Ana Carolina Etrusco Zaroni
Resumo de Farmacologia
2ª prova
FARMACODINÂMICA
A farmacodinâmica pode ser definida como o estudo dos efeitos bioquímicos e fisiológicos dos
fármacos e de seus mecanismos de ação. Fornece as bases para entendermos como os fármacos atuam no
nosso organismo (estudo da ação dos fármacos no organismo). Os fármacos não criam efeitos, mas em vez
disso, modulam funções intrínsecas. As formas pelas quais os medicamentos podem atuar são através de
receptores (proteínas ou glicoproteínas localizadas na membrana plasmática, citoplasma ou núcleo
[carioteca ou material genético]).
Os receptores são componentes do organismo com os quais o agente químico interage. Podem receber
moléculas endógenas ou exógenas (medicamentos = imitadores das substâncias endógenas). As proteínas
formam a classe mais importante de receptores de fármacos. Os ácidos nucléicos também são importantes
receptores de fármacos, especialmente para os quimioterápicos antineoplásicos. Uma propriedade
importante dos receptores fisiológicos, que também os torna excelentes alvos para os fármacos, é que eles
podem agir de modo catalítico, sendo, por conseguinte, amplificadores de sinais bioquímicos.
Se restringirmos a definição de receptores a macromoléculas, então pode-se dizer que diversos
fármacos não atuam propriamente me receptores. Alguns fármacos se ligam especificamente a pequenas
moléculas ou íons encontrados normal ou anormalmente no corpo. Um exemplo é a neutralização
terapêutica do ácido gástrico por uma base (antiácido).
Outros agentes agem de acordo com efeitos coligativos sem a necessidade de uma estrutura química
específica. Por exemplo, alguns compostos relativamente benignos, como o manitol, podem ser
administrados em quantidades suficientes para aumentar a osmolaridade de vários líquidos corporais e
assim causar alterações apropriadas na distribuição da água.
Acetilcolina: atua no endotélio estimulando a liberação de NO. O óxido nítrico atua no GMPc que age
no músculo liso produzindo vasodilatação.
Exemplos:
dramin (anticinetótico) – bloqueia a ligação da histamina aos receptores do tronco cerebral que
dariam origem ao enjôo. Bloqueia também os receptores do centro do sono-vigília e os da fome
(ausência de enjôo [cinetose → disfunção labiríntica], sono [efeitos colaterais] e fome.
plasil (metacoplamida) – é anti-hemético e bloqueia receptores de dopamina
fluoxetina – é antidepressivo e anorexígeno
Os receptores são específicos para uma substância ou para um grupo de substâncias (apresentam
características principais semelhantes). Os receptores de membrana são os mais importantes e,
normalmente, estão ligados a outras moléculas, formando o sistema de transdução de sinal. O fármaco
altera a conformação espacial do receptor e, esta mudança, reflete-se numa transformação da molécula
ligada a ele (efeito cascata). Outra função do receptor é a de ampliar a estimulação de um fármaco.
Moléculas lipídicas passam facilmente pela membrana e atuam no citoplasma e no núcleo (ex.:
estrógeno, testosterona). A exceção são os hormônios tireoidianos (são proteínas com características
lipídicas → atuam no núcleo celular).
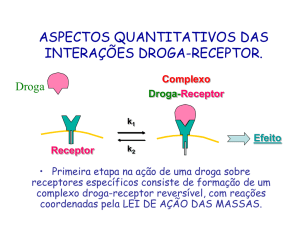
Os fármacos, em geral, podem fazer duas coisas com os receptores: (1) se ligar a eles e (2)
possivelmente alterar seu comportamento com relação ao sistema de célula hospedeira. A primeira função
é governada pela propriedade química de afinidade. A segunda é governada por uma quantidade
denominada eficácia.
Existem vários mecanismo para explicar os efeitos dos fármacos. O principal deles é o de Clarck que
criou uma fórmula que leva em conta a afinidade fármaco-receptor e a sua capacidade de mudar a
conformação do receptor (atividade intrínseca). Os medicamentos, para serem efetivos, precisavam
apresentar duas características principais: capacidade de ligação ao receptor e capacidade de alternar a
conformação do receptor (ativação/inativação).
