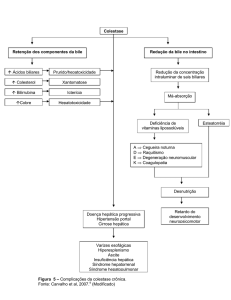
1
Hospital Servidor Público Municipal
SÍNDROME COLESTÁTICA FAMILIAR – RELATO DE CASO
BEATRIZ RODRIGUES DE ANCHIETA
São Paulo
2015
2
BEATRIZ RODRIGUES DE ANCHIETA
SÍNDROME COLESTÁTICA FAMILIAR – RELATO DE CASO
Trabalho de Conclusão de Curso apresentado
à Comissão de Residência Médica do Hospital
do Servidor Público Municipal, para obter o
título de Residência Médica.
Área: Clínica Médica.
Orientadora: Dra Renata da Silva Moutinho.
São Paulo
2015
3
AUTORIZO A DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR
QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO
E PESQUISA, DESDE QUE CITADA A FONTE.
São Paulo, ____ / ____ / ______ .
_________________________________
Beatriz Rodrigues de Anchieta
Residente de Clínica Médica
4
Beatriz Rodrigues de Anchieta
SÍNDROME COLESTÁTICA FAMILIAR – RELATO DE CASO
Trabalho de Conclusão de Curso apresentado
à Comissão de Residência Médica do Hospital
do Servidor Público Municipal, para obter o
título de Residência Médica.
Área: Clínica Médica.
Orientadora: Dra Renata da Silva Moutinho.
COMISSÃO EXAMINADORA
_____________________________________________
Dra. Christiane M. V. Boas
_____________________________________________
Dr. Sadi F. S. Navarro
_____________________________________________
Dra. Pérola G. Lerner
São Paulo, 13 de outubro de 2015.
5
RESUMO
Colestase é definida como uma redução do fluxo biliar. Em crianças, é quase
sempre devido a alterações genéticas. Pode ser por diminuição no transporte,
síntese ou obstrução biliar. As causas são divididas em intra ou extra-hepáticas.
Dentre as causas intra-hepáticas destacamos duas: colestase intra-hepática familiar
progressiva (PFIC) e colestase intra-hepática recorrente benigna (BRIC). PFIC
refere-se a um grupo heterogêneo de doenças autossômicas recessivas da infância,
secundárias a mutações em genes do sistema de transporte hepatocelular
envolvidos na formação de bile e se apresentam com colestase. Já BRIC é uma
doença rara, também de caráter autossômico recessivo, caracterizada por episódios
intermitentes de prurido e icterícia, que evolui sem sequelas funcionais hepáticas. O
objetivo do presente artigo é relatar um caso de síndrome colestática que ficou
internado no serviço de Gastroenterologia do Hospital do Servidor Público Municipal
e comentar os principais aspectos clínicos das colestases intra-hepáticas. Neste
episódio foi realizada uma investigação criteriosa, para a exclusão de outras causas
de colestase.
Palavras chave: colestase, colestase intra-hepática familiar progressiva
(PFIC), colestase intra-hepática recorrente benigna (BRIC).
6
ABSTRACT
Cholestasis is defined as a decrease of the biliary flux. In children, it is almost
always due to genetic alterations. It may be decreased in transport, synthesis or
biliary obstruction. The causes are divided into intra or extrahepatic. Among the
intrahepatic causes we highlight two: progressive familial intrahepatic cholestasis
(PFIC) and benign recurrent intrahepatic cholestasis (BRIC). PFIC refers to a
heterogeneous group of autosomal recessive disorders of childhood, secondary to
mutations in genes of hepatocellular transport system involved in bile formation and
present with cholestasis. Already BRIC is a rare disease, also autosomal recessive,
characterized by intermittent episodes of pruritus and jaundice, evolving without liver
function sequelae. The objective of this article is to report a case of cholestatic
syndrome who was admitted to the Gastroenterology service of the Hospital do
Servidor Público Municipal and comment on the main clinical features of intrahepatic
cholestasis. In this episode a judicious investigation was carried out for excluding
other causes of cholestasis.
Key words: cholestasis, progressive familial intrahepatic cholestasis (PFIC),
benign recurrent intrahepatic cholestasis (BRIC).
7
SUMÁRIO
1. Introdução ...................................................................................................... 8
2. Objetivo .........................................................................................................10
3. Relato de caso clínico ..................................................................................11
4. Discussão .................................................................................................... 14
A. Deficiência ATP8B1 ............................................................................... 15
B. Deficiência ABCB11 .............................................................................. 16
C. Deficiência ABCB4 ................................................................................ 17
D. BRIC ........................................................................................................ 19
E. PFIC e BRIC 1 ......................................................................................... 21
F. PFIC e BRIC 2 ......................................................................................... 22
G. PFIC 3 ...................................................................................................... 24
5. Conclusão..................................................................................................... 26
6. Referências .................................................................................................. 27
8
INTRODUÇÃO
Síntese
ou
obstrução
biliar,
com
consequente
retenção
de
seus
componentes no fígado e sangue1.
