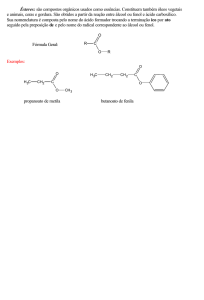
MODELAGEM MOLECULAR DO PROCESSO DE ADSORÇÃO DE
COMPOSTOS FENÓLICOS SOBRE CARVÃO ATIVADO
Antônio Edilson Sousa do NASCIMENTO([email protected])¹, Carlyle
Ribeiro LIMA ([email protected])²,Marlice Cruz MARTELLI ([email protected])¹,
Samira Maria Leão de CARVALHO([email protected])¹, Cláudio Nahum ALVES
([email protected])¹, Davi do Socorro Barros BRASIL ([email protected])¹
¹Universidade Federal do Pará
²Université Paris Diderot
RESUMO: Compostos fenólicos são considerados poluentes aquáticos altamente prejudiciais
até mesmo em baixas concentrações e o aumento da geração de efluentes ricos em tais tem
sido motivo de preocupação nos últimos anos. Isto tem proporcionado um aumento significativo
de trabalhos com intuito de desenvolver tecnologias alternativas para remoção destas
substâncias. Nesse contexto, destaca-se o processo de adsorção sobre carvão ativado, que tem
se mostrado o mais eficiente neste processo. Contudo, apesar da grande quantidade de
trabalhos envolvendo adsorção de fenol sobre carvão ativado, o processo ocorrido ainda não é
completamente compreendido. Nesse sentido, este trabalho visa estudar as interações
potenciais existentes entre compostos fenólicos e carvão ativado granular, utilizando cálculos de
dinâmica molecular, a fim de avaliar as regiões de interação das estruturas de carvão ativado
com fenol e derivados. Usou-se um modelo de carvão proposto na literatura e submeteu-se o
mesmo a cálculos de dinâmica molecular, com método B3LYP/6-31G e campo de força AMBER
FF99SB. As cargas do carvão e dos poluentes foram calculadas com o programa Gaussian 09,
cujo arquivos de saída foram convertidos para o formato necessário para cálculos com AMBER
12. O modelo usado para simular a célula unitária foi representado em monocamada com
diâmetro de poro de 20 Angstrons (mesoporo). Para a solução a ser adsorvida, foi modelada
com concentração de 8 g/L em uma caixa periódica e água como solvente. Estes modelos foram
submetidos a cálculos de aquecimento até 298 K e dinâmica molecular durante 4 ns. Dos
resultados, verificou-se que o os poluentes apresentaram maior tendência à interação com os
grupos nas extremidades e em algumas cavidades da estrutura modelada. Além disso, observouse que a distância média entre o carvão e o adsorbato após 4 ns de dinâmica molecular foi de,
em média, 3 Angstrons nas estruturas adsorvidas. Também verificou-se que a orientação da
maioria dos adsorbatos foi paralela à estrutura do carvão.
1. Introdução
As questões ambientais têm sido motivo de grande preocupação nos
últimos anos. Isto tem proporcionado um aumento significativo de trabalhos com
intuito de desenvolver tecnologias alternativas para remoção de substâncias
provenientes de efluentes industriais potencialmente nocivas ao meio ambiente
(BORBA, 2006).
Dentre as questões que causam preocupação, destaca-se a poluição de
mananciais por compostos fenólicos, contaminantes comuns contidos em águas
residuais. Estes poluentes são gerados principalmente pela indústria
petroquímica, de processamento de carvão de coque, de pesticidas, tintas,
papel, entre outras (FANG, 1997).
605
Nesse contexto, estudos apontam que a tecnologia mais eficiente na
remoção de compostos fenólicos de efluentes industriais é a adsorção sobre
carvão ativado. Por esse motivo, é importante compreender este mecanismo de
adsorção e fatores que influenciam nesse processo (NAGANO et al., 2000;
JUANG et al., 2002; DABROWSKI, et al., 2005).
