreção da amônia via epitélio branquial, que
podem ser observados na Figura 8.
11
Figura 8. Mecanismos de excreção de amônia do sangue para a água através da superfície mucosa do
epitélio branquial. Fonte: Bombardelli, 2004.
Considerações finais
De maneira geral o metabolismo proteico dos animais consiste na ingestão, digestão e
absorção e os aminoácidos absorvidos são direcionados para a síntese proteica ou para a síntese
de compostos não proteicos, ou ainda podem ser desviados para vias de degradação para a
geração de energia. Os grupos amino liberados dos processos de degradação sejam oriundos da
proteína da dieta ou degradação tissular precisam ser convertidos em compostos menos tóxicos
para serem excretados, exceto os peixes que conseguem excretar amônia junto a grande volume
de água. A amônia é um composto tóxico para o organismo por se liga aos intermediários do
ciclo de Krebs promovendo a depleção de ATP ou por precisar de íons H+ o que pode
desencadear quadros de alcalose metabólica.
Referências bibliográficas
BOMBARDELLI, R. A.; MEURER, F.; SYPERRECK, M. A. Metabolismo proteico em peixes.
Arquivos de Ciência Veterinária e Zoologia Unipar, v. 7, p. 69-79, 2004.
CHAMPE, P. C.; HARVEY, R. A.; FERRIER, D. R.; Tradução: DALMAZ, C.; et al. Bioquímica
Ilustrada. 3º ed. Porto Alegre: Artmed, 2006, 544p.
REECE, W.O. Dukes – Fisiologia dos animais domésticos. 12º Ed. Rio de Janeiro: Editora Guanabara
Koogan S.A., 2006, 926p.
GONZALEZ, F. H. D., SILVA, S. C. Introdução à bioquímica clinica veterinária. 2º Ed. Porto Alegre,
2006, 360p.
12
GOULART, C. C. Utilização de aminoácidos industriais e relação aminoácidos essenciais: não
essenciais em dietas para frangos de corte. (Tese) Programa de Doutorado Integrado em Zootecnia,
da Universidade Federal da Paraíba. Areia, Paraíba, 2010, 115p.
NELSON, D. L.; MICHAEL M. COX: Lehninger Princípios de Bioquímica. 3º ed. New York : W. H.
Freeman and Company, 2002, 907p.
SCHMIDT-NIELSEN, K. Fisiologia animal: adaptacao e meio ambiente. Editora Santos. 1996, 600p.
VIEIRA, R. Fundamentos de Bioquímica. Textos didáticos. Belém do Pará. 2003, 159p.
WALDROUP, P.W.; JIANG, Q.; FRITTS, C. A. Effects of glycine and threonine supplementation on
performance of broiler chicks fed diets low in crude protein. International Journal of Poultry
Science, v.4, n.5, p.250-257, 2005.
13