Os exames laboratoriais em pacientes com colestase revelam aumento
sérico das enzimas canaliculares (gamaglutamiltransferase – GGT e fosfatase
alcalina – FA) em graus variáveis, elevação das bilirrubinas, predominantemente da
fração conjugada, e dos níveis de colesterol.
O quadro clínico cursa com icterícia, prurido, colúria, acolia fecal, fadiga,
anorexia, náuseas, vômitos, estatorreia e dor no hipocôndrio direito. A absorção
inadequada das vitaminas lipossolúveis acrescenta ao quadro sintomas como
distúrbios visuais e da coagulação e Colestase é falha do hepatócito em secretar bile
devido à diminuição do transporte, osteodistrofia.
Podemos distinguir entre colestase intra- e extra-hepática, dependendo da
localização da alteração do fluxo biliar.
Colestase extra-hepática pode ser causada por atresia e obstrução das vias
biliares. Intra-hepática é resultado de várias doenças, incluindo as auto-imunes e
distúrbios hereditários como colestase intra-hepática familiar progressiva (PFIC) e
colestase intra-hepática recorrente benigna (BRIC)2,3.
Colestase intra-hepática familiar progressiva refere-se a um grupo
heterogêneo de doenças autossômicas recessivas da infância, secundárias a
mutações em genes do sistema de transporte hepatocelular envolvidos na formação
de bile.
9
O curso natural da PFIC provoca hipertensão portal, insuficiência hepática,
cirrose, carcinoma hepatocelular e manifestações extra-hepáticas. Três tipos de
PFIC foram identificados4,5.
A prevalência real da doença permanece desconhecida, mas a incidência
estimada é de um por 50.000-100.000 nascimentos. Ambos os sexos parecem ser
igualmente afetados4,6.
Outra forma de colestase intra-hepática é chamada de recorrente benigna
(BRIC). Trata-se de uma doença rara, de caráter autossômico recessivo,
caracterizada por episódios intermitentes de prurido e icterícia, de número e duração
bastante variáveis7. Entre as crises os pacientes permanecem assintomáticos
durante meses ou anos.
Ocorre mais frequentemente no sexo masculino, entre os 10 e os 30 anos de
idade, manifestando-se em até 80% dos casos em torno da segunda década de
vida8. O prognóstico clínico é favorável e não há o desenvolvimento de doença
hepática crônica ou cirrose9.
A
patogênese
tem
sido
elucidada
com
estudos
moleculares
dos
transportadores de sais biliares, assim como seu caráter familiar, que é reconhecido
em 50% dos casos10.
10
OBJETIVO
Os objetivos do presente artigo são:
1. Relatar um caso de síndrome colestática em paciente jovem internado no
serviço de Gastroenterologia do Hospital do Servidor Público Municipal
2. Revisar os principais aspectos clínicos, laboratoriais e histológicos das
colestases intra-hepáticas.
11
RELATO DE CASO
Identificação: G.L.S.R., 16 anos, estudante, natural e procedente de Santo
André.
Queixa principal: prurido há 15 dias.
História da moléstia atual: paciente deu entrada no Pronto Socorro do
Hospital do Servidor Público Municipal com queixa de prurido generalizado há 15
dias, com piora progressiva e associado à icterícia, colúria e acolia fecal. Negava dor
abdominal, náuseas, vômitos, diarreia, disúria e febre. Havia comparecido na
mesma unidade na semana anterior com a mesma queixa e medicado com antihistamínico, sem melhora dos sintomas.
Antecedentes pessoais: negava comorbidades, uso de medicações, drogas,
anabolizantes, energéticos e chás. Negava transfusões sanguíneas, uso de álcool e
tabagismo. Amputação bilateral de pernas após acidente automobilístico há seis
anos.
Antecedentes familiares: pais e irmã hígidos. Tio paterno falecido por cirrose
e prima paterna com doença hepática.
Exame físico: bom estado geral, consciente, contactuante, afebril, eupneico,
acianótico, descorado+/4, ictérico 3+/4+, sem estigmas de hepatopatia crônica. Sem
flapping. Pele com sinais de escoriações pelo corpo. Abdome: flácido, indolor à
palpação superficial e profunda, ruídos hidroaéreos presentes e normoativos,
descompressão brusca negativa, sinal de Murphy negativo, sem visceromegalias.
Sem alterações nos aparelhos cardiovascular e respiratório.
12
Exames laboratoriais: vide tabela 1.
Tabela 1: Exames Laboratoriais
17/05/14
Hb / Ht
23/05/14
04/06/14
16,2 / 48,9
10,4 / 32,5
Leucócitos
9.000
13.200
Plaquetas
396.000
344.000
TP (s) / AP (%)
12 /90
TTPA
14 / 84
38
AST
40
37
18
ALT
56
49
31
FA
428
391
3
9
Triglicerídeos
168
161
Colesterol total
113
97
4 / 75
6 / 59
13 / 10,2
20,8 / 18,4
GGT
HDL / LDL
Bilirrubina total / direta
Lipase
5,7 / 4,8
47
Amilase
60
Proteína total / albumina
57
6,7 / 3,7
DHL
262
279
Ck total
285
87
5,5 /3,1
Sorologias para hepatites A, B e C, HIV, toxoplasmose, citomegalovírus,
vírus Epstein Barr: negativas. VDRL: não reagente. Anti-DNA, FAN, anticorpo antimúsculo liso negativo, anticorpo anti-LKM negativo e fator reumatóide: negativos.
