CARVALHO, Hernandes F.; RECCO-PIMENTEL,
Shirlei M. Mitocôndria, In, A Célula. 2ª edição.
2007, p.211-225.
Página 211
15 MITOCÔNDRIA
Edson Rosa Pimentel
As mitocôndrias começaram a ser observadas
em 1840, em células de rim e de fígado,
coradas pelo método de Régaud. As estruturas
observadas tinham formas alongadas e
arredondadas, respectivamente. Daí o nome de
mitocôndria, junção do termo grego mitos, que
quer dizer alongado, e chondrion, que significa
pequeno grânulo, em alusão aos aspectos
morfológicos que as mitocôndrias podem
assumir na célula. As mitocôndrias podem ser
facilmente distinguidas de outras organelas,
mesmo com a célula viva, usando-se um
corante chamado verde janus. Esse corante,
por ser uma substância redox, capaz de
assumir características de um composto
reduzido ou oxidado, em contato com a
mitocôndria, pode ser oxidado para uma forma
corada pelo citocromo c oxidase, um dos
componentes da cadeia respiratória.
No geral, as mitocôndrias exibem formas
alongadas, como ocorre em tubos de Malpighi,
glândulas salivares de insetos e pâncreas de
mamíferos (Figura 15.1a), mas mitocôndrias
esféricas
também
são
encontradas
em
intestino e fígado (Figura 15.1b). Além disso,
técnicas
de
microcinematografia
têm
evidenciado que as mitocôndrias podem
assumir várias conformações em diferentes
momentos da vida da célula. O tamanho das
mitocôndrias também é variável, podendo
medir de 0,2 a 1,0 µm de diâmetro e de 2 a 8
µm de comprimento.
A quantidade de mitocôndrias também varia
para células de diferentes origens, estando
diretamente relacionada à demanda energética
da célula. Assim, temos em alguns ovócitos
uma quantidade de 300.000 mitocôndrias por
célula. Em ameba gigante, pode chegar a
10.000, em hepatócitos, de 500 a 1.600, em
células renais, em torno de 300, em
espermatozóides, cerca de 25 e algumas algas
verdes chegam a ter apenas uma mitocôndria.
As células vegetais, em geral, apresentam uma
quantidade bem menor de mitocôndrias em
relação às células animais.
A distribuição de mitocôndrias no interior da
maioria das células ocorre totalmente ao acaso,
mas há casos em que se concentram em
regiões em que a demanda energética é maior.
Em células musculares, por exemplo, as
mitocôndrias estão associadas aos filamentos
contráteis que requerem ATP (Figura 15.2); em
espermatozóides, elas se localizam na peça
intermediária, justamente para facilitar o
provimento de ATP para a movimentação da
cauda. Muitas vezes, as mitocôndrias estão
associadas com glóbulos de gordura, de forma
a expor a maior área possível de sua
superfície, em contato com os lipídios (Figura
15.3) e, conseqüentemente, aproveitar melhor
os ácidos graxos resultantes da ação das
lipases.
Página 212
A análise de imagens obtidas ao microscópio
eletrônico e estudos bioquímicos têm sido de
grande valor para melhor conhecer a ultraestrutura e fisiologia mitocondriais. Uma das
técnicas de microscopia eletrônica bem
conhecida é a contrastação positiva, que
consiste em embeber o material já fixado em
urna solução de metal pesado, como acetato
de uranila, que se acumula em algumas partes
da organela, tornando-as eletrodensas. Uma
outra técnica é a contrastação negativa, que
consiste em deixar o material embebido em
uma solução aquosa de um sal eletrodenso,
como fosfotungstato de sódio. Neste caso, as
estruturas vão aparecer como regiões claras,
em decorrência da não-penetração do sal,
contra um fundo eletrodenso. Os estudos
bioquímicos da mitocôndria só foram possíveis
após a obtenção da organela isolada, em um
procedimento
que
se
inicia
com
a
homogeneização seguida de centrifugação
fracionada (ver Capítulo 4). A partir das
mitocôndrias isoladas, após tratamento de
ultra-som,
foram
obtidas
as
partículas
submitocondriais,
ou
seja,
vesículas
mitocondriais obtidas com a selagem dos
fragmentos
da
membrana
interna
das
mitocôndrias de modo que, neste caso, os
complexos ATP sintase ficaram voltados para o
meio externo.
