Físico-Química I
Profa. Dra. Carla Dalmolin
Transformações Físicas
•
Físico-Química, cap. 4: Transformações Físicas de
Substâncias Puras
Transformações Físicas
Transformações onde não ocorrem mudança na composição química
Transformações de fase
Tendência dos sistemas
atingirem o mínimo de
Energia de Gibbs
𝐺𝑓𝑖𝑛𝑎𝑙 < 𝐺𝑖𝑛𝑖𝑐𝑖𝑎𝑙
∆𝐺 = 𝐺𝑓𝑖𝑛𝑎𝑙 − 𝐺𝑖𝑛𝑖𝑐𝑖𝑎𝑙 < 0
A T e p constantes
Fase
Forma da matéria que é homogênea no que se refere à sua composição
química e ao estado físico
Sólido
Líquido
Formas Alotrópicas
Ex.: Grafite e Diamante
Fósforo branco e fósforo vermelho
Calcita e aragonita (CaCO3)
Gás
Número de Fases (P)
𝐶𝑎𝐶𝑂3 𝑠
Gás
(P = 1)
Água + NaCl
(P = 1)
Mistura de gases
(P = 1)
Água + Óleo
(P = 2)
Água e gelo
(P = 2)
∆
𝐶𝑎𝑂 𝑠 + 𝐶𝑂2 (𝑔)
Carbonato de cálcio em decomposição
(P = 3)
Leite
(P = 2)
Transições de Fase
Conversão espontânea de uma fase em outra que ocorre numa
temperatura característica para uma dada pressão
∆𝑓𝑢𝑠 𝐺 0 = ∆𝑓𝑢𝑠 𝐻0 − 𝑇∆𝑆 0
∆𝑓𝑢𝑠 𝐺 0 = 6001 − 273 22
0
∆𝑓𝑢𝑠 𝐺 = 0
S0
⇄
⇄
T = 273 K
T = 373 K
= 22,0 J/K.mol
fusH0 = 6,01 kJ/mol
S0 = 109 J/K.mol
ebH0 = 40,1 kJ/mol
∆𝑒𝑏 𝐺 0 = ∆𝑓𝑢𝑠 𝐻0 − 𝑇∆𝑆 0
∆𝑒𝑏 𝐺 0 = 40100 − 373 109
∆𝑒𝑏 𝐺 0 = 0
Na temperatura de transição de fase, as duas fases da mesma substância estão em
equilíbrio dinâmico (G = 0)
Não há preferência termodinâmica por nenhuma direção quando duas fases estão em
equilíbrio
Equilíbrio Físico
Temperatura, T
Curva de resfriamento para uma substância pura
Tf
Resfriamento do líquido
Congelamento
Resfriamento do sólido
Tempo, t
Estabilidade das Fases
Equilíbrio: transG = 0
𝐺𝑚 𝑠ó𝑙𝑖𝑑𝑜 = 𝐺𝑚 (𝑙í𝑞𝑢𝑖𝑑𝑜)
𝜇 𝑠ó𝑙𝑖𝑑𝑜 = 𝜇(𝑙í𝑞𝑢𝑖𝑑𝑜)
⇌
Sólido ⇌ Líquido
Potencial químico
∆𝐺 = 𝜇 𝑙í𝑞𝑢𝑖𝑑𝑜 − 𝜇(𝑠ó𝑙𝑖𝑑𝑜
∆𝐺 = 0
Medida do potencial (tendência) que uma substância apresenta de sofrer uma
mudança em um dado sistema
No equilíbrio, o potencial químico de uma substância é o mesmo em toda a amostra,
qualquer que seja o número de fases presentes.
