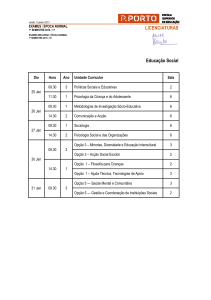
Farmacologia e Toxicologia I
1ª Frequência
1. Cite dois objectivos da Farmacologia.
A farmacologia é uma ciência experimental que estuda as propriedades das drogas ou
fármacos e a sua interacção com os seres vivos, entendendo-se como droga ou fármaco todo o
agente químico ou natural que tem acção sobre estes últimos.
São seus objectivos o conhecimento e compreensão:
Das relações que se estabelecem entre as doses e os efeitos benéficos ou indesejados,
produzidos por estes nos seres vivos;
Dos mecanismos pelos quais as drogas produzem os seus efeitos;
Os factores que podem afectar a resposta no paciente às drogas, devido à alteração na
sua disponibilidade (factores que a afectam) ou na sua eficácia no local de acção
correcto;
Das interacções que podem ocorrer quando mais do que uma droga é administrada ao
mesmo paciente (interacção medicamentosa);
Das razões de persistência de resíduos indesejáveis do medicamento nos produtos de
origem animal;
Os métodos e procedimentos para determinar os efeitos farmacológicos e
toxicológicos das drogas nos animais, e para determinar qualitativamente e
quantitativamente a presença de resíduos do medicamento nos produtos de origem
animal;
Conhecimento da sucessão de acontecimentos desde que a droga penetra no
organismo até ser totalmente destruída ou eliminada.
2. Qual o objectivo da Farmacocinética?
O objectivo da farmacocinética é estudar a descrição matemática das alterações temporais
nas concentração dos fármacos no interior do organismo, ou seja, estuda o que o organismo
faz com a droga após a sua administração. Tal acção está associada aos processos de absorção,
distribuição, metabolismo e de excreção das drogas. Trata também da relação destes
processos com a intensidade e duração dos efeitos característicos das drogas.
3. O que se entende por Farmacodinâmica?
Farmacodinâmica é o estudo dos efeitos bioquímicos e fisiológicos das drogas e seus
mecanismos de acção, ou seja, é o estudo do que os fármacos fazem ao organismo na ausência
de doença.
A farmacodinâmica estuda portanto a acção farmacológica de uma droga, isto é, a
modificação que esta produz nas funções do organismo, no sentido de aumentar ou diminuir
das mesmas. As drogas nunca criam funções novas, nem alteram as características do sistema
sobre o qual actuam, somente os modificam. O efeito farmacológico ou resposta a uma droga
é a manifestação da acção farmacológica.
4. O que se entende por fármaco?
Fármaco é toda a droga utilizada em Farmácia e que possui propriedades farmacológicas
ou, pelo menos, é de interesse médico. O conceito de droga inclui o de fármaco, ou seja, todo
o fármaco é um tipo especial de droga.
5. Quanto à origem como se classificam os fármacos?
Fármacos vegetais: componentes activos extraídos de raízes, caules, folhas, flores, sementes e
frutos das plantas.
Fármacos animais: preparados a partir de órgãos de animais, tais como pós de órgãos ou os
seus princípios activos, as hormonas.
Fármacos minerais: preparados por purificação dos minerais.
Fármacos sintéticos: obtidos por síntese total, a partir de substâncias simples e não têm
relação química com os produtos de origem natural.
Fármacos semi-sintéticos: obtidos por síntese parcial, isto é, por modificação químicas dos
produtos de origem natural. Sob o ponto de vista químico, os fármacos podem ser elementos
ou compostos, e estes podem ser de natureza orgânica ou inorgânica.
6. Definir o conceito de droga.
Droga é um ingrediente ou substância simples de natureza animal, vegetal ou mineral que
serve, em regra, à fabricação de outros produtos, estes de natureza medicinal ou química.
7. O que entende por especialidade farmacêutica de uso veterinário?
Consiste em todo o medicamento para uso veterinário, antecipadamente preparado e
apresentado sob uma denominação especial com um acondicionamento particular.
Medicamento é toda a substância ou composição que possua propriedades curativas ou
preventivas das doenças e dos seus sintomas, no Homem ou no animal , com vista a
estabelecer um diagnóstico médico ou a restaurar, corrigir ou modificar as suas funções
orgânicas.
