ABORDAGEM DAS NEOPLASIAS MALIGNAS COM ENFOQUE
NOS SARCOMAS DE PARTES MOLES DO TIPO
RABDOMIOSSARCOMA
APPROACH OF MALIGNANT NEOPLASMS WITH FOCUS ON
RHABDOMYOSARCOMA TYPE
Bianca Amanda de Oliveira SOUZA1
Carla Silva SIQUEIRA2
RESUMO
O objetivo do trabalho foi realizar uma revisão de literatura acerca das principais
características do rabdomiossarcoma para elucidar melhor a comunidade científica. O
Rabdomiossarcoma é uma neoplasia maligna de partes moles, predominantemente
encontrada em crianças, pode acometer várias regiões do corpo, porém é mais comumente
encontrado na cabeça e pescoço. Sua manifestação clínica é de difícil diagnóstico, levando
a um descobrimento e tratamento tardio. Seus subtipos variam de acordo com padrão
histopatológico e cada um leva a uma forma de tratamento, diagnóstico e prognóstico
diferente. Quanto mais precoce sua descoberta, melhor seu prognóstico para os pacientes.
UNITERMOS: Câncer; Rabdomiossarcoma; Genética.
INTRODUÇÃO
O câncer atualmente é um grave problema
enfrentado pela sociedade, mesmo diante de tantos
avanços da medicina frente a diagnóstico e tratamento
do doente. 1 Segundo Smith 2 (2002), nos Estados
Unidos, foram diagnosticados cerca de 12.400 casos
novos de câncer entre crianças e adolescentes
menores de 20 anos, constituindo a primeira causa
de óbito por doença em crianças maiores de um ano
de idade.
Segundo dados da Organização Mundial da
Saúde (OMS), em 2012 o câncer foi considerado a
segunda doença que mais causou óbitos na região
das Américas, com uma estimativa de 2,8 milhões
de novos casos e 1,3 milhões de mortes. Devido o
envelhecimento da população, e com a transição
epidemiológica na América Latina e no Caribe, a
quantidade de casos de câncer está projetada para
aumentar de forma significativa. 3
De acordo com dados científicos, baseados em
diminuição de fatores de risco e em devidas
prevenções primários, cerca de 40% dos casos de
câncer podem ser evitados, e outros 30% tem chances
de cura se houver detecção precoce e tratamento
adequado. 3
Dentre os diversos tipos de câncer, os
sarcomas de partes moles são considerados
neoplasias raras, pois sua incidência é de 1% em
adultos e 1 5% em c rianças , sendo de dif ícil
diagnóstico. Esse fato se explica pela ausência de
sintomas diferenciais para a detecção precoce da
doença. 4-5
Os sarcomas são tumores malignos, que se
iniciam na camada mesenquimal e podem acometer
várias faixas etárias, sendo mais comuns em crianças,
adolescentes, e adultos jovens. Podem ser divididos
e m di v erso s s ubt ipos hi s to ló gic os co mo :