Fases Metaestáveis
Fases termodinamicamente instáveis (𝜇𝑚𝑒𝑡𝑎 > 𝜇𝑒𝑠𝑡á𝑣𝑒𝑙 ) que
persistem porque a transição tem uma velocidade muito lenta
Ex.: Diamante e grafite
𝜇𝑑𝑖𝑎𝑚𝑎𝑛𝑡𝑒 > 𝜇𝑔𝑟𝑎𝑓𝑖𝑡𝑒
∆𝐺 < 0
Diagrama de Fases
Gráfico que mostra as fases mais estáveis em diferentes estados de p e T
Diagrama de fases da água:
Limites de Fases
Limites de fase: curvas que limitam regiões de fases diferentes
As fases vizinhas coexistem em equilíbrio dinâmico (P = 2)
Na fase vapor, a pressão em que coincide este equilíbrio é a pressão de vapor
naquela temperatura
Temperatura
Transições de Fase
Conversão espontânea de uma fase em outra
Ocorre apenas numa temperatura característica para cada pressão
Ponto triplo: encontro de 3 limites
de fase (as 3 fases coexistem em
equilíbrio dinâmico)
Propriedade característica da
substância e não pode ser
alterada mudando as condições
do sistema
Ponto crítico: final da linha líquido
– vapor
Acima da temperatura crítica, a
substância não pode ser
condensada
Fluido supercrítico
Fluidos Supercríticos
Aquecimento de um líquido num recipiente fechado:
(a) Líquido em equilíbrio com seu vapor
(b) O líquido é aquecido: aumenta a massa específica
𝑚
( 𝑉 ) de vapor e diminui a da fase líquida
(c) As massas específicas do vapor e do líquido se
igualam: não é mais observada uma interface
entre os estados líquido e vapor
Fluido supercrítico
A temperatura em que aparece o fluido
supercrítico é a temperatura crítica, Tc
A pressão de vapor em Tc é a pressão crítica, pc
Ponto Triplo
Condições de pressão e temperatura em que 3 fases diferentes de
uma substância coexistem em equilíbrio
Ponto de encontro de 3 curvas de equilíbrio, simbolizado por T3
Ponto
Crítico
Ponto
Triplo
É a pressão mais baixa em que a fase
líquida pode existir
É a temperatura mais baixa em que o
líquido pode existir
A temperatura mais alta em que o
líquido pode existir é a temperatura
do ponto crítico
Regra das Fases
F=C–P+2
Número de Fases
Variância ou Graus de Liberdade
Número de variáveis intensivas
que podem ser
independentemente alteradas
sem perturbar o número de
fases em equilíbrio
Componentes
Constituintes quimicamente
independentes do sistema necessários
para definir a composição de todas as
fases presentes no sistema
Para o diagrama de fases de uma substância:
E uma fase: F = 1 – 1 + 2 = 2
p e T podem ser alteradas independentemente
E duas fases: F = 1 – 2 + 2 = 1
Uma alteração de p e acompanhada por uma modificação definida em T
E três fases: F = 1 – 3 + 2 = 0: Ponto triplo
Diagrama de Fases do CO2
Tf aumenta com a pressão
É mais difícil fundir o sólido em
altas pressões
O ponto triplo do CO2 está acima
da pressão atmosférica
Não existe CO2 líquido na pressão
atmosférica (1 atm)
O sólido sublima com o aumento
da temperatura: gelo seco
Diagrama de Fases da H2O
Várias fases sólidas diferentes,
identificadas como gelo I, gelo II...