1
EFEITO = Kaf x α
Kaf: grau de afinidade (capacidade de ligação)
α = atividade intrínseca (0 a 1)
As alterações podem ocorrer através de:
enzimas - inibição competitiva (os fármacos se ligam à enzimas específicas e diminuem sua
atividade). Ex.: epocler (ativação das enzimas hepáticas) / ácido acetilsalicílico (se liga à
ciclooxigenase, impedindo a transformação do ácido aracdônico em prostraglandina e evitando a
vasodilatação e a dor) / IECA (a ECA transforma angiotensina I em Angiotensina II, que é
vasocontritora)
bombas – proteínas de membrana que, através de energia, fazem transporte de moléculas. Ex.:
omeprazol (inibidor de bomba de prótons → diminui a secreção de H+)
canais – estruturas que permitem, através do diâmetro de seu poro, a passagem de moléculas pela
membrana plasmática. Ex.: benzodiazepínicos [LORAX®] (abrem os canais de Cl-) / nifedipina
(anti-hipertensivos → fecham os canais de Ca+, atuam principalmente no músculo liso)
[ADALAT®]
trocadores – estruturas protéicas que trocam uma substância pela outra. Ex.: hidroclorotizida
(diurético, trocador Na/Cl)
alteração de propriedades físico-químicas. Ex.: antiácidos (hidróxido de magnésio) / manitol
(diurético osmótico muito potente facilmente filtrado pelos glomérulos, usado em casos de edema
cerebral, não é reabsorvido nem é secretado)
AGONISTA = toda substância que, quando se liga a um receptor, produz uma mudança conformacional
nele e, por conseqüência, um efeito cascata. Podem ser tanto substâncias endógenas (ex.: serotonina,
adrenalina, dopamina) quanto exógenas. Os fármacos que se ligam aos receptores fisiológicos e mimetizam
os efeitos reguladores dos componentes endógenos de sinalização são denominados agonistas.
ANTAGONISTA = toda substância que, quando ligada a um receptor, impede que um determinado
efeito ocorra. São os fármacos que se ligam aos receptores sem efeito regulador, porém sua ligação
bloqueia a ligação dos agonistas endógenos. Podem também exercer efeitos úteis pela inibição da ação de
um agonista (ex.: pela competição com os agonistas pelos locais de ligação). Existem alguns tipos:
1. competitivos – ligam-se por um breve período de tempo. São os mais comuns na indústria
farmacêutica. Possuem certa semelhança molecular com o seu agonista (“imitação”). É uma
molécula que tem alto Kaf e zero de α (não gera nenhum efeito ao receptor e se liga fortemente a
ele). Se seu α não é igual a zero, ele passa a ser chamado de antagonista/agonista parcial.
Exemplos de antagonistas competitivos com α = 0:
antagonistas de receptores muscarínicos – atropina (colírio usado para dilatar a pupila). É
utilizada também para reverter o bloqueio átrio-ventricular, nos casos de intoxicação por
drogas que estimulam o parassimpático (síndrome digitálica). Ex.: organofosforato
(agrotóxico). O tratamento é a dosagem de atropina associada à prolidoxina (atropina →
liga-se aos receptores muscarínicos / prolidoxina → reestimulam a atividade da
acetilcolinesterase)
atroveram – bloqueia a atividade da musculatura lisa (cólica)
antialérgicos – competem com a histamina (RH1)
antagonistas dos receptores AT1 de angiotensina (BRA = bloqueadores dos receptores de
angiotensina) – evitam a vasoconstrição são anti-hipertensivos. Ex.: losartan, candesartan,
telmisartan
bloqueadores/inibidores competitivos da renina – alisquiren
antagonistas dos receptores RH2 de histamina – são anti-secretórios gástricos (cimetidina,
rametidina e foditidina)
2
2. fisiológicos – atuam no seu receptor próprio, e o seu produto de ligação atuará como antagonista
em outro receptor (efeitos opostos → 2 substâncias e 2 receptores).