13
PCR: 0,13. Eletroforese de proteínas: dentro dos limites de normalidade. Urocultura
e hemocultura: negativas.
Exames de imagem:
Ultrassonografia de abdome: dentro dos limites da normalidade.
Endoscopia digestiva alta: pangastrite leve.
O paciente evoluiu com piora da icterícia com bilirrubina direta 18,4 mg/dl.
Foi realizada biópsia hepática percutânea que identificou fígado com
arquitetura lobular conservada. Espaços porta com infiltrado linfocitário moderado
envolvendo arteríolas, vênulas e ductos biliares, porém sem ultrapassar a placa
limitante. No lóbulo, os hepatócitos apresentavam colestase em zonas II e III. Os
sinusóides não apresentavam particularidades e as veias centro lobulares estavam
inalteradas. Diagnóstico: Colestase Intra-hepática.
O paciente permaneceu internado por 18 dias e recebeu alta com melhora
do prurido e diminuição dos níveis de bilirrubina após o uso de anti-histamínicos,
colestiramina e ácido ursodesoxicólico. Foi referenciado para acompanhamento
ambulatorial no mesmo serviço.
14
DISCUSSÃO
Colestase é decorrente da alteração na formação e/ou do fluxo biliar.
Clinicamente se manifesta como fadiga, prurido e, na maioria dos casos, com
icterícia. As alterações bioquímicas são aumento da fosfatase alcalina (FA) e
gamaglutamiltransferase (GGT) e hiperbilirrubinemia com predomínio da fração
conjugada. O nível de elevação das enzimas tem sido discutido: nível de FA maior
que 1,5 e GGT maior que 3 vezes o limtie superior da normalidade (LSN) tem sido
aceito. Pode ser classificada como intra-hepática ou extra-hepática. Colestase intrahepática pode resultar de defeitos funcionais ou lesões obstrutivas do trato biliar
intra-hepático distal ou dos canalículos biliares11.
O diagnóstico diferencial de colestase intra-hepática do nosso paciente,
inclui causas hepatocanaliculares, tais como hepatites virais, hepatite alcoólica, uso
de drogas/medicações, hormônios esteróides, colestase intra-hepática familiar e
colestase recorrente benigna. Dentre as causas biliares, colangite esclerosante
primária (CEP) como principal. O paciente apresentava-se ictérico, com sinais de
escoriação, eutrófico, sem estigmas de hepatopatia crônica. Negava uso de álcool,
medicações, chás e anabolizantes. Os exames laboratoriais foram compatíveis com
colestase, sem deficit de função hepática, com destaque para níveis normais de
GGT. As sorologias virais foram negativas, bem como os marcadores de doença
auto imune. A ultrassonografia de abdomen descartou causas obstrutivas de
icterícia. A biópsia hepática foi realizada para esclarecimento diagnóstico e foi
consistente com colestase hepatocelular sem injúria aos ductos biliares. Os
diagnósticos de PFIC ou BRIC com primeiro surto foram considerados.
15
A seguir, uma revisão da literatura dos principais aspectos clínicos,
laboratoriais e histológicos destas entidades que recentemente foram renomeadas.
Colestase intra-hepática familiar progressiva (PFIC) é um grupo heterogêneo
de doenças genéticas raras, autossômicas recessivas, caracterizada por colestase
intra-hepática, geralmente na infânica ou adolescência. A progressão para
insuficiência hepática pode ocorrer nas primeiras décadas de vida. Atualmente, são
reconhecidas três formas caracterizadas por defeitos nas proteínas biliares
envolvidas na formação e no fluxo de bile no fígado4.
A. Deficiência ATP8B1
A deficiência ATP8B1 é uma doença autossômica recessiva causada por
mutações no gene ATP8B1 que codifica ATP8B1 (denominada anteriormente como
FIC), uma ATPase. Atualmente, mais de 50 mutações são reconhecidas12-14.
PFIC1 e colestase intra-hepática recorrente benigna (BRIC) compartilham
das mesmas mutações. Contudo, na BRIC há um defeito parcial desta enzima.
A ATP8B1 também está expressa numa variedade de órgãos, tais como,
intestino delgado, vesícula biliar, fígado e pâncreas. Está localizada na membrana
apical das células epiteliais, incluindo a membrana canalicular dos hepatócitos. Sua
deficiência determina um posicionamento inadequado dos aminofosfolipídeos na
membrana canalicular, diminuindo a estabilidade e função dos transportadores
transmembrana, causando colestase15.
O quadro clínico caracteriza-se por colestase com prurido intenso,
geralmente na infância (PFIC1 ou doença Byler) ou com episódios recorrentes de
16
colestase em qualquer idade (BRIC1). Algunas pacientes com BRIC apresentam
progressão da doença, porém, a maioria continua a ter episódios intermitentes de
colestase de duração variável, sem evidência de dano hepático16. Pacientes com
PFIC1 apresentam colestase contínua desde o primeiro ano de vida. Na ausência de
tratamento, irão progredir para cirrose e insuficiência hepática. Complicações
decorrentes da má absorção das vitaminas lipossolúveis
podem ocorrer.