Diferentes arranjos das moléculas
de H2O na fase sólida
Tf diminui com a pressão
Comportamento anômalo
O sólido é menos denso que o
líquido
Apresenta mais de um ponto triplo
Regiões onde diferentes fases do
gelo coexistem em equilíbrio
entre si ou com a fase líquida
Diagrama de Fases do He
A fase sólida só é observada em altas
pressões
Os átomos são tão pequenos que
interagem muito pouco entre si
É preciso manter uma alta pressão
para “forçar” os átomos a se
manterem numa estrutura cristalina
Não existe o equilíbrio sólido-vapor
Há duas fases líquidas
He-I: comporta-se como um líquido
normal
He-II: superfluido – escoa sem
resistência ao fluxo
Aspectos Termodinâmicos das
Transformações Físicas
Primeira Lei da Termodinâmica
Definição de entropia:
𝑑𝑈 = 𝑑𝑞 + 𝑑𝑤
Onde:
Segunda Lei da Termodinâmica
𝑑𝑞𝑟𝑒𝑣 = 𝑇𝑑𝑆
𝑑𝑤𝑟𝑒𝑣 = −𝑝𝑑𝑉
Sistema fechado, de
composição constante que só
efetua trabalho de expansão
Eq. Fundamental da
Termodinâmica
𝑑𝑈 = 𝑇𝑑𝑆 − 𝑝𝑑𝑉
A energia interna (U) do sistema se altera quando S ou V se alteram:
𝜕𝑈
𝑑𝑈 =
𝜕𝑆
𝜕𝑈
𝑑𝑆 +
𝜕𝑉
𝑉
𝜕𝑈
𝜕𝑆
𝑑𝑉
𝑆
𝜕𝑈
𝜕𝑉
=𝑇
𝑉
= −𝑝
Propriedades da Energia de Gibbs
Quando um sistema sofre uma mudança de estado, G se altera, pois H, T e
S também se alteram:
𝐺 = 𝐻 − 𝑇𝑆
𝑑𝐺 = 𝑑𝐻 − 𝑑 𝑇𝑆 = 𝑑𝐻 − 𝑇𝑑𝑆 − 𝑆𝑑𝑇
𝐻 = 𝑈 + 𝑝𝑉
𝑑𝐻 = 𝑑𝑈 + 𝑑 𝑝𝑉 = 𝑑𝑈 + 𝑝𝑑𝑉 + 𝑉𝑑𝑝
𝑑𝐺 = 𝑑𝑈 + 𝑝𝑑𝑉 + 𝑉𝑑𝑝 − 𝑇𝑑𝑆 − 𝑆𝑑𝑇
𝑑𝑈 = 𝑇𝑑𝑆 − 𝑝𝑑𝑉
𝑑𝐺 = 𝑉𝑑𝑝 − 𝑆𝑑𝑇
Eq. Fundamental da
Termoquímica
G é uma função de p e T: consequências combinadas da Primeira e da Segunda Lei da
Termodinâmica de maneira especialmente apropriada para as aplicações químicas
Propriedades da Energia de Gibbs
A partir de 𝑑𝐺 = 𝑉𝑑𝑝 − 𝑆𝑑𝑇, tem-se que:
𝜕𝐺
𝜕𝑇
Como 𝑆 > 0, G sempre diminui com a
elevação de T
G diminui mais acentuadamente
quando a entropia do sistema é
grande (estado gasoso)
= −𝑆
𝑝
𝜕𝐺
𝜕𝑝
=𝑉
𝑇
A energia de Gibbs das fases gasosas
é mais sensível à temperatura que as
fases condensadas
Como 𝑉 > 0, G sempre aumenta com a elevação de p
G é mais sensível à pressão quando o volume do sistema é
grande
A energia de Gibbs molar de um gás é mais sensível à pressão
que nas fases condensadas
Variação da Energia de Gibbs com a
Temperatura
𝜕𝐺
𝜕𝑇
𝐺 = 𝐻 − 𝑇𝑆
= −𝑆
𝑝
−𝑆 =
𝐺−𝐻
𝑇
Equação de Gibbs-Helmholtz
𝜕𝐺
𝜕𝑇
=
𝑝
𝐺−𝐻
𝜕(∆𝐺/𝑇)
⇒
𝑇
𝜕𝑇
=−
𝑝
∆𝐻
𝑇2
A variação da Energia de Gibbs com a temperatura é determinada pela entropia
Como a entropia da fase gasosa é maior que das fases condensadas, a energia
de Gibbs se altera mais acentuadamente na fase gasosa, depois da fase líquida
e em menor grau na fase sólida
Dependência da Estabilidade de Fase com
a Temperatura
𝜕𝐺
𝜕𝑇
= −𝑆
Para uma substância pura: 𝐺𝑚 = 𝜇; então:
𝑝
𝜕𝜇
𝜕𝑇
= −𝑆𝑚
𝑝
Sempre que T aumentar, o potencial químico
() diminui.