fibrossarcoma, lipossarcoma, leimiossarcoma,
rabdomiossarcoma, sarcoma epitelióide, entre outros,
dependendo do seu tecido de origem. 6-7
Dentre
o s div ers os
subt i po s
o
Rabdomiossarcoma é o mais encontrado em crianças,
se ndo um subtipo ori gi nário da musc ula tura
e sque lét ic a e s tria da e deriv a do de c é lula s
mesenquimais primitivas. O sítio primário mais
comum em crianças e adolescentes é na região da
cabeça e pescoço, seguida pelo trato gênito-urinário,
extremidades, tórax e retroperitônio. 8
As
manif e st a çõ es
clí ni ca s
do
Rabomiossarcoma dependem de sua localização
sendo que, na região da cabeça e pescoço, os sítios
mais atacados são a órbita, sítios parameníngeos, e
sítios não parameníngeos. 9-10
Histologicamente o Rabdomiossarcoma é
dividido em cinco tipos distintos: embrionário,
botrióide, alveolar, pleomórfico, e de células fusiformes,
e em cada subtipo nota-se o predomínio de estruturas
características. 8, 11-12
O diagnóstico dos sarcomas de partes moles
de tro nco e extremidades muitas veze s, são
1 Aluna de graduação do curso de Biomedicina da Faculdade Presidente Antônio Carlos de Uberlândia.
2 Professora titular do curso de Biomedicina da Faculdade Presidente Antônio Carlos de Uberlândia.
Revista Odontológica de Araçatuba, v.35, n.2, p. 19-25, Julho/Dezembro, 2014
19
percebidos tardiamente e não são tratados com devida
importância e incluídos como diagnóstico diferencial,
assim a lesão tumoral é descoberta quando já atingiu
dimensões elevadas. 7
É de suma importância que na avaliação dos
pacientes os profissionais da saúde tenham a
percepção de que aquela determinada lesão pode ser
sarcomatosa, para que sejam feitos diagnósticos
mais precisos e um tratamento mais específico, e
não cause um prejuízo ao prognóstico do paciente. 7
O t ra t ament o pa ra pa c ie nt e s co m
Rabdomiossarcoma resulta em combinações de
cirurgia, quimioterapia ou radioterapia, sendo
escolhido individualmente devido ao estadiamento de
cada paciente em questão. 11-13
Est a pes quis a tem como finalida de,
primeiramente, abordar a raridade dos Sarcomas,
destacando o tipo Rabdomiossarcoma, em relação a
outras formas de aparecimento do câncer. Foi possível
destacar um subtipo que ocorre na maior parte em
crianças, sendo importante destacar sua forma de
diagnóstico que, nesses casos, são feitos mais tardio
devido o não aparecimento de sintomas diferenciais. 4-5
O objetivo do trabalho em questão é identificar
os principais aspectos dessa neoplasia para um
melhor conhecimento e aprofundamento da mesma.
MATERIAL E MÉTODO
Para a consolidação do mesmo, foi realizada
uma pesquisa descritiva exploratória aprofundada por
meio de materiais bibliográficos sobre o tema.
A base de pesquisa é o banco de dados da
Biblioteca Virtual em saúde (BVS), utilizando como
fontes a Literatura Latino-Americana e do Caribe em
Ciências da Saúde (LILACS), Scientific Electronic
Libray Online (SCIELO) e a Medical Literature Analysis
and Retrieval System Online (MEDLINE). As palavraschave utilizadas para pesquisa foram: Câncer;
Sarcomas; Rabdomiossarcoma, e os trabalhos
selecionados variam de 1969 a 2013.
A amostra do estudo foi constituída de todas
as publicações indexadas no banco de dados das
citadas fontes, que abordam a questão norteadora
do presente estudo. Foram incluídos textos e
resumos no idioma, inglês, espanhol e português.
Foram excluídos apenas os estudos publicados em
duplicidade em mais de uma base de dados.
A classificação epidemiológica de câncer no
Brasil indica uma transição, o que compreende um
aumento entre os tipos de câncer normalmente
associados a elevado nível sócio econômico – câncer
de mama, próstata, cólon e reto – e ao mesmo tempo,
a existência de índices constantemente elevados de
tumores normalmente associados com a pobreza –
câncer de colo de útero, pênis, estômago e cavidade
oral. 16
No Brasil, o câncer é considerado a terceira
razão de óbitos por patologias em crianças entre um
e 14 anos, sendo relatada como a primeira doença
que mais ocasiona mortes também em crianças com
idades entre cinco e 14 anos, tanto no município como
no Estado de São Paulo. 17
Sarcomas de partes moles
Os tumores mesenquimais malignos de tecido
mole são denominados de sarcomas de partes moles,
sendo considerados raros por seu acometimento
corresponder a 1% de cânceres em adultos, e 15%
em crianças. 18-19
Os sarcomas normalmente aparecem como
lesões profundas, mas que podem afetar a pele e o
tecido subcutâneo. Esses tumores podem determinar
lesões cutâneas de três formas diferentes: 1originando-se no tecido subcutâneo; 2- por extensão
direta de lesões profundas; 3- por envolvimento
metastático da pele, fenômeno extremamente raro. 20
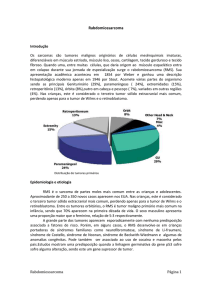
O Rabdomiossarcoma é um subtipo originário
da musculatura esquelética e é o mais comum em
crianças, correspondendo a 50% destes tumores,
diferentemente dos adultos, onde essa porcentagem
cai pra 10%. Nesses, outros sarcomas de partes
moles dev em s e r co ns iderado s, t ai s co mo :
li po ss arco ma , his ti oc it oma, angi os sa rc oma,
leiomiossarcoma, conforme podemos observar na
figura 1. 21-22
Epidemiologia do Câncer
Segundo o Instituto Nacional do Câncer (INCA),
a estimativa do número de novos casos de câncer no
Brasil aponta para 576 mil, incluindo os casos de pele
não melanoma, para os anos de 2014 e 2015. Esses
dados mostram a magnitude desse problema no país.