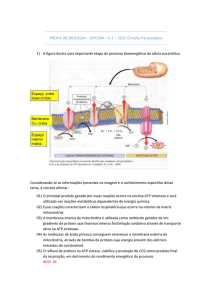
Ultra-estrutura
As mitocôndrias podem ser detectadas com
microscopia óptica comum, mas detalhes da
sua estrutura só são observados com o uso de
um microscópio eletrônico. Essas organelas são
constituídas de duas membranas, estrutural e
funcionalmente distintas. Elas definem dois
compartimentos na mitocôndria, o espaço
intermembrana, que separa as membranas
interna e externa, e a matriz mitocondrial, que
está circundada pela membrana interna
(Figuras 15.4 e 15.5). Na matriz podem ser
observados ribossomos e alguns glóbulos
eletrodensos de fosfato de cálcio.
A membrana interna se invagina para o
interior da mitocôndria, constituindo as cristas
mitocondriais. Essas projeções para o interior
da organela permitem um considerável
aumento da área da membrana interna, que,
como veremos a seguir, é o local em que estão
os componentes da cadeia respiratória e o
complexo enzimático F1F0 responsável pela
síntese de ATP. As membranas interna e
externa são estrutural e funcionalmente
diferentes. A utilização da técnica de freezeetching permitiu visualizar o aspecto bastante
particulado da membrana interna, enquanto a
membrana externa exibia um aspecto mais
liso.
Logicamente, as diferenças estruturais entre
as duas membranas são conseqüências diretas
de suas composições químicas e das interações
entre alguns de seus componentes. A
composição química das membranas, em geral,
é de lipídios e proteínas, mas a quantidade
relativa desses dois componentes pode variar.
Assim, a membrana externa contém 50% de
lipídios e 50% de proteínas, enquanto na
interna encontramos apenas 20% de lipídios e
80% de proteínas. Entre essas proteínas estão
os citocromos, que fazem parte da cadeia
respiratória, a ATP sintase, que participa da
síntese do ATP, NADH desidrogenase, que
libera um par de elétrons para a cadeia
respiratória, a succinato desidrogenase, que
catalisa urna das reações do ciclo de Krebs, a
carnitina aciltransferase, que participa da
transferência de ácido graxo do espaço interPágina 213
membrana para a matriz mitocondrial, entre
muitas outras proteínas, que têm função de
transporte de vários metabólitos.
A presença de proteínas na membrana
interna com funções bem definidas, como
mencionado anteriormente, já confere uma
maior seletividade dessa membrana à entrada
dos mais diversos componentes, até mesmo de
dimensões submoleculares, como os íons. Já a
membrana externa, além de apresentar uma
maior fluidez, dada sua maior quantidade de
lipídios em relação à membrana interna,
também apresenta uma proteína conhecida
como porina, que forma verdadeiros canais
transmembrânicos, permitindo a passagem
livre de íons e moléculas de até 10.000 daltons
de peso molecular. Curiosamente, a porina
encontrada
na
membrana
externa
de
mitocôndrias é muito semelhante às proteínas
que formam poros na membrana externa de
bactérias do tipo Gram-negativa.
Composição química
Além dos componentes já mencionados no
item anterior, essa organela contém ácidos
nucléicos e várias enzimas, que participam do
metabolismo de carboidratos, de ácidos graxos
e de compostos aminados. É interessante
observar que a molécula de DNA é circular,
semelhante àquela encontrada em bactérias, e
corresponde a apenas 1 % do DNA contido no
núcleo. Embora poucas proteínas (apenas 13)
sejam codificadas pelo DNA mitocondrial, a
mitocôndria contém todo o mecanismo para
replicação e transcrição do DNA e tradução de
proteínas.
Fisiologia
Respiração celular
A respiração, em um sentido mais amplo,
pode ser definida como sendo o processo de
oxidação de moléculas orgânicas acompanhado
da liberação de energia, que é aproveitada na
síntese de ATP.
Entre os compostos que, após oxidação,
resultam em alto rendimento de ATP estão os
carboidratos e os lipídios, mas não se pode
esquecer que os compostos aminados, como os
aminoácidos, também podem ser oxidados e
liberar energia para produzir ATP (Figura 15.6).
Uma das vias metabólicas mais importantes e
mais conhecidas é a glicólise aeróbica, que
também faz parte da respiração. Contudo, há
cerca de 3,5 bilhões de anos, a ausência de
oxigênio na atmosfera levava as bactérias a
realizarem a glicólise anaeróbica, como parte
de um processo chamado fermentação.