Manifestações extra hepáticas como diarréia, pancreatite e perda de audição
também são observadas17-19.
Em pacientes com deficiência ATP8B1, as aminotransferases e ácidos
biliares estão elevados, atividade da GGT e concentração de colesterol são normais.
Na histologia, observa-se colestase intra-canalicular com grânulos biliares identificados
na microscopia eletrônica18,20.
B. Deficiência ABCB11
Doença autossômica recessiva decorrente da mutação no gene ABCB11
que codifica a bomba exportadora de sais biliares (BSEP), uma enzima
transportadora específica do fígado. A BSEP transporta ativamente sais biliares
conjugados para o canalículo biliar , produzindo fluxo biliar. Foram identificadas
mais de 100 mutações21,22.
Quando a função da BSEP está diminuída, a doença é denominada BRIC 2
e quando estiver ausente, PIFIC 2 (Síndrome Byler). Na BRIC 2, a colestase é
episódica. Na PFIC 2 , ocorre colestase permanente desde a infância23.
17
Os pacientes, geralmente, apresentam icterícia colestática no período
neonatal. Prurido é uma característica dominante da doença. As manifestações das
deficiências de vitaminas lipossolúveis são bem frequentes.
Manifestações extra
hepáticas da doença não são observadas, uma vez que a proteína é específica do
fígado. O surgimento de colelitíase é comum, em até um terço dos pacientes, uma
vez que há menor concentração de sais biliares na bile, provocando aumento da
saturação de colesterol. O risco para tumores hepatobiliares está aumentado nestes
pacientes,
devendo
ser
realizado
rastreamento
para
hepatocarcinomas
e
colangiocarcinomas24,25.
As alterações laboratoriais encontradas são aminotransferases elevadas,
colesterol normal e GGT normal ou baixa26. A doença pode progredir rapidamente
para cirrose, mas a doença hepática e os sintomas podem melhorar com o
tratamento cirúrgico. A necessidade de transplante é comum.
Na histologia, observa-se hepatite de células gigantes e canalículo biliar
afilado. A imuno histoquímica caracteriza-se por ausência ou redução da BSEP.
C. Deficiência ABCB4
Doença autossômica recessiva causada por mutações no gene ABCB4 que
codifica uma glicoproteína transportadora (MDR3). Ela se localiza na membrana
canalicular dos hepátócitos, translocando fosfatidilcolina de dentro para fora da
membrana. Neste processo, são formadas micelas que protegem as células da
membrana da árvore biliar das propriedades detergentes dos ácidos biliares. Assim,
na ausência ou diminuição da atividade MDR3 ocorre um desequilíbrio na
18
composição biliar com falta de fosfatidilcolina
e predomínio dos sais biliares ,
levando à inflamação e morte do epitélio biliar25,27.
Esta deficiência pode causar seis doenças hepáticas: PFIC3, cirrose biliar,
colelitíase, colestase intra-hepática da gravidez, colestase neonatal transitória e
colestase induzida por drogas28.
O prurido é menos intenso. Hipertensão portal pode ocorrer na adolescência
ou na fase adulto jovem. Os primeiros sintomas podem se manifestar como
colestase induzida por droga ou mais tardiamente como colestase na gravidez. Não
há manifestações extra hepáticas, nem evolução para malignidade24.
A atividade da GGT está elevada, acima de 10 vezes LSN. As
aminotransferases, bilirrubina conjugada e fosfatase alcalina estão aumentadas.
Fosfolipídeos biliares estão bem reduzidos. Pacientes com pequenas mutações
podem responder ao tratamento com ácido ursodesoxicólico. Entretanto, transplante
hepático pode ser necessário24, 26.
Na histologia, inflamação portal com proliferação ductular são identificados
na fase inicial. Nas fases tardias, cirrose biliar é observada. A ausência ou
diminuição, na imunohistoquímica, da expressão MDR3 pode auxiliar no diagnóstico,
embora possa estar normal em até 50% dos pacientes.
19
Tabela 2: Características dos três subtipos de colestase familiar
Etiologia
Deficiência
ATP8B1
Deficiência
ABCB11
Deficiência
ABCB4
Mecanismo
presumido: prejuízo na
translocação de
aminofosfolípides
através da membrana
celular
Mau funcionamento de
BSEP:
comprometimento do
transporte canalicular
de sais biliares
Comprometimento da
translocação
canalicular de
fosfatidilcolina
Características
clínicas
BRIC1 e PFIC1
Possíveis
características extra
hepáticas (diarreia,
doença pancreática e
deficiência auditiva)
BRIC2 e PFIC2
Incidência
aumentada de
colestase
Risco de malignidade
hepatobiliar
PFIC3
Características
diagnósticas
Elevação sérica de
sais biliares
GGT sérica normal
Colestase branda
Elevação sérica de
sais biliares
GGT sérica normal
Elevação sérica de
sais biliares
GGT sérica elevada
Proliferação ductular
D. BRIC
É uma doença rara, sendo encontrados 77 casos relatados na literatura, e desses
apenas cinco casos brasileiros. Ocorre mais frequentemente no sexo masculino,
entre os 10 e os 30 anos de idade, manifestando-se em até 80% dos casos em torno
da segunda década de vida 8,29.