É considerado como um grande problema de saúde
pública, tanto em países desenvolvidos, como nos
países em desenvolvimento, se tornando a causa de
mais de seis milhões de mortes a cada ano, retratando
em torno de 12% a razão de óbitos no mundo. 14-15
Revista Odontológica de Araçatuba, v.35, n.2, p. 19-25, Julho/Dezembro, 2014
Figura 1 – Diferenciação de células mesenquimais e
desenvolvimento de diversos tipos de sarcomas.
Fonte: Neves et al. 22, 2003.
20
Etiopatogenia
Com a finalidade de buscarem informações de
principais causas de alguns tipos de câncer os
pesquisadores Li e Fraumeni 23 (1969) revisaram 280
prontuários e 418 atestados de óbito de crianças que
tiveram o diagnóstico de rabdomioss arcoma.
Pesquisando mais sobre acontecimentos na família
dos pacientes em questão, foi observado que dentre
as famílias estudadas, cinco exibiam um perfil de
elevada incidência de tumores, inclusive as com
histórico de sarcoma na infância e casos de câncer
de mama em idade jovem. Diante do levantamento
proposto pode ser observado que os tumores malignos
são menos frequentes em idade precoce, sugerindo
então um padrão de propagação hereditária. Assim
foi intitulada a síndrome de Li-Fraumeni. 23
Outra alteração genética comum identificada
nos cânceres é a mutação do gene TP53, o qual é
responsável por inativar as funções regulatórias de
crescimento celular, também ocasiona a perda de
atividade supressora do tumor, e em alguns casos
confere uma função promotora do tumor, assim
estimula os genes envolvidos na proliferação celular,
e no progre sso de sobrev ida da célula e na
angiogênese.24-25
Como um auxílio na prevenção do câncer, a
mutação de p53 tem como consequência a inativação
de suas funções. O aparecimento de apenas uma
cadeia alterada é considerado o suficiente para a
perda de função, e para a formação de um fenótipo
alterado, onde o indivíduo passa a apresentar dentro
de seu ‘‘pool’’ proteico 80% de moléculas anormais,
e 20% normais. 25
Em casos de pacientes diagnosticados com
ra bdomios sarc oma , supõe -se que 9% serão
portadores de casos de alterações germinativas na
célula TP53, estes podem ou não possuírem relato
familiar de outros tumores. 26
Em 70% dos casos de rabdomiossarcomas do
t ipo a lv e ola r, o c orre a t ra nsloc a çã o
t(2;13)(q35:q14),que faz a fusão entre 5’ do gene PAX3
com 3’ de FOXO1A. Em outros 10%, ocorre fusão
entre o gene PAX7 ao FOXO1A. No restante dos 20%,
não há detecção de fusão de genes em exames de
rotina (Mehra et al. 27, 2008)
O Rabdomiossarcoma do tipo pleomórfico
apresenta carióticos complexos, não-específicos.