Em determinada fase da glicólise (Figura
15.7), que consiste na degradação de glicose
até ácido pirúvico, ocorre redução de duas
moléculas de NAD, resultando em NADH+H+
(Figura 15.8). No entanto, essas moléculas de
NADH+H+ são reoxidadas, transferindo seus
elétrons a um outro aceptor, para que o estado
de equilíbrio seja mantido. Também existe um
consumo de duas moléculas de ATP e uma
produção de 4 moléculas, possibilitando um
rendimento líquido de 2 moléculas de ATP para
cada molécula de glicose degradada até
piruvato.
Uma das formas empregadas por bactérias
para reoxidar as moléculas de NADH+H+ é usar
esses nucleotídeos para reduzir piruvato em
lactato, constituindo a fermentação láctica:
Outro tipo de fermentação envolve a
decomposição do piruvato em acetaldeído e
CO2, com o acetaldeído sendo reduzido a
etanol, caracterizando a fermentação alcoólica:
Página 214
No caso dos organismos aeróbios, o
NADH+H+, produzido durante a glicólise, é
reoxidado pelos componentes da cadeia
respiratória presentes na membrana interna e
cristas mitocondriais, como será visto mais
adiante.
Ciclo de Krebs
Como pode ser observado na Figura 15.6, o
acetil-CoA pode se originar da degradação de
aminoácidos, lipídios e carboidratos. No caso
deste último, o piruvato formado, ao entrar na
mitocôndria,
sofre
descarboxilação
e
desidrogenação em um processo catalisado por
um complexo enzimático, chamado piruvato
desidrogenase (Figura 15.9). Nesta reação,
uma molécula de NADH+H+ é formada e o
radical acetil se liga à coenzima A, originando o
acetil-CoA. A molécula de coenzima A, um
carreador de grupos acil, é geralmente
representada como CoA - SH, porque o grupo
tiol -SH é a parte da molécula que reage e se
liga ao grupo acil, que no caso da descarboxilação do piruvato, é o grupo acetil.
Desse modo, a coenzima A se liga ao radical
acetil formando o acetilCoA (Figura 15.9), que
reage com o oxaloacetato, formando o ácido
cítrico, um ácido tricarboxílico, dando início a
uma seqüência de reações que regenera a
moléPágina 215
cula de ácido oxaloacético. Esse conjunto de
reações (Figura 15.10) é conhecido como ciclo
do ácido tricarboxílico ou ciclo de Krebs, em
homenagem ao bioquímico Hans Krebs (19001981). Neste ciclo, devido às reações de
desidrogenação, 4 pares de átomos de
hidrogênio são liberados, sendo que 3 serão
utilizados para reduzir 3 moléculas de NAD,
resultando em NADH+H+, e 1 irá reduzir 1 FAD
a FADH2 (Figura 15.11). Um outro produto
desse ciclo é uma molécula de ATP, produzida
a partir de ADP e Pi, utilizando energia de
hidrólise
de
GTP.
Uma
questão
que
normalmente surge quando se estuda o ciclo
de Krebs é sobre a necessidade de tantas
reações para decompor um radical tão pequeno
como o acetil. A razão se deve ao fato do
grupo acetil ser altamenPágina 216
te resistente à oxidação. Um caminho mais
simples foi encontrado pela natureza quando o
radical acetil reagiu com uma molécula de
oxaloacetato, produzindo um composto mais
suscetível à oxidação.