Numa determinada temperatura, o potencial
químico do líquido torna-se menor que o do
sólido
O sólido funde
Tf
Numa temperatura ainda mais elevada, o
potencial químico do gás torna-se menor que o
do líquido
O líquido evapora
Tb ou Tvap
Variação da Energia de Gibbs com a
Pressão
onde 𝑑𝑇 = 0
𝑑𝐺 = 𝑉𝑑𝑝 − 𝑆𝑑𝑇
𝑝𝑓
𝑑𝐺 = 𝑉𝑑𝑝 =
𝑉𝑑𝑝
𝑝𝑖
p/1 mol:
𝑝𝑓
𝐺𝑚 (𝑝𝑓 ) = 𝐺𝑚 (𝑝𝑖 )
𝑉𝑚 𝑑𝑝
𝑝𝑖
Para sólidos e líquidos 𝑉𝑚 é praticamente
independente da pressão e ∆𝐺 = 𝑉𝑚 ∆𝑝
Nos gases, 𝑉𝑚 varia com a pressão. Para
gases perfeitos 𝑉𝑚 =
𝑅𝑇
𝑝
e ∆𝐺 = 𝑅𝑇 ln
𝑝0 : pressão inicial padrão (1 bar)
𝑝
𝑝0
Resposta da Fusão à Pressão Aplicada
𝜕𝐺
𝜕𝑝
=𝑉
𝑇
Para uma substância pura: 𝐺𝑚 = 𝜇; então:
𝜕𝜇
𝜕𝑝
= 𝑉𝑚
𝑇
p : na maioria dos casos
𝑉𝑚 𝑙 > 𝑉𝑚 (𝑠), e uma elevação da
pressão causa um aumento do
potencial químico do líquido em
maior proporção que no sólido
Aumento da temperatura de fusão
Exceção: água
𝑉𝑚 𝑙 < 𝑉𝑚 (𝑠)
Curvas de Equilíbrio
No equilíbrio: 𝜇(𝛼) 𝑇,𝑝 = 𝜇(𝛽) 𝑇,𝑝
𝑑𝑝
𝑑𝑇
⇒ coeficiente angular
𝑑𝜇 𝛼 = 𝑑𝜇(𝛽)
𝑑𝐺 = 𝑉𝑑𝑝 − 𝑆𝑑𝑇
𝑑𝜇 = −𝑆𝑚 𝑑𝑇 + 𝑉𝑚 𝑑𝑝
(−𝑆𝑚 𝑑𝑇 + 𝑉𝑚 𝑑𝑝)∝ = (−𝑆𝑚 𝑑𝑇 + 𝑉𝑚 𝑑𝑝)𝛽
𝑉𝑚 𝛽 − 𝑉𝑚 (𝛼) 𝑑𝑝 = 𝑆𝑚 𝛽 − 𝑆𝑚 (𝛼) 𝑑𝑇
𝑑𝑝 ∆𝑡𝑟𝑠 𝑆𝑚
=
𝑑𝑇 ∆𝑡𝑟𝑠 𝑉𝑚
Equilíbrio Sólido-Líquido
Na fusão: ∆𝑓 𝑆 =
∆𝑓 𝐻
𝑇
∆𝑓 𝐻 1
𝑑𝑝 =
𝑑𝑇
∆𝑓 𝑉𝑚 𝑇
∆𝑓 𝑆
∆𝑓 𝐻
𝑑𝑝
=
=
𝑑𝑇 ∆𝑓 𝑉𝑚 𝑇∆𝑓 𝑉𝑚
∆𝑉 > 0
∆𝐻 > 0
∆𝑝 =
𝑝 = 𝑝∗ +
∆𝑓 𝐻
∆𝑓 𝑉𝑚
𝑑𝑝
>0
𝑑𝑇
∆𝑓 𝐻 𝑇2
1
𝑑𝑇 =
ln
𝑇
∆𝑓 𝑉𝑚 𝑇1
∆𝑓 𝐻
(𝑇 − 𝑇 ∗ )
𝑇∆𝑓 𝑉𝑚
Eq. De uma reta com
alto coef. angular
Equilíbrio Líquido-Vapor
Vaporização: ∆𝑣𝑎𝑝 𝑆 =
∆𝑣𝑎𝑝 𝐻 1
𝑑𝑝 =
𝑑𝑇
∆𝑣𝑎𝑝 𝑉𝑚 𝑇
∆𝑣𝑎𝑝 𝐻
𝑇
∆𝑉 > 0
∆𝐻 > 0
∆𝑣𝑎𝑝 𝑉𝑚 ≈ 𝑉𝑚 (𝑔) e 𝑉𝑚 𝑔 =
𝑑𝑝 ∆𝑣𝑎𝑝 𝐻
=
𝑑𝑇 𝑇 𝑅𝑇
𝑝
𝑅𝑇
𝑝
𝑑𝑝
Mas menor que no equilíbrio