Ano rma li da des es trutura i s numéri ca s e
des ba lance a da s s ão co muns co mo ganho s
(cromossomos 1, 5, 8, 14, 18, 20, e 22) e perdas
(cromossosmos 2, 5, 6, 10, 11, 13, 14, 15, 16, 17,
18, 19 e Y) de cromossomos, sendo que a perda dos
cromossomos 2, 13, 14, 15,16, e 19 são os mais
frequentes. Esse tipo de lesão não contem a
translocação t(2;13) ou t(1;13) que é característica
ao subtipo alveolar. 28
Características clínicas
O Rabdomiossarcoma é um tumor maligno
provenientes de células mesenquimais primitivas, são
constantemente encontrados em crianças podendo
acometer várias partes do corpo. Porém é comumente
encontrado na região da cabeça e pescoço (Figura
2), seguidos pelo trato-gênito urinário e extremidades
do corpo. 29-31
Figura 2 – Rabdomiossarcoma em criança, em região de asa
de nariz. Fonte: Lima et al. 45, 2011.
Em alguns casos de tumores encontrados na
região da cabeça e pescoço, é possível sua
observação devido à dor ou distúrbio funcional que
ele causa, não conseguindo ser detectado pelo exame
físico. São diversas as formas que o tumor pode
apresentar, sendo mais comuns por ter massa cervical
indolor, por causar obstrução nasal, rinorreia e otites
médias frequentes. 32- 34
Rabdomiossarcoma do tipo pleomórfico
usualmente ocorre em extremidades, especialmente
na coxa, em homens de meia-idade. É um tumor
bastante agressivo, propenso à recorrência e
metástase nos pulmões. 28
Características histopatológicas e Imunohistoquímicas
O Rabdomiossarcoma histologicamente é
classificado em subtipos como: alveolar, pleomórfico
Revista Odontológica de Araçatuba, v.35, n.2, p. 19-25, Julho/Dezembro, 2014
21
e embrionário sendo este subdividido em botrióide e
spindle c ell. Segundo o I NCA, em criança s,
aproximadamente 60% dos tumores são embrionários,
20% alveolares, 15% não classificados e 5%
pleomórficos. 8, 11-12
Histopalogicamente, o rabdomiossarcoma do
tipo embrionário é composto por células primitivas
mesenquimais, com núcleo central e ligeira eosinofilia
citoplasmática e, a medida que vão se diferenciando,
tornam-se mais alongadas e com o citoplasma mais
eosinofílico. A diferenciação ocorre com mais evidência
depois da quimioterapia. Células diferenciadas
marcam mioglobina, miosina e, em alguns casos,
desmina e actina músculo específica. 3 (Figura 3A).
Já o subtipo alveolar exibe células pequenas e
redondas com características citológicas que lembram
o linfoma, mas com diferenciação mioblástica. A
maioria apresenta estroma fibroso e histologia
embrionária, com produção de septos fibrovasculares
que separam as células tumorais.3 (Figura 3B). A
marcação imuno-histoquímica ocorre com expressão
de vimentina, actina músculo específica, desmina,
miogenina e MyoD1. 27
Por f i m, o ra bdo mi o ss a rc oma do t ipo
pleomórfico é composto por células pequenas e
redondas, indiferenciadas, permeadas por células
poligonais com citoplasma eosinofílico denso (Figura
3C), expressando os mesmos marcados imunohistoquímicos dos tipos anteriores. 3
Figura 3 – Características histopatológicas de
Rabdomiossarcoma do tipo embrionário (A), alveolar (B) e (B)
e pleomórfico (C). Fonte: WHO 3, 2002.