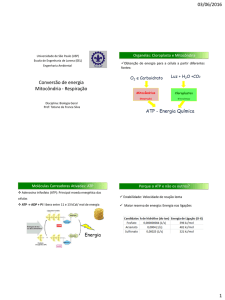
Transdução de energia
Embora a mitocôndria tenha sido detectada
no século XIX, só em 1960 foram realizados
estudos bioquímicos que permitiram conhecer
como ocorre a respiração celular. Essa
organela pode aproveitar a energia presente
em ligações químicas covalentes, entre átomos
de carbono (-C - C-), e transformá-la em
energia elétrica, para novamente armazená-la
em ligações químicas também covalentes,
como ocorre entre ADP e fosfato, na formação
da molécula de ATP. A molécula de ATP
formada pode ser facilmente decomposta em
ADP e Pi, liberando energia para o
aproveitamento imediato pela célula. Na
verdade, ocorrem duas transformações de
energia, primeiramente química em elétrica, e
depois elétrica em química novamente. A
primeira está diretamente relacionada com a
quebra de ligações C - C dos componentes do
ciclo do ácido tricarboxílico, ou com a
degradação dos ácidos graxos ou dos
aminoácidos. Em todos os casos, ocorre perda
de elétrons que são captados pelos nucleotídeos NAD e FAD, conhecidos em suas formas
reduzidas como NADH + H+ e FADH2,
respectivamente (Figuras 15.8 e 15.11). Esses
equivalentes redutores vão ceder seus elétrons
para os componentes da cad respiratória,
presentes
na
membrana
interna
das
mitocôndrias. À medida que esses elétrons vão
sendo transferidos na cadeia respiratória até
chegar ao oxigênio, aceptar final de elétrons,
ocorre ejeção de prótons para o espaço
intermembranas, e até mesmo para fora da
mitocôndria, já que a membrana externa é
permeável a prótons. Esse fato resulta em uma
diferença de pH (pH) entre os meios externo
e interno da mitocôndria. Também ocorre uma
diferença de potencial () entre as faces
interna e externa da membrana interna da
mitocôndria (Figura 15.12). A diferença de pH
e o potencial de membrana são importantes no
processo da fosforilação oxidativa, como será
visto mais adiante.
Página 217
Cadeia respiratória
É constituída de diversos componentes
formados, em sua maioria, por complexos
protéicos
contendo
grupos
heme,
que
permitem a transferência de elétrons graças à
possibilidade dos átomos de ferro se reduzirem
(aceitando elétrons) e se oxidarem (doando
elétrons), até ceder elétrons ao oxigênio com
conseqüente
formação
de
água.
Esses
compostos são os cito cromos (Figura 15.13),
que estão dispostos na bicamada lipídica da
membrana interna da mitocôndria (Figura
15.14). Além dos citocromos, os complexos
protéicos
também
possuem
estruturas
polipeptídicas contendo Fe ou S e nucleotídeos
como FMN ou FAD (Tabela 15.1).
Os componentes da cadeia respiratória
diferem em suas tendências de perder
elétrons.
Essas
tendências
podem
ser
expressas pelos seus potenciais padrão de
oxidorredução, que são medidos em condições
especiais, fora do meio celular. Essa medida é
feita pela diferença de potencial gerada quando
uma solução contendo 1M de agente oxidante
e 1M de agente redutor, em 25°C e pH 7,
estiver em equilíbrio com um eletrodo capaz de
aceitar elétrons do agente redutor. O valor
encontrado é o potencial de oxidorredução.
Quanto maior esse valor, maior será a
tendência de um determinado composto perder
elétrons. Assim, considerando os valores
mostrados na Tabela 15.2, temos o ubiquinol,
a forma reduzida da ubiquinona, com uma
maior tendência em ceder seu par de elétrons
para o citocromo c1. Este tem uma maior
tendência em ceder elétrons para o citocromo
c, este para o citocromo a, em seqüência para
o citocromo a3, e deste para o aceptor final da
cadeia respiratória, o oxigênio.
Página 218
Essa transferência de elétrons resulta em dois
acontecimentos: (1) ejeção de prótons para
fora da mitocôndria com conseqüente formação
de um gradiente de H+. (2) Formação de um
potencial de membrana entre as faces externa
(espaço intermembranas) e interna (matriz) da
membrana interna. Ambos os eventos são
fundamentais para que ocorra a fosforilação
oxidativa do ADP.
Tabela 15.1 Características dos complexos
protéicos presentes na cadeia respiratória.
É preciso ficar bem claro que, ao contrário do
que parece ocorrer especialmente quando se
observa a Figura 15.14, os complexos de
transferência de elétrons não se situam
linearmente na membrana mitocondrial e os
diferentes complexos não estão presentes em
quantidades equimolares. Alguns estudos sobre
componentes da cadeia respiratória, junto ao
conhecimento que hoje se tem do caráter
dinâmico do atual modelo de membrana, têm
sugerido que o citocromo c pode se difundir
rapidamente de um complexo para outro, não
ficando, portanto, fixo em um complexo, e que
as movi-
Tabela 15.2 Valores de potenciais redoxi para
componentes da cadeia respiratória e número
de elétrons envolvidos.
Página 219
mentações dos citocromo c, ubiquinona e dos
próprios complexos como um todo ocorrem em
velocidades diferentes, o que significa que não
podem estar fincados todos juntos na
bicamada lipídica. Essas informações nos
ajudam a ter uma visão dinâmica da cadeia
respiratória na mitocôndria.