>0
Sólido-Líquido
𝑑𝑇
p/ gases perfeitos
𝑑 ln 𝑝 ∆𝑣𝑎𝑝 𝐻
=
𝑑𝑇
𝑅𝑇 2
Eq. De Clausius – Clapeyron
Pressão de Vapor
𝐻2 𝑂(𝑙) ⇄ 𝐻2 𝑂(𝑔)
É a pressão exercida pelo vapor que está em equilíbrio dinâmico com a
fase condensada
Vaporização
A pressão de vapor de um líquido aumenta com a temperatura
Quando a temperatura aumenta, as moléculas tem mais energia para
vencerem as atrações intermoleculares
Quando a pressão de vapor se iguala à pressão do ambiente, o líquido entra
em ebulição
A temperatura em que a pressão de vapor de um líquido se iguala à pressão, é a
temperatura de vaporização do líquido naquela pressão
Líquidos com alta pressão de vapor vaporizam mais facilmente e são
chamados de voláteis
Quanto menores as interações moleculares entre as moléculas do líquido, mais
volátil ele será
Estimativa do Ponto de Ebulição de um Líquido
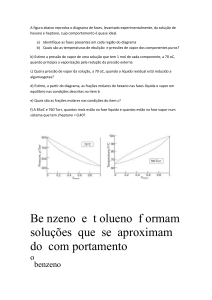
A pressão de vapor do etanol em 34,9 oC é 13,3 kPa. Estime seu ponto de
ebulição normal (p = 1 atm = 101,325 kPa)
Dedução da equação de Clausius-Clapeyron
Variação da Energia de Gibbs de um gás na pressão padrão (1 bar) e em outra pressão
𝐺 𝑔, 𝑝 − 𝐺 0 𝑔 = 𝐻 𝑔, 𝑝 − 𝐻0 𝑔
− 𝑇 𝑆 𝑔, 𝑝 − 𝑆 0 (𝑔)
A entalpia de um gás ideal é independente da pressão: 𝐻 𝑔, 𝑝 − 𝐻0 𝑔 = 0
A variação de entropia de um gás ideal é: 𝑆 𝑔, 𝑝 −
Para um gás ideal: 𝐺 𝑔, 𝑝 = 𝐺 0 𝑔 + 𝑅𝑇 ln
𝑝
𝑝0
𝑆 0 (𝑔)
= 𝑅 ln
𝑝0
𝑝
Se p for exatamente a pressão de vapor do líquido, forma-se o equilíbrio
Líquido ⇄ Gás: ∆𝑒𝑏 𝐺 = 0
𝑝
𝑝
∆𝑒𝑏 𝐺 = 𝐺 𝑔, 𝑝 − 𝐺 0 𝑙 = 𝐺 0 𝑔 + 𝑅𝑇 ln 0 − 𝐺 0 𝑙 = ∆𝑒𝑏 𝐺 0 + 𝑅𝑇 ln 0
𝑝
𝑝