Exemplos:
adrenalina x acetilcolina - midríase/miose
histamina x adrenalina (usada em caso de choque anafilático) – histamina causa diminuição da
PA e broncoconstrição, enquanto que a adrenalina causa aumento da PA (α1) e
broncodilatação (β2)
Em caso de choque anafilático, o pacote de emergência é: aplicação de (1) adrenalina, (2) corticóide e
(3) anti-histamínico. Pode-se optar por não utilizar os 3 passos (usar medicamento até a melhora do
paciente).
3. competitivos irreversíveis – ligam-se por tempo muito prolongado (não são mais utilizados pois
produzem muitos efeitos colaterais/tóxicos). A molécula se liga de maneira extremamente forte ao
receptor (não traz benefícios).
Exemplos: chumbo (se acumula principalmente no osso e é altamente tóxico).
Em caso de intoxicação, são utilizados quelantes (EDTA) para a reversão da situação.
ANTAGONISTA/AGONISTA PARCIAL = tem afinidade pelo receptor e tem α, porém, o seu α é
maior que zero e menor do que o do agonista verdadeiro (ex.: heroína → droga que produz euforia,
alegria, paz de espírito, além de dependência física / metadona → agonista parcial dos receptores
opióides; apresenta os mesmos efeitos da heroína, porém, não é tão potente).
Quando atingem níveis tóxicos, os opióides causam depressão cardio-respiratória. A natorfina
(NARCAN®) é agonista parcial com α muito pequeno (alguns a consideram um agonista competitivo).
Pode ser usada para reverter casos de intoxicação por opióides.
Doenças atribuídas ao mau-funcionamento dos receptores:
diabetes tipo II: comum em pessoas acima de 40 anos (pessoas obesas apresentam predisposição à
resistência à insulina). Neste caso, os receptores não respondem mais à insulina. Diminuindo-se o
peso corporal, ocorre uma melhora da resposta dos receptores.
Obs.: diabetes tipo I → doença auto-imune (destruição das ilhotas de Langherans), comum em jovens.
diabetes insipidus nefrogênica (doença autossômica recessiva): relaciona-se com o metabolismo da
água. Divide-se em dois tipos: central (retirada da glândula no caso de presença de tumores → [↓]
vasopressina e [↑] diurese) e nefrogênica (os receptores V2 da vasopressina, para a absorção de
água, localizados no rim, estão deformados devido a fatores genéticos ou estão ausentes → haverá
poliúria com grande perda de água). Os núcleos hipotalâmicos produzem ADH (vasopressina) e
ocitocina. A vasopressina atua na neurohipófise e daí para a eminência média (circulação
sanguínea). A diabetes insipidus central é tratável com a administração de vasopressina (DDAVP)
sintética instilada no nariz através de uma bombinha. A DDAVP aumenta a permeabilidade do
ducto coletor (poliúria). Já no caso de diabetes do tipo nefrogênico, o tratamento pode ser feito
através de: (1) hidroclorotiazida, que em diabéticos tem um efeito paradoxal (diminui a diurese),
mas apresenta muitos efeitos colaterais e (2) endometacina, um antiinflamatório não-esteroidal
que inibe a ciclooxigenase (responsável pela síntese de prostaglandinas que são diuréticos); seu
principal efeito colateral é a cefaléia.
MECANISMOS DE TRANSDUÇÃO DE SINAL
As ações reguladoras de um receptor podem ser exercidas diretamente em seus alvos celulares, as
proteínas efetoras, ou podem ser conduzidas por moléculas intermediárias de sinalização denominadas
transdutores. O receptor, seu alvo celular e quaisquer moléculas intermediárias são denominados sistema
receptor-efetor ou via de transdução de sinal.
3
Receptores acoplados à proteína G: uma grande família de receptores utiliza diferentes proteínas
reguladoras de ligação ao GTP, conhecidas como proteínas G, como transdutores para conduzir sinais
para suas proteínas efetoras. Os efetores regulados pela proteína G são as enzimas, como a adenililciclase e
a fosfolipase C, e os canais iônicos seletivos da membrana plasmática para Ca 2+ e K+.