Fosforilação oxidativa
A transferência de um par de elétrons do
NADH+H+ para o O2 envolve a liberação de
grande quantidade de energia G0', que está
diretamente relacionada com a variação de
potencial redoxi E0'. Considerando os valores
de E0, mostrados na Tabela 15.2, a variação de
EO' (E0’), quando um par de elétrons caminha
de NADH+H+ ao O2 é:
Eo’ = Eo’(O2/H2O) - Eo’(NADH+H+/NAD)
= 0,82 - (- 0,32)
= 1,14 volts
A variação de energia livre (Go’) pode ser
obtida pela fórmula Go’ = -nFEo’, na qual n é
o número de elétrons e F é a constante de
Faraday = 23.060
cal-1mol-1. Assim, quando um par de elétrons é
transferido, temos:
Go’ = -2 x 23.060 x 1,14 = -52,6 Kcal/mol
Esse valor negativo significa que, quando um
par de elétrons passa do NADH+H+ para o O2,
ocorre uma diminuição de quase 53 Kcal. Se
comparado esse valor com a variação de
energia livre na formação do ATP:
ADP + Pi
Kcal
ATP + H2O
Go’
=
+7,3
verifica-se que a quantidade de energia
liberada, durante a transferência de 2 elétrons
do NADH+H+ para o O2, é bem maior do que a
quantidade de energia necessária para a
síntese de 1 molécula de ATP. Sabe-se,
contudo, que apenas 3 moléculas de ATP são
sintetizadas para cada 2 elétrons transferidos
na cadeia respiratória. Portanto, apenas 42%
da energia liberada na cadeia respiratória é
aproveitada para a síntese de ATP, sendo o
restante da energia parte utilizada para o
transporte através da membrana mitocondrial,
e parte perdida na forma de calor.
A explicação físico-química para a síntese de
ATP, acoplada à transferência de elétrons na
cadeia respiratória, teve início na Inglaterra
com os estudos de P. Mitchell em 1961,
quando propôs a hipótese quimiosmótica,
testada
experimentalmente
por
vários
pesquisadores durante as décadas de 1960 e
1970. Mitchell ganhou o Nobel de Química pelo
seu trabalho nesta área, em 1978.
A teoria quimiosmótica afirma que, com a
passagem de elétrons na cadeia respiratória,
ocorre uma ejeção de prótons da matriz para o
espaço intermemPágina 220
brana e mesmo para fora da mitocôndria,
gerando um gradiente de H+ (ou gradiente de
pH) entre o meio externo e interno da
mitocôndria. Este gradiente de H+ e o potencial
de membrana () somados resultam em uma
força chamada força próton-motiva (fpm): fpm
= pH + .
A fpm pressiona o H+ a retornar para a matriz
mitocondrial.
A
membrana
interna
é
impermeável a H+, contudo os prótons podem
passar para o interior da mitocôndria via
complexo ATP sintase (Figura 15.15). Esse
complexo, também chamado de complexo F0F1,
é constituído de um pedúnculo, F0 (complexo
protéico sensível ao antibiótico oligomicina),
embutido na bicamada lipídica, e de uma
estrutura denominada F1, contendo o sítio
catalítico, constituída de 5 subunidades
diferentes, com a seguinte composição
estequiométrica (Figura 15.16). A
porção F0 é composta também de várias
subunidades, cujo número pode variar de
espécie para espécie. No caso mostrado da
Figura 15.16, a F0 é composta de 3
subunidades diferentes representadas como
ab2c11. Recentemente foi demonstrado que
existe um acoplamento químicomecânico entre
F1 e F0. Com a passagem de prótons através de
F0, ocorre rotação de , e c11, que é traduzida
em
mudanças
conformacionais
nas
subunidades catalíticas presentes em F1, que
resultam na síntese de ATP. Provavelmente
essas
mudanças
conformacionais
são
transmitidas
com
a
participação
dos
componentes estacionários ab2, que não
sofrem torção à medida que c11 estão em
rotação. Vários trabalhos com o complexo F0F1
têm sugerido que a rotação ocorre em três
etapas, com um pequena pausa entre elas, que
corresponderiam
a
três
estados
conformacionais,
respectivamente
nas
subunidades
da
F1 .
Esses
estados
conformacionais poderiam representar: os
sítios que acomodam ADP e Pi em uma das
partículas , o ATP já formado em outra
subunidade , e em uma terceira subunidade, a
liberação do ATP formado (Figura 15.17).