8. O que são medicamentos etiotrópicos e organotrópicos?
Medicamentos etiotrópicos são medicamentos que se destinam a combater
essencialmente os agentes animados causadores de doenças, por exemplo pesticidas,
antibióticos, etc.
Medicamentos organotrópicos são medicamentos que actuam sobre os tecidos, órgãos e
suas funções no organismo.
9. Compare as cápsulas com as drageias quanto ao modo de obtenção e vantagens de
utilização.
As cápsulas são formas farmacêuticas sólidas, num recipiente normalmente semi-rígido,
que contém um pó ou um líquido no seu interior. A tecnologia dos comprimidos é
incompatível com certas fórmulas que não permitam processos de compressão ou que são
líquidos. A cápsula permite a sua correcta administração por via oral, garantindo que no local
onde se pretende que ocorra a absorção, esteja a correcta quantidade de fármaco.
Enquanto as drageias consistem num comprimido envolvido por um revestimento especial
que lhe confere algumas propriedades importante relativamente ao comprimido.
10. Compare os comprimidos com as drageias quanto ao modo de obtenção e vantagens
da sua utilização.
Os comprimidos são preparações farmacêuticas de consistência sólida e forma variada,
obtidas agregando-se por meio e pressão, várias substâncias medicamentosas secas, e
podendo ou não encontrar-se envolvidas por revestimentos especiais, tomando nestes casos a
designação de drageias.
As drageias têm a vantagem em relação aos comprimidos de se poder administrar o
princípio medicamentoso desprovido de qualquer aroma ou sabor desagradáveis; tornar
possível o emprego de substâncias agressivas para as mucosas evitando a acção emética que
possam provocar; permitir por meio de envolvimento adequado que os comprimidos resistam
à acção do suco gástrico; promover a mais fácil ???? do comprimido e permitir a eficaz
protecção e conservação dos princípios activos medicamentosos.
11. O que entende por depuração e tempo de meia-vida?
Depuração ou clearance de um fármaco é a quantidade total de plasma depurada de uma
determinada substância por unidade de tempo, ou seja, a quantidade de plasma que fica
liberto da substância com actividade farmacológica.
O tempo de meia-vida é o tempo necessário para que a concentração de um fármaco no
plasma diminua para metade da concentração inicial administrada.
12. O que entende por biodisponibilidade dos medicamentos?
Consiste na fracção da dose administrada que é realmente posta à disposição do
organismo para exercer a sua acção – relação entre a dose da droga e a intensidade de acção.
Uma biodisponibilidade <1 pode resultar de diversas razões, cuja natureza pode ser física,
química e/ou fisiológica, incluindo deficiente dissolução do fármaco, quando apresentado na
forma sólida. Nos líquidos intestinais pode ocorrer instabilidade ou inactivação da substância
medicamentosa, a passagem através da mucosa ser difícil e ocorrer metabolismo na parede
intestinal ou no fígado antes da penetração da droga na circulação sistémica.
Com I.V. a biodisponibilidade é sempre 1.
13. Cite factores que a possam influenciar.
Depende da dose, da via de administração, do grau de ionização, da lipossolubilidade, da
ligação com a proteína plasmática, da biotransformação e da excreção.
14. O que se entende por biotransformação dos fármacos e xenobióticos?
Os mecanismos de eliminação das drogas são a biotransformação (metabolismo) e a
excreção.
A biotransformação das drogas tem como principal objectivo a formação de metabolitos
com propriedades físicas e químicas favoráveis à sua excreção do organismo. Os produtos da
biotransformação são em geral menos lipossolúveis e de natureza polar, sendo que esta última
característica os torna apropriados para os processos de excreção mediada por
transportadores.
O padrão geral da biotransformação das drogas costuma ser bifásico. A fase inicial consiste
em reacções que podem ser classificadas como oxidativas, redutoras, hidrolíticas; ao passo
que as reacções de 2ª fase incluem reacções de síntese (conjugação).
