UNIVERSIDADE FEDERAL DA BAHIA – INSTITUTO DE FÍSICA
DEPARTAMENTO DE GEOFÍSICA NUCLEAR SEMESTRE: 2003.2
Notas de Aula (18-21)
TERMODINÂMICA
PROFESSOR: JOSÉ GARCIA VIVAS MIRANDA
Aula 18
TEORIA CINÉTICA
Para melhor compreendermos os sistemas microscópicos é fundamental conceituar sua
composição.
Teoria atômica da Matéria
Início no século V A.C. com Leucipo e Demócrito cunharam o termo
átomo=indivisível.
Século I A.C. com a poesia de Lucrécio sobre o movimento das partículas de poeira
refletidas por feixes solares. ... “Esta dança é uma indicação de movimentos
subjacentes invisíveis da matéria ... grande número de partículas minúsculas, sob o
impacto de choques invisíveis, mudam de direção e se agitam...”
Império romano destrói a biblioteca de Alexandria. (Início da época negra da
ciência). Recentemente ocorreu algo similar com a cultura indígena.
1800 anos depois (séc. 17) ainda prevalecia a idéia dos elementos primordiais: água,
fogo, terra e ar.
Robert Boyle, em seu livro “O químico céptico” foi o primeiro a dar uma definição
mais próxima à atual.
1811 Hipóteses de Avogadro:
o Gás simples (1 elemento) pode ser formado por + de um átomo (ex. H2)
o Nas mesmas CNTP volumes iguais de todos os gases contém o mesmo
número de partículas. (isótopos não estão considerados).
A TEORIA CINETICA e a TERMODINÂMICA ESTATISTICA surgiram inicialmente a
partir da mecânica clássica.
T .C . dinâmica corpuscula r
T .E . consideraç ões probalisti cas
HIPÓTESES BÁSICAS – TEORIA CINÉTICA
Número grande de moléculas;
Tamanho das moléculas muito menor do que a distância que as separam;
Moléculas não interagem exceto quando colidem;
Colisões perfeitamente elásticas;
As moléculas estão distribuídas uniformemente;
As direções e sentidos das velocidades estão uniformemente distribuídos.
1
TEORIA CINÉTICA DA PRESSÃO
Baseados nas hipóteses descritas no item anterior desenvolveremos um modelo para explicar, do
ponto de vista microscópico, a medida macroscópica da pressão. Consideremos um modelo
bidimensional onde a medida da pressão de estabelece mediante a transferência de momento às
paredes do reservatório. As partículas se chocam com às paredes e, de acordo com a hipótese 4,
voltam com a mesma velocidade em módulo contudo com sentido oposto como mostra a ilustração
abaixo,
v 'y
vx
vy
v 'x
Desta forma o momento transferido para a parede será dado por:
Py mv,y mvy 2mvy
(1)
Força exercida sobre a parede devido aos choques das partículas pode ser resumida a uma força
média atuando sobre a parede:
F
É equivalente a
F Px N x
(2)
onde Nx é o número de vezes por segundo que uma partícula se choca em x.
Considerando uma caixa cúbica de lado L.
2
L
vx
L
L
L
o intervalo de tempo entre choques será dado por:
t
2L
vx
(3)
logo o número de partículas pode ser reescrito de forma que
Nx
v
1
x ,
t 2 L
(4)
sendo assim,
Px
2m v x v x m v x2
Fx
vx
2L
2L
L
(5)
A força total resultante do choque de todas as partículas sobre a parede na direção x será:
Fx
m v x21 m v x2i
n
m v x2n
i 1
L
L
(6)
Pela definição de pressão e área
P
F
,
A
(7)
A L2
Podemos contabilizar a pressão do ponto de vista microscópico
n
m v x2n
i 1
L3
Px
(8)
Contudo massa e velocidade são grandezas microscópicas, podemos reescrever (8) de forma que,
Nm
L3
m
3
N
L
(9)
n
v x2n
i 1
N
Px
Sendo a densidade.