O gradiente de prótons não serve somente
para a síntese de ATP em mitocôndrias, mas
permite o transporte ativo de Ca+2 e
metabólitos e serve também para o transporte
de carboidratos e aminoácidos em bactérias.
Em cloroplastos, é fundamental para a
fotofosforilação do ADP, em processo muito
semelhante ao que ocorre em mitocôndria.
Rendimento de ATP
Para cada molécula de NADH+H+ que cede 2
elétrons na cadeia respiratória são produzidas
3 moléculas de ATP, enquanto cada molécula
de FADH2 que cede 2 elétrons vai gerar 2
moléculas de ATP. Assim, temos que, dentro da
mitocôndria, 2 moléculas de NADH+H+ são
produzidas pela desidrogenação de 2 moléculas
de piruvato, 3 NADH+H+ e 1 FADH2 são
formadas, para
Página 221
cada molécula de acetil-CoA que entra no ciclo
de Krebs, e 1 GTP que imediatamente é
transformado
em
ATP
produzido
metabolicamente, em uma das reações do ciclo
de Krebs. Dessa forma, teremos uma produção
de 15 moléculas de ATP para cada molécula de
piruvato que entra na mitocôndria. É preciso
considerar que fora da mitocôndria também
são produzidas moléculas de NADH+H+ (ver
etapas 5 e 6 da glicólise na Figura 15.7), que
não conseguem atravessar a membrana interna da organela. Por outro lado, existem
mecanismos que podem transferir elétrons do
NADH+H+ citoplasmático para a cadeia
respiratória. Um desses mecanismos envolve a
ação da enzima glicerol-3-fosfato desidrogenase citoplasmática, que utilizando os H
do
NADH,
catalisa
a
redução
da
dihidroxiacetona em gliceraldeído-3-fosfato
(Figura 15.18), que alcança a membrana
interna da mitocôndria. Nessa membrana
interna, voltada para o espaço intermembrana,
uma
outra
enzima,
a
glicerol-3-fosfato
desidrogenase mitocondrial, que tem o FAD
como grupo prostético, vai transferir elétrons
do glicerol-3-fosfato, para a ubiquinona, na
cadeia transportadora de elétrons.
Uma outra forma de se aproveitar os H do
NADH citoplasmático é pela ação da enzima
malato desidrogenase, que transfere elétrons
do NADH para o oxaloacetato, formando
malato (Figura 15.19). Este atravessa a
membrana interna mitocondrial com a ajuda de
um transportador de membrana, e na matriz
mitocondrial sofre desidrogenação pela ação da
malato desidrogenase mitocondrial que tem
NAD
como
coenzima,
regenerando
oxaloacetato e formando NADH+H+, que vai
ceder elétrons no início da cadeia respiratória.
O
oxaloacetato
formado
não
consegue
atravessar a membrana interna da mitocôndria
para retomar ao citoplasma, mas ao sofrer
uma reação de transaminação, é convertido em
aspartato, e para esse aminoácido existe um
transportador que permite a sua passagem
para o citosol. No citosol, também por uma
reação de transaminação, o oxaloacetato é
regenerado e assim está pronto para receber
os H do NADH citoplasmático, completando-se
o ciclo. O efeito final desse processo é como se
o NADH produzido no cito sol passasse diretamente para o interior da mitocôndria e fosse
ceder seus elétrons no início da cadeia
respiratória, e levar à produção de três
moléculas de ATP. Todo esse mecanismo é
conhecido como o dispositivo malato-aspartato,
pois malato é a molécula que "carrega" os H
dos NADH citoplasmáticos e aspartato é o
componente que retorna ao citoplasma e
regenera o oxaloacetato (Figura 15.19).
Outras atividades metabólicas
Após a ação de lipases e fosfolipases, os
ácidos graxos resultantes podem atravessar a
membrana mitocondrial, graças à complexação
com carnitina. Esse complexo se desfaz tão
logo chegue no interior da mitocôndria,
liberando novamente a longa molécula de ácido
graxo. Dentro da organela, os ácidos graxos
são degradados por uma seqüência de reações,
chamada de -oxidação de ácidos graxos.
Neste processo, os ácidos graxos vão perdendo
seus carbonos na forma de acetil-CoA e
também ocorre redução de NAD e FAD, que
acabam cedendo seu par de elétrons na cadeia
respiratória. O acetil-CoA liberado é, por sua
vez, aproveitado no ciclo de Krebs.