Diagnóstico
Para realização de um diagnóstico mais preciso
e correto, deve ser acrescentado um relato clínico
completo do paciente, incluindo exame físico,
hemograma, perfil bioquímico sanguíneo incluindo
enzimas hepáticas, nasofibroscopia, tomografia
computadorizada, ressonância magnética e biópsia
com anatomopatológico. 8
Os exames de imagem são responsáveis por
fazer uma identific ação mais precis a para o
diagnóstico, a tomografia computadorizada exibi a
dimensão e as relações das doenças de acordo com
as estruturas vitais, o remodelamento ósseo é
responsável por preconizar se é um tumor benigno ou
uma patologia de propagação lenta, por sua vez, a
destruição óssea e perda de tecidos moles indicam
malignidade. A ressonância magnética possibilita uma
melhor resposta para tecidos moles e supressão de
gordura e m vo lt a do s s ei os da fa ce , fo ss a
pterigopalatina e fossa infratemporal sensibiliza o
exame para a extensão tumoral. A distinção entre
tumor, musculatura, secreções e espessamento
muc os o t a mbém é me lho r i de nti fi c ada pela
ressonância, bem como a análise de estruturas
perineurais, perivasculares e de invasão intracraniana.
32, 35
Contudo, após ter sido realizado o diagnóstico
e constatado o Rabdomiossarcoma, é feita uma
análise para verificar e atestar a área acometida pela
doença, e assim dar início ao tratamento. Nos exames
a serem executados estão inclusos tomografia
computadorizada de tórax, punção de medula óssea,
cintilografia óssea e ressonância magnética de base
de crânio e cérebro. 9
Exa me s c omo RT-PCR e FI SH sã o
essenciais para as análises dos tipos de alterações
moleculares, principalmente em estágios precoces.
Revista Odontológica de Araçatuba, v.35, n.2, p. 19-25, Julho/Dezembro, 2014
22
Quanto menos usuais forem as características
his topa toló gicas , ma is i ndic ada é a a náli se,
principalmente nos tipos alveolares e embrionários. 28
Tratamento
O tipo de tratamento a ser utilizado depende
de fatores como sítio de recorrência de cada paciente.
É preconizado em casos de micrometástases, a
ressecção cirúrgica e posterior combinação de
quimioterapia auxiliar. É necessário complementar o
tratamento com radioterapia, em casos que não é
possível a retirada total do tumor. Esse tipo de terapia
multimodal faz com que a sobrevida dos pacientes
tenha um aumento de cinco anos. 32, 36-37
O Intergroup Rhabdomyosarcoma Study (IRS)
indica como tratamento inicial a retirada do tumor
caso não ocorra dano funcional ao paciente em
questão, e depois sessões de quimioterapia como
complementação. Em pacientes onde não é possível
a remoção deste como método introdutório, é
a co ns e lhá ve l s e ini ci a r o t ra ta ment o co m
quimioterapia. 38-39
Por fim, como padrão ouro, o tratamento
principal continua sendo a cirurgia para retirada do
tumor primário. Antigamente a regra era a amputação
do membro onde havia se instalado a lesão, após
avanços na medicina o tratamento consiste em
preservação do membro em mais de 90% dos casos.
40-41
Prognóstico
O pro gnó s ti co pa ra pa ci e nt e s co m
Rabdomiossarcoma é estipulado mediante vários
parâmetros, como o tamanho do tumor, o sítio primário,
o subtipo histológico, estágio tumoral, idade do
paciente, bem como extensão da doença e sua
ressecabilidade. 33, 42-43
Outros fatores a serem destacados são que
tumores invasivos apenas na órbita apresentam um
melhor prognóstico, ao mesmo tempo em que tumores
de sítios parameníngeos sendo incorporados também
cavidade nasal, seios paranasais, orelha média,
nasofaringe, mastoide, fossa infratemporal, e fossa
pterigopalatina exibem pior prognóstico. 8, 11, 32, 37
O subtipo embrionário apresenta melhor
prognóstico em crianças, sendo mais agressivo em
adultos. O subtipo alveolar apresenta sobrevida baixa
por possuir maior risco de metástase à distância. Já
o pleomórfico ocorre em maior quantidade nos adultos.
38-39
CONCLUSÃO
O Rabdomiossarcoma é uma neoplasia que
pode ser encontrada em diversas regiões do corpo,
com características diferentes e acometimento maior
em crianças e adultos jovens. Possui vários subtipos
que fazem com que sua agressividade, tratamento e
prognóstico sejam variados. Pode ser notada em
virtude do aparecimento de uma massa indolor, assim
seu tratamento inicial consiste em ressecção
cirúrgica, mas na maioria das vezes devido ao local
acometido pode ser considerado de difícil diagnóstico,
tornando seu tratamento tardio. Quanto mais tardio o
tratamento, pior o prognóstico e maiores as sequelas
aos pacientes.