O termo no somatório da equação (9) representa a média do quadrado das velocidades na direção x,
ou seja
3
v x21 v x22 ... v x2n
N
v x2
(10)
logo
Px v x2
(11)
más pela hipótese da distribuição homogênea das velocidades, v 2 3v x2 (explicaremos melhor
mais adiante).
E considerando que a pressão será a mesma sobre todas as paredes concluímos que
v2
P
3
(12)
Podemos definir uma grandeza, a Velocidade quadrática média
vrms v 2
(13)
3P
(14)
Logo
v rms
O que representa uma associação inédita entre o macro (representado pela pressão P) e micro
(representado pela velocidade quadrática média das partículas)
Multiplicando por V ambos os termos da equação temos
v2
PV V
3
(15)
em que V é a massa total, que pode ser escrita como nM , onde n é o número de moles e M
massa molar. Com isso temos
v2
v2
nM
3
3
(16)
v2
2 1
n Mv 2
3
3 2
(17)
PV V
Ou
PV V
1
2
Contudo o termo Mv 2 podemos definir como a energia interna de translação de um mol das
partículas. E assim
PV
2n
u
3
(18)
em que u energia interna molar do gás que só depende de T,
P
2n u T
3 V
(19)
Para levantarmos a dependência de u e T deveremos lançar mão das relações TdS.
4
Sabemos que
du PdV
T
T
C
2n u
v dT T dV
T
3 VT
(20)
S
S
dS
dv dT
V T
T V
(21)
dS
Contudo sendo S V , T
Comparando temos
2 uT
S
n
3 VT
V T
e
(22)
C
S
v
T
T V
Mas sabemos que
V
S S
T V T V T
(23)
V
S Cv
T V V T
(24)
e
0
Logo
n
V
2 uT
0
T 3 T V
(25)
0
O que nos leva a
2 uT
R
3 T
(26)
2
uT RT
3
PV nRT
(27)
e assim finalmente temos que
5
Aula 19
PRINCIPIO DA EQÜIPARTIÇÃO DA ENERGIA
Sabemos que
2
u RT
3
( 28)
Onde u é a energia cinética média das partículas em um mol e R a constante universal dos
gases. Utilizando a equação ( 28) e a definição de energia cinética média de um mol temos que:
RT
21
2
v
32
( 29)
Sendo a massa molar do gás e <v2> a média da velocidade quadrada das partículas, logo:
Nam
21
2
Nam v
32
RT 2 1
m v2
Na 3 2
R
Sendo
K
Na
RT
( 30)
1
3
m v 2 KT
2
2
Onde se explicita a associação entre temperatura e energia cinética.
Na dedução do resultado obtido em ( 30) tivemos que utilizar a hipótese de isotropia, ou seja:
v 2 3 v x2 3 v y2 3 v z2
logo
1
1
1
1
m v x2 m v y2 m v z2 KT
2
2
2
2
( 31)
Ou seja cada termo em ( 31) associado a uma coordenada de translação contribui de forma
igualitária a energia. Este fato exprime-se a partir do princípio da eqüipartição da energia: “cada
termo quadrático no Hamiltoniano acrescenta 1/2KT a energia total do sistema.
Desta forma é possível generalizar a relação da energia ( 28) e ( 30) para:
U
f
KT
2
( 32)
Este é o PRÍNCIPIO DA EQUIPQRTIÇÃO DA ENERGIA. Veremos que é apenas um caso
limite. Enunciado: “Se a energia associada a qualquer grau de liberdade for uma função quadrada
6
da variável que especifica o grau de liberdade, o valor médio correspondente a este grau será de
1
KT ”.
2
E partindo das relações da termodinâmica para o calor específico a volume constante
obtemos de forma similar:
f
f
u
Cv K nR
2
T v 2
( 33)
Exemplo:
Gás monoatômico
f 3
.
3
u nRT
2
Gás diatômico sem vibração
f 3 de translaçã o 2 de rotação
.