Outra
participação
importante
das
mitocôndrias é no ciclo da uréia. A formação
desse composto ocorre no fígado de animais
ureotélicos, isto é, animais como muitos
moluscos e anfíbios terrestres e mamíferos em
geral que vivem em ambiente em que a água é
limitada e que tem a uréia como principal
produto final nitrogePágina 222
nado. Nesse processo, dois grupos amino,
derivados de aminoácidos, e uma molécula de
CO2, dão origem a uma molécula de uréia, que
é transportada pela corrente sangüínea até os
rins e então, excretada. Um daqueles grupos
amino se origina no interior da mitocôndria de
célula hepática por desaminação de glutamato.
O grupo amino vai fazer parte da molécula de
citrulina que sai da mitocôndria (Figura 15.20)
e, no citosol, resulta na formação de uréia, que
transportará o NH3 formado na matriz
mitocondrial.
As mitocôndrias também participam da
produção de hormônio esteróide. O colesterol
produzido
nas
membranas
do
retículo
endoplasmático é lançado no cito plasma,
atravessa a mitocôndria e, na sua membrana
interna, é transformado em pregnenolona, que
então retorna ao retículo, onde é finalmente
transformado em testosterona.
Uma outra função das mitocôndrias ocorre em
alguns tecidos adiposos de recém-nascidos e
também de alguns animais durante a
hibernação. Nestes casos, ocorre a expressão
de uma proteína chamada termogenina, que
torna a membrana externa permeável aos
prótons, desacoplando o transporte de elétrons
da síntese de ATP. A energia liberada durante o
transporte de elétrons é então perdida na
forma de calor, que nos casos citados é
altamente benéfico, tanto para o recémnascido como para o animal que está
hibernando.
Biogênese
As mitocôndrias são formadas a partir da
divisão
e
crescimento
de
mitocôndrias
preexistentes (Figura 15.17). Embora as
mitocôndrias contenham DNA, este não
Página 223
Figura 15.19 Dispositivo do Malato-Aspartato.
O malato transporta os H do NADH
citoplasmático para o interior da mitocôndria.
Na matriz mitocondrial o malato se oxida,
reduzindo o NAD que passa a ser NADH+H+.
Observe que nesse processo estão envolvidas
outras
reações
importantes.
No
meio
extracelular o aspartato sofre transaminação,
perdendo o seu grupamento -NH3+ para o cetoglutarato (KG), assim o aspartato é
convertido em oxaloacetato, que então é
reduzido pelo NADH e pela enzima Malato
Desidrogenase,
formando
malato,
que
atravessa a membrana interna da mitocôndria
por
um
sistema
de
transporte
que
simultaneamente coloca KG para fora da
mitocôndria. O malato, já dentro da organela
ao ser oxidado pela Malato Desidrogenase e
pelo NAD, forma oxaloacetato que sofre
transaminação, regenerando aspartato. Nessa
reação de transaminação o -NH3+ é transferido
do glutamato para o oxaloacetato, regenerando
aspartato e formando KG. Note que glutamato
tem que ser continuamente transportado para
dentro da mitocôndria, e KG, continuamente
transportado para fora da mitocôndria.
Página 223
contém todas as informações necessárias para
que a organela possa viver independentemente
do resto da célula. Embora a organela tenha
condições de realizar todos os processos de
replicação, transcrição e tradução, apenas 13
proteínas
são
codificadas
pelo
DNA
mitocondrial. Vários componentes necessários
para a própria expressão gênica, como
proteínas ribossomais, aminoácidos e a maioria
das enzimas do ciclo de Krebs, são
provenientes do citosol (Tabela 15.3).
Para que as proteínas possam entrar na
mitocôndria, primeiro é necessário que elas
sejam reconhecidas pela organela, o que é
possível devido à presença de seqüências
sinais, que dirigem as proteínas para o
compartimento
adequado
dentro
da
mitocôndria. Essas seqüências são geralmente
removidas por proteases, impedindo, assim,
que
as
proteínas
portadoras
daquelas
seqüências possam retomar para o citosol. Um
aspecto que também não pode ser ignorado na
importação de proteínas pela mitocôndria é a
presença de chaperones, ou moléculas
companheiras, que, às custas de ATP,
impedem
que
as
proteínas
assumam
conformações inadequadas. Duas chaperones
particularmente importantes são a hsp 70 e
hsp 60. Um outro fato fundamental para a
translocação de proteínas é a existência de um
potencial de membrana (), como visto
anteriormente.