ABSTRACT
The objective was to conduct a literature review of the
main rabdomiossrcoma features to better elucidate
the scientific community. The Rhabdomyosarcoma is
a malignant neoplasm of soft tissues, predominantly
found in children, can affect various parts of the body,
but is most commonly found in the head and neck.
Their clinical presentation is difficult to diagnose,
leading to discovery and delayed treatment. Its
subtypes vary according to standard histopathological
and each leads to a form of treatment, diagnosis and
prognosis different. The earlier finding their better
prognosis for patients.
U NITERMS: Canc er; Rhabdomy os a rc o ma ;
Genetics.
REFERÊNCIAS
1. Cardoso FT. Câncer infantil: aspectos emocionais
e atuação do psicólogo. Rev SBPH, 2007;
10(1):25-52.
2. Smith MA, Ries LAG. Childhood cancer: incidence,
survival and mortality. In: Pizzo PA, Poplack DG,
editors. Principles and practice of pediatric
oncology. 4rd ed. Philadelphia: Ed. LippincontRaven, 2002.p. 1-12.
3. W HO. National cancer control programs:policies
and managerial guidelines. 2nd
ed. 2002 Disponível em: <http://www.paho.org/hq/
i n d e x . p h p ? o p t i o n = c o m c
ontent &vie w=ar tic le&i d=29 2:
cancer&Itemid=3855&lang=en> Acesso em 10
de Abril 2014.
4. Lin GY, Sun X, Badve S. Pathologic Quiz Case.
Vaginal wall mass in a 47-year-old W oman.
Vaginal rhabdomyoma. Arch Pathol Lab Med
2002; 126(10):1241-2.
5. Burningham Z, Hashibe M, Spector L, Shiffman JD.
The epidemiology of sarcoma. Clin Res Sarcoma
2012;2(1):14.
6 . Ama nkwah EK, Ant hony PC, Re ed DR.
Epi de mio logy
a nd
thera pi e s
fo r
metastatic sarcoma.Clin Epidemiol 2013; 5: 14762.
7. Verweij J, Baker LH. Future treatment of soft tissue
sarcomas will be driven by histological subtype
and molecular aberrations. Eur J Cancer 2010;
46(5): 863-68.
Revista Odontológica de Araçatuba, v.35, n.2, p. 19-25, Julho/Dezembro, 2014
23
8. Anderson GJ, Tom LWC, Womer RB, Handler SD,
Wetmore RF, Potsic WP. Rhabdomyosarcoma
of the he ad and nec k i n Children. Arch
Otolaryngol Head Neck Surg 1990; 116(4): 42831.
9. Moretti G, Guimaraes R, Oliveira KM, Sanjar F,
Voegels RL. Rhabdomyosarcoma of the head
and neck: 24 cases and literature review. Braz
J Otorhinolaryngol 2010; 76(4): 533-7.
10. W olden SL, Alektiar KM. Sarcomas across the
age spectrum. Semin Radiat Oncol 2010; 20(1):
45-51.
11. Wurm J, Constantinidis J, Grabenbauer GG, Iro
H. Rhabdomyosar­comas of the nose and
paranasal sinuses: treatment results in 15
cases. Otolaryngol Head Neck Surg 2005;
133(1): 42-50.
12. Luu QC, Kasky JL, Moore TB, Nelson S, Wang
MB. Trea t me nt of e m­bryo na l
rhabdomyosarcoma of the sinus and orbit with
chemotherapy, radiation, and endoscopic
surgery. J Pediatr Surg 2006; 41(6): e15-7.
13. Kraus DH, Saenz NC, Gollamud S, Heller G,
Moustakis M, Gardiner S, et al. Pediatric
Rhabdomyosarcoma of head and neck. Am J
Surg 1997; 174(5): 556-60.
14. INCA. Estimativa 2014: incidência de câncer no
Brasil. Rio de Janeiro: Instituto Nacional de
Câncer/ Ministério da Saúde, 2014.