5
u nRT
2
Exemplo para Gás Diatômico:
No exemplo da figura ao lado, ao contabilizarmos os
graus de liberdade com representação quadrática no
hamiltoniano, obtemos:
z
Para translação temos:
½ mvx2+½ mvy2+½ mvz2
3 termos quadráticos
y
Para rotação:
½ I1wy2+½ I1wz2
2 termos
x
Para vibração:
cinético = ½ I1(r’)2
potencial=½ Kr2
2 termos
Num total de 7 termos, ou seja, f=7 e conseqüentemente Cv=3,5R.
7
Gás poliatômico sem vibração
f 2 de translaçã o 3 de rotação
.
6
u nRT 3nRT
2
Gás diatômico com vibração
f 3 de translaçã o 2 de rotação 2 de vibração
.
7
u nRT
2
9.7) Calor específico
Para uma gás ideal C P Cv R . C P
para um gás monoatômico C v
f 2
f 2
R,
2
f
3
R 1,5 R
2
para gás de He
Cv
1,506 , valor experimental.
R
Para o H2 C v
C
7
R 2,5 R , sendo o valor experimental v 2,47 !!
2
R
9.8) CALOR ESPECÍFICO EM SÓLIDOS
8
Não existe translação
apenas vibração.
Em z
Em x
Em y
para cada grau teremos energia potencial e cinética logo C v
6
R 3R
2
Aula 20
FORÇAS INTERMOLECULARES
Nas hipóteses anteriores consideramos que as moléculas eram massas puntiformes, V 0 .
Podemos incrementar este modelo considerando o volume das partículas. De fato Hirn (18151890) foi o que primeiro propôs um modelo considerando o volume accessível das partículas. A
hipótese levantada considera que o volume real associado à acessibilidade das partículas deve ser o
resultado do volume total do recipiente menos o volume de todas as partículas, o que implica em
uma simples modificação na equação de estado deduzida anteriormente.
PV b RT
( 34)
Onde b representa o volume das partículas que compõe o gás.
Outra aproximação pode ser realizada ao consideramos a interação entre as partículas. Esta
interação deve ser de curto alcance e deve existir um limite mínimo para a distância entre
partículas, representado pelo limite do volume das partículas. Van der Waals propôs uma função
potencial na forma descrita no diagrama abaixo,
9
F
FORÇA DE VAN der WAALS
Sobreposição de nuvem eletrônica implica em repulsão
r
Partículas “duras”.
Esta força atrativa entre as moléculas é do tipo r 6 , as moléculas exteriores são atraídas no centro
exercendo uma pressão menor que a no centro do gás logo a pressão real no interior do gás, deve
ser maior que a medida nas paredes P' P A , como mostra a ilustração abaixo,
Desta forma a expressão corrigida para a equação de estado será :
P Av b RT
Baseado neste potencial a constante A é definida como: A
a
P 2 v b RT
v
( 35)
a
,
v2
( 36)
LIVRE CAMINHO MÉDIO
Se colocarmos um frasco de perfume em um dos cantos de uma sala de aula e abrirmos sua
tampa o odor agradável do perfume não se propagará de forma instantânea pela sala. Esta relativa
10
demora na propagação dos gases constituiu o maior entrave para a aceitação da teoria cinético
como modelo para a dinâmica dos gases.
Movimento Browniano, primeira vez observado por Robert Brown em 1827.
Roberto Brown em 1827 descobre o movimento Browniano (MB).
Einstein em 1905, utilizando os conceitos de MB, finaliza a formalização dos conceitos e elimina
as dúvidas a respeito da teoria cinética com sua teoria sobre o MB das partículas de um gás.
Definição: é a distância média que uma partícula percorre antes de uma colisão.
Na base dos conceitos de viscosidade e difusão o conceito de livre caminho médio pode ser
deduzido facilmente a partir de idéias geométricas simples:
L
l = L / No. de choques.