Os receptores acoplados à proteína G atravessa, a membrana plasmática como um feixe de sete αhélices. Os agonistas se ligam a uma fenda na superfície extracelular do feixe. As proteínas G se ligam à
superfície citoplasmática dos receptores. Os receptores desta família respondem aos agonistas promovendo
a ligação de GTP à proteína G. O GTP ativa a proteína G e permite que esta, por sua vez, ative a proteína
efetora. A proteína G permanece ativa até hidrolisar o GTP ligado para GDP, que não ativa. As proteínas
G são compostas por uma subunidade α de ligação do GTP e por um dímero associado de subunidades β
e γ. A ativação da subunidade alfa pelo GTP permite que ela regule uma proteína efetora e conduza a
liberação das subunidades beta e gama, que podem regular seu próprio grupo de efetores.
Os sinais fisiológicos também são integrados no interior da célula como resultado e interações entre
vias de segundo mensageiros. Os segundo mensageiros mais conhecidos são AMPc, GMPc, Ca2+, fosfatos
de inositol, diacilglicerol e óxido nítrico.
1) mecanismo ligado ao AMPc – o AMPc é sintetizado pela adenilil ciclase sob o controle de muitos
receptores acoplados à proteína G (ATP → ADP + AMPc); é eliminado por uma associação de hidrólise,
catalisado por fosfodiesterases. O AMPc é responsável pela ativação dos receptores β2 da adrenalina
(broncodilatador). Os receptores são acoplados à proteína G (α+β+γ) e a ativam (parte α se descola do
grupo). Assim, a parte alfa ativa a adenilil ciclase) e esta converte o ATP em AMPc (liberação de ADP).
O AMPc atua sobre as PKs (proteínas quinase), que fosforilam outras proteínas, através das quais o efeito
pretendido é obtido. O AMPc dentro da célula é quebrado pela 5-fosfodiesterase em AMP (produto
inativo).
Exemplos: cafeína/teofilina são broncodilatadores. Eles inibem a enzima 5-fosfodiesterase e prolongam o
efeito pretendido pelo AMPc.
2) mecanismo vasodilatador (GMPc) – proteína G ativa a guanilatociclase que atua na transformação de
GTP em GMPc. O GMPc atua sobre as PKs provocando vasodilatação (GMPc → [5’-fosfodiesterase]
→ GMP). A acetilcolina atua sobre o endotélio provocando a liberação de NO (vasodilatador).
Exemplos: nitroglicerato é vasodilatador (liberação de NO).
3) mecanismo vasoconstritor – receptores α1 adrenérgicos (ex.: prazozim), receptores de AT2 (ex.:
losartan).
Temos receptores do s. parassimpático nos vasos, porém eles não são inervados por fibras parassimpáticos
(enigma: como a acetilcolina chega até os vasos???). Mecanismo: proteína G → atuação sobre a fosfolipase
C (PLC) → quebra do fosfatidil de inositol pela PLC em: IP3 (aumento de Ca2+, ativação do complexo
Ca-calmodulina que tem a mesma ação do ATP [contração muscular]) + DAG (diacilglicerol) →
provocam a ativação das PKc [proteína quinase C] que fosforilam proteínas → estas, por sua vez,
provocam vasoconstrição.
4) mecanismo dos compostos mercuriais – eficiente diurético (efeito colateral: necrose dos túbulos renais).
Estes compostos eram utilizados para casos de hipertensão arterial.
Aquaporinas:
A água passa pela membrana plasmática celular através de poros protéicos (aquaporina). As
aquaporinas são encontradas nos túbulos contorcidos distais, e em lugares com receptores V2 (AVP). Os
hormônios antidiuréticos são liberados devido ao aumento da PO (osmorreceptores presentes murcham
→ potencial de ação é deflagrado)
AVP → V2 → G
ATP → [G] → AMPc → [ativação] → PKA → AQP2 (aquaporina 2) → (entrada de água na
célula) → AQP3 → AQP1 (retorno da água ao sangue)
O AVP atua sobre o receptor V2 nas células do túbulo distal e do ducto coletor, que ativa a proteína
G. Esta quebra o ATP em AMPc, responsável pela ativação das PKAs que abrem os canais de AQP 2 para
4
a entrada de água na célula. A água passa então da célula ao interstício através da AQP 3 e entra nos vasos
através da AQP1.