Mutações no gene ATP8B1 tem sido descritas como responsáveis pela
BRIC. Baseados na diferença no número de mutações identificadas no gene
ATP8B1 nestas duas condições, acredita-se que o produto proteico do FIC1 na
BRIC poderia ser mais disfuncional ou susceptível a outras formas de injúria,
exlicando a natureza recorrente da doença. A predisposição genética está bem
descrita na literatura: 50% dos casos tem caráter familiar. Entretanto, formas
esporádicas têm sido relatadas30.
20
O quadro clínico se manifesta por episódios recorrentes de icterícia e
prurido. Outros sintomas são fadiga, anorexia, hipocolia fecal, colúria e esteatorréia.
Menos frequentemente, são observadas febre, artralgia e cefaléia. Recentemente,
tem sido descrito o surgimento de tosse intratável e alteração nas provas de função
pulmonar. Estas anormalidades desaparecem espontanemente após o término da
colestase.
Os surtos da doença podem durar semanas ou meses com recuperação
total espontânea e não estão relacionados com a idade. Apesar da sintomatologia
ser intensa durante a crise, com prejuízo temporário da qualidade de vida, o
paciente evolui sem sequelas funcionais hepáticas9,31.
Fatores ambientais têm sido aventados como possíveis desencadeadores
dos episódios de colestase, como gastroenterocolites, infecções por influenza, otite
média e erupções cutâneas.
As principais alterações laboratoriais são ácidos biliares elevados,
hiperbilirrubinemia e GGT normal32. A biópsia hepática é normal nos períodos sem
crise e no surto da doença é identifica colestase canalicular sem fibrose, cirrose ou
demonstração de obstrução ducto biliar extra hepático. Em estudo de seguimento de
30 anos de pacientes com BRIC, nenhum sinal de doença crônica foi observado.
Contudo, alguns pacientes com BRIC podem progredir para doença crônica com
fibrose e formação de septos porta-porta, caracterizando PFIC1.
Tygstrup e Jensen, em 1969, propuseram critérios clínicos para o
diagnóstico desta entidade33, sendo os seguintes: (1) pelo menos dois episódios de
icterícia e prurido separados por períodos assintomáticos com duração de vários
meses a anos; (2) alterações bioquímicas consistentes com colestase; (3)
21
demonstração de colestase centrilobular em espécime hepático; (4) vias biliares intra
e extra-hepáticas normais à colangiografia; (5) ausência de outras causas
conhecidas de colestase.
Embora o diagnóstico possa ser suspeitado após a resolução do primeiro
episódio de colestase, o diagnóstico definitivo de BRIC não pode ser feito antes que
um segundo ou terceiro episódio de colestase sintomática se desenvolva e se
resolva espontaneamente.
Tratamentos com corticóide, fenobarbital, colestiramina, rifampicina, ácido
ursodesoxicólico não se mostraram eficazes. A plasmaferese pode auxiliar no
prurido e diminuição dos níveis de bilirrubina.
E. PFIC e BRIC 1
PFIC1 é causada por mutações no gene ATP8B1 (designado FIC1)14,16. Este
gene codifica
uma ATPase do tipo P, designada proteína FIC1, localizada na
membrana canalicular de hepatócitos e cuja função é desconhecida. Como
mutações nesta proteína causam colestase não é claro. Postula-se que
indiretamente a função da proteína anormal poderia perturbar a secreção de ácidos
biliares, explicando a baixa concentração biliar destes encontrada em pacientes com
PFIC1 16,34. O gene FIC1 está localizado no cromossoma humano 18 12,35.
Mutações neste gene têm sido confirmadas no fenótipo
mais suave de
colestase intra-hepática recorrente benigna tipo 1 (BRIC1)14,16,36. É muito provável
que a deficiência de FIC1 representa um processo contínuo com fenótipos
intermediários entre o fenótipo benigno BRIC1 e o fenótipo grave PFIC116. As
22
mutações em PFIC1 perturbam gravemente a função da proteína, enquanto em
BRIC 1 esta funçaõ é apenas parcialmente prejudicada37.
Pacientes com PFIC1 mostram um nível normal de GGT no soro. A
colestase geralmente aparece nos primeiros meses de vida, e causa episódios
recorrentes de icterícia que se tornam mais tarde permanente no curso da doença.
Prurido grave é geralmente observado.
Características extra-hepáticas têm sido descritas em PFIC1, tais como
baixa estatura persistente, surdez e pancreatite, sugerindo uma função biológica
celular geral para FIC1
16,18, 38
. O gene FIC1 é expresso em vários órgãos, incluindo
fígado, pâncreas, intestino delgado e rim 34.
A histopatologia hepática é caracterizada pela colestase canalicular e a
ausência de uma verdadeira proliferação ductular com metaplasia biliar só periportal
de hepatócitos18,24.