15. W orld Health Organization. Polic ies and
managerial guidelines for national cancer control
programs. Rev Panam Salud Publica 2002;
12(5): 366-70.
16. Koifman S, Koifman R. Environment and cancer
in Brazil: an overview from a public health
perspective. Mutat Res 2003; 544(2-3): 305-11.
17. Rodrigues KE, Camargo B. Diagnóstico precoce
do câncer infantil: responsabilidade de todos.
Rev Assoc Med Bras 2003; 49(1): 29-34.
18. Rajput A, Kraybill WG. Clinical trials and soft
tissue sarcomas. Surg Oncol Clin N Am 2003;
12(2): 485-97.
19. Shmookler B, Bickels J, Jelinek J, Sugarbaker
P, Malawer M. Bone and soft-tissue sarcomas:
epidemio lo gy, ra di ology, pat ho lo gy a nd
fundamentals of surgical treatment. In: Malawer
MM, Sugarbaker PH. Musculoskeletal Cancer
Surge ry. W ashingto n: Kluwe r Aca demic
Publishers; 2001. 1761-74.
20. Fletcher CDM, McKee PH. Sarcoma s a
clinic opat hologic al guide wi th part icular
ref ere nc e t o c ut ane ous manif e st a ti on.
Dermatofibrossarcoma protuberans, malignant
fibrous histiocytoma and epthelioid sarcoma of
Enzinger. Clin Exp Dermatol 1984; 9(5): 45165.
21. Callander TA, Weber BS, Janjan N, Benjamin R,
Zaher M, Wolf P, et al. Rhabdomyosarcoma of
nose and paranasal sinuses in adults and
children. Otolaryngol Head Neck Surg 1995;
112(2): 252-7.
22. Neves BMJ, Pontes PAL, Caran EM, Figueiredi
C, Weckx LLM, Fujita RR. Rabdomiossarcoma
de cabeça e pescoço na infância. Rev Bras
Otorrinolaringol 2003; 69(1): 24-8.
23. Li FP, Fraumeni JFJr. Rhabdomyosarcoma in
children: epidemiologic study and identification
of a familial cancer syndrome. J Natl Cancer Inst
1969; 43(6): 1365-73.
24. Frebourg T, Barbier N, Yan YX, Garber JE, Dreyfus
M, Fraumeni J Jr, et al. Germ-line p53 mutation
in 15 families with Li-Fraumeni syndrome. Am J
Hum Genet 1995; 56(3): 608-15.
25. Cadwell C, Zambetti GP. The effects of wild-type
p53 tumor supressor activity and mutant p53 gainof-function on cell growth. Gene 2001; 277(1-2):
15-30.
26. Diller L, Sexsmith E, Gottlieb A, Li FP, Malkin D.
Germline p53 mutations are frequently detected
in young children with rhabdomyosarcoma. J Clin
Invest 1995; 95(4): 1606-11.
27. Mehra S, de la Roza G, Tull J, Shrimpton A, Valente
A, Zhang S. Detection of FOXO1 (FKHR) gene
break-apart by fluorescence in situ hybridization
in formalin-fixed, paraffin-embedded alveolar
rhabdomyosarcomas and its clinicopathologic
correlation. Diagn Mol Pathol 2008; 17(1): 1420.
2 8. J a in S, Xu R, Pri e to VG, L ee P.
Molec ula r c la ss i fi ca t io n o f s of t t is sue
sarcomas and its clinical applications. Int J Clin
Exp Pathol 2010; 23(3-4): 416-28.
29. Maurer HM, Beltangady M, Gehan EA, Crist W,
Hammond D,Hays DM, et al. The intergroup
rhabdomyosarcoma study-I. A final report. Cancer
1988; 61(2): 209-220.
30. Maurer HM, Gehan EA, Beltangady M, Crist W,
Dickman PS, Donaldson SS, et al. The intergroup
rhabdomyosarcoma study-II. Cancer 1993; 71(5):
1904-22.