O álcool inibe a liberação de vasopressina e, conseqüentemente, aumenta a micção (age sobre os
osmorreceptores, diminuindo sua sensibilidade e, conseqüentemente, a liberação de AVP nos sangue).
O mercúrio inibe a aquaporina I (presente nos túbulos renais). Ele se liga a AQP1 de maneira
irreversível (inibição não-competitiva). Com isso, a água não retorna ao sangue e seu acúmulo acaba por
lesionar de maneira irreversível as células renais (necrose tubular renal).
MECANISMOS DE DESSENSIBILISAÇÃO DE RECEPTORES (quando o remédio não faz mais
efeito)
A estimulação contínua das células pelos agonistas geralmente leva a um estado de dessensibilização
(estado refratário ou modulação negativa), de modo que o efeito que se segue à exposição contínua ou
subseqüente a uma mesma concentração de fármacos é reduzido (ex.: uso repetido de agonistas βadrenérgicos, como os broncodilatadores no tratamento da asma).
Doenças resultantes do mau-funcionamento dos receptores: síndrome de feminilização testicular (receptor
androgênico), miastenia gravis (receptores colinérgicos), diabetes melito resistente à insulina (receptores
insulínicos).
Exemplo: doença de Parkinson – ocorre devido ao desequilíbrio dos níveis de acetilcolina e de dopamina
(hiperexcitabilidade muscular). A dopamina é destruída. Logo, seu efeito inibidor é reduzido. O
tratamento é feito pela administração de L-dopa e carbi-dopa (inibe a quebra precoce de L-dopa no
intestino; normalmente, ela deve ser quebrada no SNC; caso isso ocorra, o indivíduo sente tontura) . De
início, a dose utilizada deve ser mínima, para que, no caso de tolerância farmacológica, exista uma faixa
extra de aplicação para um possível aumento na dosagem.
Todo medicamento tem potencial de causar tolerância.
Drogas de abuso - ex: heroína (opióide) – o grande problema é que, ao invés de causar o efeito esperado, a
droga é internalizada. Os principais mecanismos que ocorrem após sua administração são: (1)
internalização da droga, (2) fosforilação de receptores (passam a responder menos aos ligantes) e (3)
destruição dos receptores.
OVERDOSE = destruição dos receptores pela droga → [↑] da ingestão para fazer o mesmo efeito →
abstinência → normalização dos receptores → alta dose → alto efeito → morte
Diabetes insipidus nefrogênico (farmacológico – “pseudo DIN”): uso de medicamento a base de lítio, que
“entorta” os receptores V2 do organismo. Ex.: carbolítio (usado para o tratamento de transtorno bipolar).
Diabetes melitus (tipo II): medicamento hipoglicemiante (uso de insulina em alguns casos).
Alimento → jejuno (intestino com função endócrina) → GLP-I
pâncreas: produção de insulina
hipotálamo: saciedade
INCRETINOMIMÉTICOS = são medicamentos que imitam as incretinas (exenatide, sitagliptina,
vildagliptina, rimonabanto)
Exenatide - análogo de GLP-1.
Sempre que comemos carboidratos, estimulamos a liberação de GLP-I, que é uma incretina.
O GLP-1 é destruído por várias peptidases (ex.: DPP-4, principal enzima), produzidas no próprio
intestino, e transformado em metabólitos inativos.
Foram criados inibidores da DPP-4 (exenatide, sitagliptina, vildagliptina) que permitem que a GLP-I
permaneça mais tempo no corpo. Este mecanismo de ação é mais barato e apresenta menos efeitos
colaterais do que o uso de exenatide. Para o tratamento do diabetes, o mais importante não é a diminuição
da glicemia, mas sim a redução dos níveis de hemoglobina glicosilada.
Rimonabanto – uso em tratamentos de obesidade. Diminui a glicose e a hemoglobina glicosilada do
sangue sem que o peso seja alterado. Apresenta graves efeitos colaterais (alto índice de suicídios entre os
pacientes que o utilizam). É um antagonista de receptores canabinóides (Δ9-THC → maconha),
especificamente, o receptor EC1 (maconha gera fome). Estes receptores são responsáveis pela estimulação
do SNC (se são inibidos, o SNC passa para uma fase de depressão, levando ao suicídio).
5