F. PFIC e BRIC 2
PFIC2 é causada por mutações no gene ABCB11 (designado BSEP). O
gene BSEP codifica a proteína hepática da membrana canalicular BSEP ATPdependente e está localizado no cromossoma humano 2 21,39.
Mutações nesse gene levam a uma falta de expressão da proteína BSEP,
que é a maior exportadora de ácidos biliares primários contra gradientes extremos
de concentração, o que leva a diminuição do fluxo de bile e acúmulo destes sais
dentro dos hepatócitos, resultando, assim, em grave dano hepatocelular 40.
23
A deficiência de BSEP representa um fenótipo contínuo entre BRIC2 e
PFIC2.
Pacientes com PFIC2 tem atividade sérica normal de GGT e alta
concentração de ácidos biliares no soro. Também tem maiores níveis séricos de
transaminase e alfa-fetoproteína do que pacientes com PFIC1 24, 34, 40,41.
A evolução inicial de colestase é mais grave do que nos outros tipos de
PFIC, com icterícia permanente desde os primeiros meses de vida e aparecimento
rápido de insuficiência hepática dentro dos primeiros poucos anos.
Os achados histopatológicos revelam arquitetura hepática mais perturbada
do que PFIC1, com fibrose e inflamação lobular portal mais pronunciadas. Colestase
canalicular, ausência de uma verdadeira proliferação ductular, lesão lobular grave,
necrose hepatocelular mais óbvia e
transformação de células gigantes mais
evidente são confirmados com exame histopatológico18,24,41.
Para PFIC1 e PFIC2, medicação como ácido ursodesoxicólico
é
considerada durante a gestão terapêutica inicial das crianças42. Alguns pacientes
podem também se beneficiar de desvio biliar43,44. No tratamento do prurido tem sido
utilizado colestiramina, que é uma resina de troca aniônica que se liga a sais biliares
no intestino aumentando a eliminação fecal desses45.
Em PFIC2, ainda é incerto se transplante de hepatócitos ou terapia gênica
com hepatócitos modificados são uma boa abordagem terapêuticas. De fato, pode
haver um risco de deixar as células pré-malignas do fígado no local46.
24
Pacientes com deficiência de BSEP estão em risco significativo para
malignidade hepatobiliar (15% podem desenvolver carcinoma hepatocelular ou
colangiocarcinoma)46.
G. PFIC 3
PFIC3 é causada por mutações no gene ABCB4 (designado MDR3),
localizado no cromossoma 7. Tal gene codifica uma proteína multidroga resistente
classe 3, denominada MDR3, que é uma proteína translocadora de fosfolípidos
(fosfatidilcolina) envolvida na excreção biliar desses e é predominantemente
expressa na membrana canalicular de hepatócitos4,47.
Os fosfolipídeos cumprem a função de solubilizar os sais biliares e proteger
o epitélio ductal. Colestase resulta da toxicidade de bile em que sais biliares
detergentes não são inativados por fosfolipídeos. O mecanismo dos danos no fígado
em PFIC3 está provavelmente relacionado com a ausência de fosfolipídeos
biliares24.
Pacientes PFIC3 mostram um elevado nível sérico de GGT, normal de
colesterol e concentrações moderadamente elevadas de sais biliares primários.
Raramente se apresenta com icterícia no período neonatal, e, em vez, ocorre mais
tarde na infância e até mesmo na idade adulta jovem. O prurido é geralmente leve.
No entanto, adolescentes e pacientes adultos jovens têm sintomas de cirrose devido
a hipertensão portal, que pode resultar em insuficiência hepática.
25
A histopatologia do fígado obtido na fase inicial
mostra fibrose portal e
verdadeira proliferação ductular com infiltrado inflamatório misto. Num estágio mais
avançado, existe extensa fibrose portal e uma imagem típica da cirrose biliar 24.
O espectro fenotípico da PFIC3 varia de colestase neonatal para cirrose em
adultos jovens48.
Deficiência MDR3 pode também representar uma clínica contínua, em que um
único paciente pode experimentar diferentes fenótipos durante o curso da doença.
Essa deficiência está envolvidade em colestase intra-hepática da gravidez tipo 3,
doença biliar do colesterol, colestase induzida por drogas,
colestase neonatal
transitória e cirrose idiopática adulta6.
Nos candidatos a PFIC, o diagnóstico molecular pode ser proposto. A análise
genética é geralmente realizada por sequenciamento de DNA.
26
CONCLUSÃO
A presença de colestase em paciente jovem, sem estigmas de hepatopatia
crônica, com as causas mais comuns de colestase intra-hepática excluídas, e níveis
normais de GGT sugeriram o diagnóstico de BRIC. A histologia hepática
identificando a presença de colestase hepatocelular sem fibrose e sem
anormalidades dos ductos biliares foi fundamental para esclarecimento da patologia.
Pelo fato de ter sido o primeiro episódio de colestase do paciente, o diagnóstico de
BRIC fica ainda sendo provável. Apesar de rara, BRIC deve ser considerada nos
diagnósticos diferenciais das colestases. O esclarecimento do caráter recorrente e
benigno da doença devem ser informados aos pacientes, uma vez que não estão
bem estabelecidos os fatores que determinam os episódios de colestase, assim
como um tratamento eficaz.