𝑝
0 = ∆𝑒𝑏 𝐺 0 + 𝑅𝑇 ln 0
𝑝
𝑝
∆𝑒𝑏 𝐺 0
ln 0 = −
𝑝
𝑅𝑇
Estimativa do Ponto de Ebulição de um Líquido
𝑝
∆𝑒𝑏 𝐺 0
∆𝑒𝑏 𝐻0 − 𝑇∆𝑒𝑏 𝑆 0
ln 0 = −
=−
𝑝
𝑅𝑇
𝑅𝑇
As pressões de vapor p1 e p2 para duas temperaturas T1 e T2 diferentes podem ser
relacionadas:
𝑝2
𝑝1
∆𝑒𝑏 𝐻0 ∆𝑒𝑏 𝑆 0
∆𝑒𝑏 𝐻0 ∆𝑒𝑏 𝑆 0
ln 0 − ln 0 = −
+
− −
+
𝑝
𝑝
𝑅𝑇2
𝑅
𝑅𝑇1
𝑅
Considerando que H e S são praticamente constantes com T:
𝑝2
𝑝1
∆𝑒𝑏 𝐻0 1
1
ln 0 − ln 0 =
−
𝑝
𝑝
𝑅
𝑇1 𝑇2
𝑝2
∆𝑒𝑏 𝐻0 1
1
ln
=
−
𝑝1
𝑅
𝑇1 𝑇2
Eq. De Clausius – Clapeyron
Usando ebH0(etanol) = 43,5 kJ/mol
1
1
𝑅
𝑝2
1
8,314
13,3
= +
ln
=
+
ln
= 2,86 × 10−3
0
4
𝑇1 𝑇2 ∆𝑒𝑏 𝐻
𝑝1
308 4,35 × 10
101,3
T1 = 350 K
Equilíbrio Sólido-Vapor
Sublimação: ∆𝑠𝑢𝑏 𝑆 =
∆𝑠𝑢𝑏 𝐻
𝑇
𝑑𝑝
∆𝑠𝑢𝑏 𝐻
=
𝑑𝑇 𝑇∆𝑠𝑢𝑏 𝑉𝑚
∆𝑠𝑢𝑏 𝐻 > ∆𝑣𝑎𝑝 𝐻
𝑑𝑝
𝑑𝑇
𝑠𝑢𝑏
𝑑𝑝
>
𝑑𝑇
𝑣𝑎𝑝
Classificação de Ehrenfest
Classificação dos diferentes tipos de transformações de fase
Transições de primeira ordem
Sólido – líquido
Líquido – vapor
Transições de segunda ordem
Condutor – semicondutor
Sólido – sólido
Derivadas das propriedades
termodinâmicas com a temperatura
são descontínuas
Derivadas das propriedades
termodinâmicas com a temperatura
são contínuas
Classificação de Ehrenfest
𝐶𝑝 é infinito na transição de primeira ordem, pois o calor absorvido
provoca a transição ao invés de aumentar T
Transições de
Primeira Ordem
Transições de
Segunda Ordem
𝑑𝑞
𝐶𝑝 =
𝑑𝑇
Interpretação Molecular
Transições de primeira ordem: envolvem realocação de átomos /
moléculas com variação das energias de interação
Transições de segunda ordem: envolvem a modificação da estrutura
cristalina
Transição de fase