Defeitos mitocondriais
Defeitos mitocondriais têm sido detectados
em várias doenças, especialmente aquelas que
envolvem tecidos que necessitam de uma alta
demanda energética proveniente de respiração,
como tecido muscular, onde a miopatia
mitocondrial leva a uma fraqueza do músculo,
ou como no tecido nervoso, onde uma
neuropatia ou encefalopatia decorrentes de
mutações em genes para síntese de proteína
mitocondrial podem resultar
Página 225
em epilepsia e/ou cegueira. Nestes últimos 10
anos, mais de 150 registros de mutações em
DNA mitocondrial têm sido relacionados com
uma variedade de doenças degenerativas.
Outras doenças também foram relacionadas
com mutações em genes nucleares, afetando
especialmente a fosforilação oxidativa ou a
biogênese
mitocondrial.
Os
defeitos
relacionados
com
a
bioenergética
da
mitocôndria
são
acompanhados
de
um
aumento na taxa de lactato na circulação,
sendo por isso um importante dado para a
diagnose. Além disso, outras evidências de
natureza
estrutural
também
têm
sido
identificadas, como mitocôndrias vacuolizadas
com poucas cristas ou com cristas semelhantes
a favos de mel ou a redemoinhos concêntricos.
Os defeitos mitocondriais também podem ser
rapidamente detectados por meio de ensaios
enzimáticos para algumas enzimas específicas,
como
piruvato
desidrogenase,
citocromo
oxidase e ATP sintase. O seqüenciamento de
DNA mitocondrial também permite que se
detecte precisamente o sítio do defeito
genético.
Origem
Existem duas teorias para explicar a origem
das mitocôndrias. Uma delas afirma que a
mitocôndria
surgiu
de
uma
associação
simbiótica entre um eucarioto anaeróbico e um
procarioto aeróbio. A outra teoria, não
simbiótica, defende a idéia de que a
mitocôndria deve ter surgido de um processo
que envolvia invaginação de membrana de um
procarioto, contendo componentes da cadeia
respiratória e complexo ATP sintase, seguido
do desprendimento daquele segmento de
membrana e subseqüente incorporação de
fragmento de molécula de DNA.
Hoje, há fortes indicações de que as
mitocôndrias tenham origem simbiótica. Alguns
aspectos que favorecem essa teoria são: (1) a
dupla
hélice
de
DNA
encontrada
em
mitocôndrias é circular, como ocorre em
bactérias; (2) os mitorribossomos têm um
coeficiente de sedimentação em torno de 55S,
sendo, portanto, mais próximo daquele
encontrado em bactérias, que é de 70S,
enquanto os ribossomos encontrados no citosol
de eucariotos têm 80S; (3) a síntese de
proteínas em mitocôndrias é inibida por
cloranfenicol, o mesmo antibiótico que inibe a
síntese de proteína em procarioto, porém, no
citosol, a síntese é inibida por cicloheximida;
(4) o aminoácido iniciador da síntese de
proteínas em mitocôndria é o formil-metionina,
da mesma forma que ocorre em bactérias.
O genoma mitocondrial dos animais, em
geral, carrega uma quantidade mínima de
genes, juntamente com algumas seqüências
não-codificadoras, mas que servem para
regular a atividade gênica. O tamanho
compacto do genoma das mitocôndrias de hoje
provavelmente é resultado de uma gradual
transferência de genes da mitocôndria para o
genoma nuclear.
Referências bibliográficas
1. Gray MW, Burger G, Lang BE. Mitochondrial
evolution. Science 1999; 283: 1476-81.
2. Race HL, Herrman RG and Martin W. Why
have organelles retained genomes? Trends in
Genetics 1999; 15: 364-70.
3. Sarasate M. Oxidative phosphorilation at
the fin de siècle. Science 1999; 283: 1488-93.
4. Wallace DC. Mitochondrial diseases in man
and mouse. Science 1999; 283: 1482-8.
5. Yaffe MP. The machinery of mitochondrial
inheritance and behavior. Science 1999; 283:
1493-7.
6. Yasuda R, Noji H, Yoshida M, Kinosita K,
Itoh H. Resolution of distinct rotational
substeps by submillisecond kinetic analysis of
F1- ATPase. Nature 2001; 410: 898-904.
7. Kaim G, Prummer M, Sick B, Zumofen G,
Renn A, Wild UP, Dimroth P. Coupled rotation
within single F0P1 enzyme complexes during
ATP synthesis or hydrolysis. FEBS Letters
2002;525: 156-63.