27
REFERÊNCIAS
1. Erlinger S. Medical management of chronic cholestasis. In: Schiff E, Sorrel MM,
Mandrey WC, editors. Schiff's Diseases of the liver. 8a ed. Philadelphia: LippincootRaven Publishers; 1999. p. 611-29.
2. Reshef R, Sbeit W, Lachter L. The chronic cholestasis enigma in adults. IMAJ
2002; 4: 449-53.
3. Blum H.Chronic cholestatic liver diseases. Journal of Gastroenterology and
Hepatology (2002) 17,S399–S402.
4. Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial
intrahepatic cholestasis. Orphanet J Rare Dis 2009;4:1.
5. Kullak-Ublick GA, Beuers U, Paumgartner G. Hepatobiliary transport. J Hepatol
2000;32:3-18.
6. Hori T, Nguyen JH, Uemoto S. Progressive familial intrahepatic cholestasis.
Hepatobiliary Pancreat Dis Int. 2010; Dec; 9: 570-8.
7. Cissarek T, Schumacher B, Schwöbel H, Sarbia M, Neuhaus H. Follow-up of
benign recurrent intrahepatic cholestasis (Summerskill-Walshe-Tygstrup syndrome)
over 46 years. Z Gastroenterol 1998; 36(5):379-83. German.
8. Bruni L, Amato R, Tozzi MC, Tarani L, Vignetti P. Benign recurrent intrahepatic
cholestasis. Description of a clinical case. Pediatr Med Chir 1991; 13(2):189-91.
Italian.
9. Parolin MB, Langowiski AR, Ioshii SO, Maggio EM, Coelho JC. Colestase
intrahepática benigna recurrente benigna: seguimento de um caso por 7 anos. Arq
Gastroenterol 2000; 37(4):231-34.
28
10. van Mil SW, van der Woerd WL, et al. Benign recurrent intrahepatic cholestasis
type 2 is caused by mutations in ABCB11. Gastroenterology 2004; 127(2):379-84.
11. Chazouille`res O, Housset C. Intrahepatic cholestasis. In: Rode´s J, editor.
Textbook of hepatology: from basic science to clinical practice. Oxford: Blackwell;
2007. p. 1481–1500.
12. Ujhazy P, Ortiz D, Misra S, Li S, Moseley J, Jones H, et al. Familial intrahepatic
cholestasis 1: studies of localization and function. Hepatology 2001;34:768–75.
13. van Mil SW, van Oort MM, van den Berg IT,et al. Fic1 is expressed at apical
membranes of different epithelial cells in the digestive tract and is induced in the
small intestine during postnatal development of mice. Pediatr Res 2004;56:981–7.
14. Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, et al. A
gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat
Genet 1998;18:219–24.
15. Verhulst P, van der Velden L, et al. A flippase-independent function of ATP8B1,
the protein affected in familial intrahepatic cholestasis type 1, is required for apical
protein expression and microvillus formation in polarized epithelial cells. Hepatology
2010;51:2049–60.
16. van Mil SW, Klomp LW, Bull LN, Houwen RH. FIC1 disease: a spectrum of
intrahepatic cholestatic disorders. Semin Liver Dis 2001;21:535–44.
17. Tygstrup N, Steig BA, Juijn JA, Bull LN, Houwen RH. Recurrent familial
intrahepatic cholestasis in the Faeroe Islands. Phenotypic heterogeneity but genetic
homogeneity. Hepatology 1999;29:506–8.
18. Lykavieris P, van Mil S, Cresteil D, et al. Progressive familial intrahepatic
cholestasis type 1 and extrahepatic features: no catch-up of stature growth,
exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation.
J Hepatol 2003;39:447–52.
29
19. Stapelbroek JM, Peters TA, van Beurden DH, Curfs JH, Joosten A, Beynon AJ, et
al. ATP8B1 is essential for maintaining normal hearing. Proc Natl Acad Sci U S A
2009;106:9709–14.
20. Davit-Spraul A, Fabre M, Branchereau S, et al. ATP8B1 and ABCB11 analysis
in 62 children with normal gamma-glutamyl transferase progressive familial
intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2
and natural history. Hepatology 2010;51:1645–55.
21. Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al. A gene
encoding a liver-specific ABC transporter is mutated in progressive familial
intrahepatic cholestasis. Nat Genet 1998;20:233–8.
22. Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, et al. The
sister of P-glycoprotein represents the canalicular bile salt export pump of
mammalian liver. J Biol Chem 1998;273:10046–50.
23.
Kagawa T, Watanabe N, et al. Phenotypic differences in PFIC2 and BRIC2
correlate with protein stability of mutant Bsep and impaired taurocholate secretion in
MDCK II cells. Am J Physiol Gastrointest Liver Physiol 2008;294:G58–67.
24. Jacquemin E, de Vree JM, Cresteil D, Sokal EM, Sturm E, Dumont M, et al. The
wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to
cirrhosis of adulthood. Gastroenterology 2001;120:1448–58.