Sabe-se que a capacidade de adsorção do carvão é determinada pela sua
estrutura de poro e pela química de sua superfície. O carvão é um adsorvente
microporoso obtido de uma variedade de materiais carbonáceos, cujo poder
adsorvente é proveniente da sua alta área superficial e da presença de grupos
funcionais nessa superfície. (TARKOVSKAYA, 1981; BAÇAOUI, et al., 2001;
SNOEYINK, et al., 1967).
No entanto, a grande quantidade de estudos sobre os fatores que
influenciam o processo de adsorção de fenol ainda não explica completamente
seu mecanismo. Por essa razão, a modelagem molecular é uma ferramenta útil
para tais estudos, visto que pode auxiliar na compreensão desse mecanismo no
que se refere à sua interpretação a nível molecular (DABROWSKI, et al., 2005).
Os métodos de modelagem molecular visam submeter cálculos de
mecânica clássica e quântica e fornecer uma representação tridimensional das
estruturas resultantes. Nesse sentido, esse trabalho visa realizar um estudo, a
nível molecular, do mecanismo de adsorção de compostos fenólicos. Busca-se
entender, portanto, os grupos funcionais e diâmetros de poro que favorecem a
adsorção dessa classe de compostos (SANT’ ANNA, 2002).
2. Metodologia
O modelo assumido para a representação molecular do carvão ativado foi
proposto em Bourke et al. (2007). Esse modelo foi desenhado utilizando o
programa Marvin Scketch e otimizado por cálculo DFT, a nível de teoria B3LYP
e base 6-31G pelo programa Gaussian versão 9.
Os arquivos obtidos da otimização contém dados de coordenadas
tridimensionais e carga. Com estes dados, usou-se o programa AMBER para
gerar os dados de topologia, ângulos e diedros de ligação. Estes arquivos serão
necessários para a execução de cálculos de dinâmica molecular utilizando o
pacote AMBER 12.
606
Todas essas etapas foram repetidas com a estrutura de cada um
poluentes: fenol, orto-cresol, meta-cresol e para-cresol.
O campo de força ff99SB foi utilizado na execução dos cálculos pelo
programa AMBER. Este é um método semi-empírico aplicado em cálculos de
dinâmica molecular. Além de apresentar boa acuracidade química e velocidade
de cálculo, este campo de força tem apresentado resultados mais próximos aos
obtidos experimentalmente e por cálculo ab initio em relação aos demais
métodos semi-empíricos (BARREIRO, 1997).
3. Resultados
A figura 1 apresenta o estado inicial da dinâmica molecular omitindo as
moléculas de água. Nesse estado, 10 moléculas de fenol foram usadas na
simulação, representando uma concentração de aproximadamente 8 g/L. A
figura 2 mostra o resultado da dinâmica molecular após 4 ns de cálculos.
Figura 1: Estado inicial do sistema antes da dinâmica molecular. Em verde: Fenol. Em
cinza: carvão.
Figura 2: Estado final da dinâmica molecular após 4 ns.
607
É possível observar que há interação entre as moléculas dos poluentes e
a superfície do carvão. Cavidades na estrutura do carvão, formada pela ausência
de alguns anéis em alguns trechos da estrutura, apresentaram potencial de
atração sobre os poluentes (figura 3). Além disso, a camada grafítica da estrutura
também apresentou capacidade de retenção dos poluentes (figura 4). Grupos
oxigenados, por sua vez, não apresentaram nenhuma interação com as
moléculas de fenol.
Verifica-se também pela figura 5 que não foram todas as moléculas de
poluentes retidas pela superfície do adsorvente. Ou seja, há uma quantidade
máxima de adsorção de fenol por estrutura de carvão. Nesse caso, é necessário
o estudo da interferência do processo de oxidação na ativação do carvão com
fim de adsorção de fenol, pois de acordo com a literatura tal processo inibe a
adsorção de fenol sobre carvões ativados.
Figura 3: Interação entre as moléculas de fenol e as cavidades na superfície da estrutura
de carvão.
608
___________________________________________________
Figura 4: Interação entre as moléculas de fenol e a região grafítica da superfície do
carvão.
Figura 5: Estado final da dinâmica molecular com ênfase nas moléculas não retidas
pela superfície do carvão.