As transformações da fase I geralmente põem a descoberto ou introduzem na molécula
grupos polares, que capacitam a conjugação do composto com substâncias endógenas. Os
conjugados formados são hidrossolúveis e invariavelmente inactivos em termos
farmacológicos. A via metabólica mais provável pode ser prevista com base no grupo funcional
presente no composto. A divergência entre os baixos níveis plasmáticos e os efeitos de uma
droga, sugerem que um ou mais dos seus metabolitos possam ter actividade farmacológica.
15. Quais as principais características dos fármacos que os tornam mais sujeitos a
biotransformação?
A lipossolubilidade, o grau de ionização (polaridade) e também os grupos constituintes de
um fármaco.
16. Cite os principais tipos de receptores de membrana.
Há 4 tipos de receptores celulares:
Proteínas nucleares e citoplasmáticas, que são activadas com factores de transcrição
de proteínas específicas.
Proteínas transmembranárias, cujos domínios intracitoplasmáticos só são activados ou
medeiam a activação de proteínas quando o ligando se une a uma porção exterior.
Proteínas transmembranares, que quando ligadas estimulam a síntese e/ou libertação
de 2º mensageiros.
Canais iónicos transmembranares que abrem ou fecham com a ligação do ligando, logo
iniciam ou inibem o fluxo de iões específicos para uma célula ou organito.
17. Qual a finalidade da utilização de adjuvantes na preparação de formas farmacêuticas?
Existem substâncias directamente compressíveis, mas a maioria necessita da presença de
um adjuvante que garanta que o estado de agregação se mantenha.
Os adjuvantes podem ser diluentes, absorventes, aglutinantes, desagregantes,
lubrificantes, molhantes, corantes, tampões, aromatizantes e edulcorantes.
18. Cite duas vias de administração e duas razões que nos levem a optarmos por uma em
relação à outra.
A escolha da via de administração depende muitas vezes do objectivo do fármaco, do seu
percurso no organismo e da região que deve atingir.
A via digestiva ou entérica (oral ou rectal) tem a vantagem de ser mais segura, económica
e menos perigosa no caso de sobredosagem. Tem os inconvenientes de por vezes não ser
adequada para algumas espécies animais ou idades; o facto de existirem substâncias irritantes,
destrutíveis pelo suco digestivo ou instáveis a pH gástrico. A absorção irregular depende da
velocidade de esvaziamento do estômago, motilidade de intestino, circulação sanguínea do
tubo digestivo, ausência da secreção devida a estados patológicos, presença de outros
medicamentos ou alimentos, e da natureza dos excipiente dos medicamentos. As substâncias
reabsorvidas entram pela veia porta-hepática, sendo levadas ao fígado, onde podem sofrer
uma degradação importante ou ser excretadas pela bílis.
As vias parentéricas permitem a administração de medicamentos que por outra via não
eram absorvidos, eram destruídos ou demasiado irritantes. Têm como inconvenientes a
possível irritação local, perigo de reacções gerais (particularmente em injecções via I.V.),
impossibilidade de eliminar o excesso de medicamento no caso de sobredosagem, necessidade
de empregar material estéril e custo mais elevado.
19. Como é que a via de administração pode afectar a actividade farmacológica de um
fármaco?
Pode afectar pois uma vez que os fármacos têm propriedades específicas que podem ser
alteradas por acção do organismo no qual irão actuar, a via de administração é um factor que
intervém na biodisponibilidade de um fármaco.
Deste modo verifica-se que através da via digestiva o fármaco aplicado pode interagir
com outros medicamentos também administrados ou até mesmo com os alimentos, pode
sofrer metabolização no fígado após absorção intestinal, sendo necessário maior dose a
aplicar. Esta via apresenta efeitos mais tardios relativamente a uma administração por via
parentérica, a qual permite uma acção do fármaco mais cedo, com doses mais reduzidas, dado
que o fármaco entra em circulação mais rapidamente.
20. Discuta a facilidade de absorção a nível da mucosa gástrica de duas substâncias
orgânicas com carácter ácido e diferentes valores de pKa.
A maioria das drogas consiste em bases ou ácidos orgânicos fracos, e existem em solução
tanto na forma ionizada como na não ionizada. Esta última costuma ser lipossolúvel e pode
difundir-se prontamente através da membrana celular para alcançar a mesma concentração
de equilíbrio do outro lado. Em contraste com isso, a porção ionizada costuma ser
praticamente excluída da difusão transmembranar, em virtude da sua baixa lipossolubilidade.