25. de Vree JM, Jacqemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, et al. Mutations
in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad
Sci U S A 1998;95:282–7.
26. Maggiore G, Bernard O, Hadchouel M, Lemonnier A, Alagille D. Diagnostic value
of serum gamma-glutamyl transpeptidase activity in liver diseases in children. J
Pediatr Gastroenterol Nutr 1991;12:21–6.
30
27. Oude Elferink RP, Paulusma CC. Function and pathophysiological importance of
ABCB4 (MDR3 P-glycoprotein). Pflugers Arch 2007;453:601-10.
28. Gonzales E, Davit-Spraul A, Baussan C, Buffet C, Maurice M, Jacquemin E. Liver
diseases related to MDR3 (ABCB4) gene deficiency. Front Biosci 2009;14:4242-56.
29. da Silva Jr, J;, Argente,JS; Antunes,GN; Basso, FO; Ricardo Tonial, R. Colestase
intra-hepática benigna recorrente. Revista da AMRIGS, Porto Alegre, 52 (3): 209211, jul.-set. 2008.
30. Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Clin Liv Dis
1999;3:509-28.
31. Summerskill WH, Walshe JM. Benign recurrent intrahepatic obstructive
“jaundice”. Lancet 1959; 31(2):686-90.
32. Whitington PF, Freese DK, Alonso EM, Schwarzenberg SJ, Sharp HL. Clinical
and biochemical findings in progressive familial intrahepatic cholestasis. J Pediatr
Gastroenterol Nutr 1994; 18(2):134-41.
33. Tygstrup N, Jensen B. Intermittent intrahepatic cholestasis of unknown etiology in
five young males from the Faroe Islands. Acta Med Scand 1969; 185(6):523-30.
34. Bull LN, Carlton VEH, Stricker NL, et al. Genetic and morphological findings in
progressive familial intrahepatic cholestasis (Byler disease [PFIC-1] and Byler
syndrome): Evidence for heterogeneity. Hepatology 1997, 26:155-164.
35. Demeilliers C, Jacquemin E, Barbu V, Mergey M, Paye F, Fouassier L, et al.
Altered hepatobiliary gene expressions in PFIC1: ATP8B1 gene defect is associated
with CFTR downregulation. Hepatology 2006;43:1125-1134.
36. Klomp LW, Bull LN, Knisely AS, van Der Doelen MA, Juijn JA, et al. A missense
mutation in FIC1 is associated with greenland familial cholestasis. Hepatology
2000;32:1337-1341.
31
37. Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in
ATP8B1 associated with hereditary cholestasis. Hepatology 2004;40:27-38.
38. Egawa H, Yorifuji T, Sumazaki R, Kimura A, Hasegawa M, Tanaka K: Intractable
diarrhea after liver transplantation for Byler's disease: successful treatment with bile
adsorptive resin. Liver Transplant 2002, 8:714-716.
39. Thompson R, Strautnieks S. BSEP: function and role in progressive familial
intrahepatic cholestasis. Semin Liver Dis 2001;21:545-550.
40. Jansen PL, Strautnieks SS, Jacquemin E, Hadchouel M, Sokal EM, Hooiveld GJ,
et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive
familial intrahepatic cholestasis. Gastroenterology 1999;117:1370-1379.
41. Chen HL, Chang PS, Hsu HC, Ni YH, Hsu HY, Lee JH, et al. FIC1 and BSEP
defects in Taiwanese patients with chronic intrahepatic cholestasis with low
gammaglutamyltranspeptidase levels. J Pediatr 2002;140:119-124.
42. Jacquemin E, Hermans D, Myara A, Habes D, Debray D, Hadchouel M, et al.
Ursodeoxycholic acid therapy in pediatric patients with progressive familial
intrahepatic cholestasis. Hepatology 1997;25:519-523.
43. Modi BP, Suh MY, Jonas MM, Lillehei C, Kim HB. Ileal exclusion for refractory
symptomatic cholestasis in Alagille syndrome. J Pediatr Surg 2007;42:800-805.
44. Bustorff-Silva J, Sbraggia Neto L, et al. Partial internal biliary diversion through a
cholecystojejunocolonic anastomosis-- a novel surgical approach for patients with
progressive familial intrahepatic cholestasis: a preliminary report. J Pediatr Surg
2007;42:1337-1340.
45. Jung C, Driancourt C, Baussan C, et al: Prenatal Molecular Diagnosis of Inherited
Cholestatic Diseases. J Pediatr Gastroenterol Nutr 2007; 44: 453-8.
32
46. Knisely AS, Strautnieks SS, Meier Y, Stieger B, Byrne JA, Portmann BC, et al.
Hepatocellular carcinoma in ten children under five years of age with bile salt export
pump deficiency. Hepatology 2006;44:478-486.
47. Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver
disease: one gene for three diseases. Semin Liver Dis 2001;21:551-562.
48. Ziol M, Barbu V, Rosmorduc O, Frassati-Biaggi A, Barget N, Hermelin B, et al.
ABCB4 heterozygous gene mutations associated with fibrosing cholestatic liver
disease in adults. Gastroenterology 2008;135:131-141.