Para a determinação do no. de choques se faz necessário a definição de seção de choque:
Seção de choque
d
11
Qualquer partícula cujo centro esteja a esta distância se chocará. Assim considerando que as
partículas alvo estão paradas, a quantidade de partículas que se chocará uma outra partícula em
movimento será dada pelo volume associado à sua seção de choque e ao caminho percorrido pela
partícula vezes a densidade de partículas no meio, como ilustra a figura abaixo.
Partículas alvo
Seção de choque
Onde No. de choques pode ser estimado mediante:
No. de choques = L d2n.
Onde d2 é a seção de choque, e o volume L d2 representa um cilindro em onde toda partícula
alvo que esteja nele se chocara com a partícula.
Nesta aproximação consideramos que as partículas alvo estão PARADAS.
Com isso a equação definida para o livre caminho médio será:
l
1
( 37)
d n
2
Aula 21
(cap. 11.2 e 11.3 Sears)
Termodinâmica Estatística
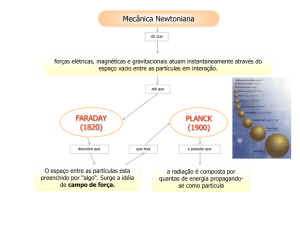
Os princípios da mecânica quântica conduzem ao resultado de que a energia de uma partícula sob
um campo conservativo, como o gravitacional, magnético ou elétrico, não podem assumir qualquer
valor continuamente, se diz que a energia é quantizada, ou seja, a partícula só pode existir em
alguns dos ESTADOS que têm uma energia bem específica.
Podemos utilizar como ilustração do conceito quântico o exemplo clássico da onda estacionária em
uma corda, como se ilustra na figura abaixo,
12
L
Sabemos que, nem todos os comprimentos de onda são possíveis em uma corda vibrante com as
extremidades fixas. De fato os comprimentos de onda possíveis são dados por:
i
1
2L .
ni
( 38)
Onde ni é um número inteiro que representa o número de antinodos da vibração da corda.
De acordo com a mecânica quântica, uma onda de Schrödinger estacionária é completamente
equivalente a uma partícula presa. E o comprimento de onda da onda estacionária está relacionado
com o momento da partícula por:
p
h
( 39)
.
Sendo h a famosa constante de Planck.
De forma que o momento de dita partícula também estará quantizado, e a expressão que determina
como, vem dada por:
pi
h
i
ni
h
.
2L
( 40)
E para o caso de uma partícula confinada em uma caixa cúbica de aresta L teremos:
p x nx
h
h
, p y ny
2L
2L
e p z nz
h
.
2L
( 41)
Onde nx, ny e nz são números inteiros chamados de números quânticos.
A energia cinética desta partícula será dada por:
E
p2
,
2m
h2
Ej n
8mL2
( 42)
.
2
j
De forma que a energia total do sistema pode ser estimada através de
13
E j (nx2 n y2 nz2 )
h2
8mL2
( 43)
.
Assim os números quânticos definirão os estados possíveis de encontrar o sistema.
Relembrando do conceito clássico introduzido por Boltzmann, não existiam restrições para a
energia do sistema, ele poderia se encontrar em qualquer valor, assim as média deveriam ser
efetuadas sobre todo o universo de parâmetros possíveis. No caso quântico não poderemos fazer o
mesmo, a contabilização dos estados acessíveis é imprescindível para a operacionalização da
estatística (valores médios, desvios, etc.). Por exemplo, a menor energia possível assumida pelo
sistema, a que denominaremos E1, será dada para os valores nx=ny=nz = 1, assim
E1
3h 2
8mL2
( 44)
.
Contudo se tentamos determinar o segundo estado energético veremos que diferentes valores de nx,
ny e nz podem levar ao mesmo valor de energia, como ilustramos na tabela abaixo:
Estado
1
nx
2
ny
1
nz
1
Energia
2
1
2
1
3h 2
4mL2
3
1
1
2
3h 2
4mL2
3h 2
4mL2
Tabela 1
Ou seja, as três configurações terão o mesmo estado energético. A isso chamamos de estado
degenerado, ou seja, o estado referente a energia
3h 2
4mL2
é DEGENERADO
e com
DEGENERESCÊNCIA 3 (g2=3), pois existem três configurações possíveis que levam a este
mesmo valor.