31. Karakas Z, Agaoglu L, Biner B, Devecioglu O, Anak
S, Ya lma n N, e t a l. Re sults o f
rhabdomyosarcoma treatment in a developing
country. Acta Med Okayama 2000; 54(4): 173-7.
32. Bonila JA, Healy GB. Management of malignant
head and neck tumors in children. Pediatr Clin
North Am 1989; 36(6): 1443-50.
33. Hermann BW, Sotelo-Avila C, Eisenbeis JF.
Pediatric sinonasal rha­bdomyosarcoma: three
cases and a review of the literature. Am J
Otolaryngol 2003; 24(3): 174-80.
34. Walterhouse D, Watson A. Optimal management
strategies for rha­bsomyosarcoma in children.
Pediatric Drugs 2007; 9(6): 391-400.
35. Burkat CN, Lucarelli MJ. Rhabdomyosarcoma
masquerading as acute dacryocystitis. Ophthal
Plast Reconstr Surg 2005; 21(6): 456-8.
Revista Odontológica de Araçatuba, v.35, n.2, p. 19-25, Julho/Dezembro, 2014
24
36. Daya H, Chan HS, Sirkin W, Forte V. Pediatric
rhabdomyosarcoma of the head and neck: is there
a pla ce f or s urgi ca l management? Arc h
Otolaryngol Head Neck Surg 2000; 126(4): 46872.
37. Brecher AR, Reyes-Mugica M, Kamino H, Chang
MW. Co nge ni ta l pri mary c ut a ne ous
rhabdomyosarcoma in a neonate. Pediatric
Dermatology 2003; 20(4): 35-38.
38. Ferman SE. Análise de sobrevida de pacientes
pediátricos portadores de rabdomiossarcoma: 18
anos de experiência do Instituto Nacional de
Câncer - RJ. Tese apresentada à Faculdade de
Medicina da Uni­versidade de São Paulo, 2005.
39. Raney B, Anderson J, Breneman J, Donaldson
SS, Huh W, Maurer H, et al. Results in pacients
wit h cranial para meni ngeal sarc oma and
met as t as e s
tre at ed
in
I nte rgroup
Rhabdomyosarc oma Study Group (IRSG)
Protocols II-IV, 1978-1997: report from the
Children´s On­cology Group. Pediatr Blood
Cancer 2008; 51(1): 17-22.
40. Lawrence W Jr, Donegan W, Natarajan N, Mettlin
C, Beart R, W inchester D. Adult soft tissue
sarcomas. A pattern of care survey of the
American College of Surgeons. Ann Surg 1987;
205(4): 349-59.
41. Shiu MH, Castro EB, Hajdu SI, Fortner JG. Surgical
treatment of 297 soft tissue sarcomas of the lower
extremity. Ann Surg 1975; 182(5): 597-602.
42. Agarwala S. Pediatric rhabdomyosarcomas and
non rhabdomyosarcoma soft tissue Sarcoma. J
Indian Assoc Pediatr Surg 2006; 11(1): 15-23.
43. Tari AS, Amoli FA, Rajabi MT, Esfahani MR, Rahimi
A. Cutaneous embryonal rhabdomyosarcoma
presenting as a nodule on cheek; a case report
and review of literature. Orbit 2006; 25(3): 235-8.
44. Dantonello TM, Int-Veen C, Winkler P, et al.: Initial
patient characteristics can predict pattern and
risk of relapse in localized rhabdomyosarcoma.
J Clin Oncol 2008; 26(3): 406-13.
45. Lima LL, Rodrigues CAC, Pereira PMR, Schettini
APM, Tupinambá W L. Rabdomiossarcoma
alveolar cutâneo primário em paciente pediátrico.
An Bras Dermatol 2011; 86(2): 363-5.
Revista Odontológica de Araçatuba, v.35, n.2, p. 19-25, Julho/Dezembro, 2014
ENDEREÇO PARA CORRESPONDÊNCIA
Carla Silva Siqueira
Rua Planalto, 120 - apto. 1203 A
CEP: 38408-064
Bairro: Santa Mônica
Uberlândia – MG - Brasil
[email protected]
25