4. Conclusão
Verificou-se que a estrutura de carvão utilizada apresentou boa
capacidade de retenção de fenol em soluções aquosas. Contudo, um estudo de
uma estrutura contendo menores quantidades de grupos oxigenados na
superfície é necessário para identificar o grau de interferência destes no
processo. Desse modo, evidencia-se também a capacidade de adsorção nas
regiões de cavidade formadas, características de algumas superfícies de
grafeno.
609
Referências
BANAT, F. A.; AL-BASHIR, B.; AL-ASHEH, S.; HAYAJNEH, O., Adsorption of
phenol by bentonite. Environ. Pollut. 107 (2000) 391–398.
BARREIRO, E. J.; RODRIGUES, C. R.; ALBUQUERQUE, M. G.; SANT’ANNA,
M. R.; ALENCASTRO, R. B.; Modelagem molecular: uma ferramenta para o
planejamento racional de fármacos em química medicinal. Química Nova,
20(1)(1997).
BAÇAOUI, A.; YAAVOUBI, A.; DAHBI, A.; BENNOUNA, C.; TAN LUU, R. P.;
MALDONADO-HODAR, F. J.; UTRILLA, J. R.; CASTILLA, C. M.; Carbon, v. 39,
p. 425, 2001.
BORBA, Carlos Eduardo. Modelagem da Remoção de Metais Pesados em
Coluna de Adsorção de Leito Fixo. Dissertação (Mestrado). Universidade
Estadual de Campinas. Campinas, 2006.
BOURKE, Jared; MANLEY-HARRIS, Merilyn; FUSHIMI, Chihiro; DOWAKi,
Kiyoshi; NUNOURA, Teppei; ANTAL JR, Michael Jerry. Do All Carbonized
Charcoals Have the Same Chemical Structure? A Model of the Chemical
Structure of Carbonized Charcoal. Ind. Eng. Chem. Res. 2007, 46, 5954-5967
COSTA, Wanessa Almeida, Simulação computacional da adsorção dos
poluentes benzeno, tolueno e xileno sobre carvão ativado. Dissertação
(mestrado), Universidade Federal do Pará.
DABROWSKI, A.; PODKOSCIELNY, P.; HUBICKI, Z.; BARCZAK, M.;
Adsorption of phenolic compounds by activated carbon—a critical review.
Chemosphere 58 (2005) 1049–1070.
FANG, H. H. P., CHAN, O.C., Toxicity of phenol towards anaerobic
biogranules, Water Res. 31 (1997) 2229–2242.
GUILARDUCI, V. V. S.; MESQUITA, J. P.; MARTELLI, P. B.; GORGULHO, H.
F.; Adsorção de fenol sobre carvão ativado em meio alcalino, Química nova,
vol. 29, No. 6, 1226-1232, 2006.
610
JUANG, R.; WU, F.; TSENG, R.; Colloids Surf., A 2002, 201, 191.
LUCENA, S. M. P.; PAIVA, C. A. S.; SILVINO, P. F. G.; AZEVEDO, D. C. S.;
CAVALCANTE, C. L.; The effect of heterogeneity in the randomly etched graphite
model for carbon pore size characterization, Carbon, v. 48, p. 2554- 2565, 2010.
NAGANO, S.; TAMON, H.; ADZUMI, T.; NAKAGAWA, K.; SUZUKI, T.; Carbon,
2000, 38, 915.
NANAME, A.; HELLAL, A.; The dynamic adsorption characteristics of phenol
by granular activated carbon. Journal of Hazardous Materials B137 (2006)
618–625.
SANT’ANNA, C. M. R., ALENCASTRO, R. B., BARREIRO, E. J. & FRAGA, C. A.
M. J. Mol. Struct. (Theochem) 1995, 340, 193.
SCHNIDER, Eduardo Luiz. Adsorção de compostos fenólicos sobre carvão
ativado. Dissertação (Mestrado). Universidade Estadual do Oeste do Paraná.
Toledo, 2008.
SCHINDLER, B. J.; LEVAN, M. D.; The theoretical maximum isosteric heat of
adsorption in the Henry’s law region for slit-shaped carbon nanopores.
Carbon, v. 46 p. 644 – 648, 2008.
SNOEYINK, V. L.; WEBER, W. J.; Environ. Sci. Technol. 1967, 1, 228.
611