Obviamente, não poderemos fazer estatística com apenas uma partícula, assim que consideremos
um sistema com N partículas onde em cada estado de energia j teremos uma população de Nj
partículas, a energia total deste sistema será dada por:
U EjN j
.
( 45)
j
De forma que, se conhecemos como estão distribuídas as partículas sobre seus estados energéticos
teremos toda a informação do sistema. ESTE É O PROBLEMA CENTRAL DA MECÂNICA
ESTATÍSTICA.
Macroestados e Microestados
Para que possamos melhor compreender os conceitos associados a como às distribuições de
partículas em seus estados energéticos podem definir o estado do sistema, é imprescindível definir
melhor o conceito de estado.
14
No esquema abaixo ilustramos o exemplo dado anteriormente (Tabela 1) em um sistema com 11
partículas idênticas.
g3=3, N3=3
E3
(2,2,1)
(1,2,2)
(2,1,2)
(2,1,1)
(1,2,1)
(1,1,2)
g2=3, N2=5
E2
g1=1, N1=3
E1
(1,1,1)
onde cada caixinha representa um estado energético. Estando as caixas na mesma altura significa
que têm a mesma energia, ou seja, constituem estados degenerados.
A energia deste sistema, COM ESTA CONFIGURAÇÃO, será dada por:
3h2
3h2
9h 2
E 3
5
3
8mL2
4mL2
8mL2
( 46)
.
Contudo vale ressaltar que ao trocar de caixa uma partícula no mesmo nível de energia estaremos
modificando a configuração do sistema, apesar de manter a energia constante. Desta forma, vem-se
à necessidade de subdividirmos o conceito de estado.
Macroestado:
É caracterizado pelo número de partículas para cada nível. No exemplo anterior temos: N1=3,
N2=5 e N3=3.
Microestado:
Definido por cada nível e cada degenerescência. No exemplo anterior temos.
Nível
Nj
gj
1
3
(1,1,1)
2
2
(2,1,1)
1
(1,2,1)
2
(1,1,2)
2
(2,2,1)
1
(2,1,2)
3
Note que para uma mesma energia total teremos diversos macroestados possíveis e para cada
macroestado pode conter uma infinidade de microestados!!!
15
Postulado fundamental da M.E.: “Todo microestado possível de um sistema isolado em equilíbrio
é igualmente provável”
O que significa que se fazemos N réplicas de um conjunto de sistemas (ensamble), e contamos ao
freqüência com que cada microestado ocorre, obteremos que todo o microestado terá a mesma
freqüência!
O número de microestados igualmente prováveis, que corresponde a um macroestado k é chamado
de PROBABILIDADE TERMODINÂMICA (Wk). Ou seja, quanto mais microestados tenha um
macroestado maior será sua probabilidade termodinâmica. Funciona, exatamente como o exemplo
do dado, quanto mais possibilidades existam de que um evento ocorra maior será a sua
probabilidade ocorrência.
A Probabilidade termodinâmica de todo o ensamble será a soma de todas as probabilidades
termodinâmicas de cada macroestado.
( E ,V )
1
WK
N k
( 47)
.
Assim que para uma mesma energia teremos diversos macroestados e respectivamente diversos
microestados. O valor de energia mais provável será aquele que MAXIMIZE a quantidade de
configurações possíveis, ou seja, que maximize a probabilidade termodinâmica.
De fato a probabilidade termodinâmica do ensamble dependerá das propriedades do sistema e dos
vínculos internos que o configuram (E,V,N,{Xi}).
Logo, podemos concluir que a probabilidade de encontrarmos um sistema no estado definido por
(E,V,N,{Xi}) será:
P( E,V , N ,{ X i }) ( E,V , N ,{ X i }) .
( 48)
Interpretação estatística da entropia
A partir da lei de conservação da energia sabemos que:
Ts u Pv .
( 49)
Ou seja, a entropia específica é função apenas da energia u e do volume específico v. Ao
multiplicarmos a entropia por N, temos que S=f(U,V,N). Contudo sabemos a partir da teoria
desenvolvida no item anterior sobre a probabilidade termodinâmica que =f(U,V,N). De forma
que podemos escrever uma função J que estabeleça a ligação entre S e .
S J () .
( 50)
Sabemos que S é aditiva e é probabilística, logo:
S S1 S 2
1 2
( 51)
.
Desta forma a função J será dada por:
J (1 ) J (2 ) J (12 ) .
( 52)
Derivando ( 52) em relação a 1 temos:
16
dJ (1 ) dJ (1 2 ) dJ (1 2 ) d (1 2 )
d1
d1
d (1 2 ) d1 .
( 53)
O que nos leva a:
dJ (1 )
2 J (1 2 ) .
d1
( 54)
Fazendo o mesmo para 2 obtemos:
dJ ( 2 )
1 J (1 2 ) .
d 2
( 55)
Comparando as equações ( 54) e ( 55),
dJ ( 2 ) 1
dJ ( 1 ) 1
d 2 1
d 1 2
dJ ( 2 )
dJ ( 1 )
2
1
d 2
d 1
( 56)
.
Sendo 1 e 2 independentes esta equação só poderá ser satisfeita para
dJ ()
KB .
d
( 57)
O que nos leva finalmente a expressão para a função J.
S K B ln( ) .
( 58)
Que é a conhecida definição da entropia de Boltzmann.
Aplicação do formalismo estatístico à mudança de vínculos
Vimos que a função potencial termodinâmico é uma função da energia interna, volume e número
de partículas. Más como deve estar afetada ao se modificarem os vínculos do sistema? A
resposta a esta pergunta pode ser facilmente obtida ao seguirmos a idéia de que o sistema, uma vez
modificado o vínculo tenderá para um estado cuja probabilidade seja máxima. Abaixo veremos o
exemplo para um sistema composto isolado em cujos subsistemas estão separados por uma parede
diatérmica.
Interação térmica entre 2 sistemas macroscópicos:
Consideremos o sistema ilustrado abaixo
17
1
E1,N1,V1.
2
E2,N2,V2.
De forma que podemos definir uma função (E,V,N)
subsistema independente temos que
para cada subsistema. Sendo cada
12 .
( 59)
A energia de cada subsistema pode variar livremente contudo estará sujeita ao vínculo
E1 E2 Eo .
( 60)
As demais variáveis permanecerão constantes.
Aplicando o vínculo ( 60) na equação ( 59) temos que a Prob. Termod. (PT) será função apenas de
uma das energias.
( E1 , Eo ) 1 ( E1 ) 2 ( Eo E1 ) .
( 61)
Como estamos interessados em maximizar a probabilidade, devemos antes defini-la melhor.
Sabemos que esta é proporcional a P.T.
P( E1 ) C( E1 , Eo ) .
( 62)
Como a probabilidade é normalizada a constante de proporcionalidade C será dada por:
1 Eo
1 ( E1 ) 2 ( Eo E1 ) .
C E1 0
( 63)
A maximização de uma função também será máxima para o logaritmo desta função.
f ( E1 ) ln P( E1 ) ln C ln 1 ln 2 .
( 64)
De forma que a máxima probabilidade será dada por:
d ln P( E1 ) d ln 1 d ln 2
0.
dE1
dE1
dE1
( 65)
Contudo uma variação em E1 será seguida de uma variação contrária de E2, e assim:
d ln P( E1 ) d ln 1 d ln 2
0.
dE1
dE1
dE2
( 66)
Pela definição da entropia temos
18
ln
S
KB
( 67)
.
Que substituindo em ( 66) temos
1 dS1
1 dS 2
K B dE1 K B dE 2
( 68)
.
O que implica em
T1 T2 .
( 69)
19