O grau de ionização de um electrólito orgânico depende do seu pKa e do pH o meio. O
valor de pKa é constante e a maioria dos agentes terapêuticos tem valores de pKa entre 3 e 11,
e surgem nas suas formas ionizadas e não ionizadas dentro dos parâmetros de pH fisiológico.
Pela forma indicada podemos perceber que quanto maior for o pKa, em pH ácido (estômago),
menor será a % de dissociação, logo mais facilmente é absorvido.
21. Qual é o papel do glutationo transferase na biotransformação dos fármacos?
O papel deste enzima na biotransformação dos fármacos consiste na sua intervenção em
reacções de conjugação – reacções de adição ou de substituição, entre o xenobiótico em causa
e uma molécula de glutationa (GSH), levando à formação de mercapturato, o qual pode ser
então facilmente excretado através da urina.
22. Caracterize o citocromo P450 quanto à composição química, estrutura e localização
celular.
O citocromo P450 é um sistema enzimático que pertence à família dos isoenzimas, com as
quais partilha o tipo de estrutura 3D. Trata-se de uma hemoproteína existente no sistema
enzimático microssómico dos hepatócitos, onde se comporta como enzima oxidante.
A capacidade dos enzimas microssómicos que metabolizam drogas e medeiam uma ampla
variedade de reacções oxidativas, pode ser atribuída ao mecanismo de hidroxilação.
O NADPH reduz o citocromo P450, o qual por sua vez, na sua forma reduzida, reage com o
O2 formando um oxigénio intermediário activo. A interacção entre este complexo e uma droga
lipossolúvel produz um substracto hidroxilado, o citocromo P450 e uma molécula de água.
23. Como é que a indução destes enzimas pode influenciar a actividade dos fármacos?
A indução enzimática é o processo pelo qual se aumenta a expressão das proteínas
enzimáticas que actuam no metabolismo das drogas, devido ao aumento da biossíntese dessas
proteínas. Assim, a metabolização dos fármacos será maior e mais rápida, pelo que a sua
actividade será menor e desaparecerá mais rapidamente. A indução enzimática pode ocorrer
por exposição a inúmeras substâncias e é uma característica muito importante dos isoenzimas
citocromo P450 e de alguns enzimas das reacções de conjugação da fase II.
24. Qual o papel do glucoronil transferase na biotransformação dos fármacos?
A glucoronidação é uma das principais vias de conjugação das substâncias endógenas e
dos xenobióticos. As reacções desta via passam pela transformação do uridina de difosfatoglucoronato, seguida de transferência do glucoronato para o xenobiótico e libertação de
uridina difosfato (UDP), catalizada por uma UDP-glucoronil transferase.
1ªetapa: glucose glucose-6-fosfato glucose-1-fosfato UDP-glucose UDP-glucoronato.
2ªetapa: UDP-glucoronato + xenobiótico
xenobiótico-glucoronato + UDP
25. Justifique a importância de se conhecer o mais claramente possível o destino dos
medicamentos e seus resíduos nos animais de carne.
É importante conhecer o destino dos medicamentos no organismo animal, uma vez que
cada fármaco tem locais, tecidos ou órgãos de selecção. Ao longo do tempo, as concentrações
presentes nas diversas regiões do organismo podem variar em função das propriedades do
fármaco (lipossolubilidade e grau de ionização), biotransformação e excreção.
A presença de resíduos em animais de carne pode levar ao transporte do próprio fármaco
ou seus metabolitos a outros animais, os quais poderão reagir a tal componente.
26. Quais são os três parâmetros mais importantes para o ajustamento da posologia?
O esquema posológico é estabelecido com o objectivo de manter a concentração
plasmática da droga dentro da amplitude terapêutica no decorrer do tratamento. A posologia
estabelece a quantidade da dose e o intervalo entre as doses sucessivas. A manutenção da
concentração plasmática da droga acima de certo limiar – concentração mínima eficaz, é um
requisito básico para a terapia anti-microbiana bem sucedida.
Os três factores mais importantes para o ajustamento da posologia são o volume de
distribuição, clearence e a biodisponibilidade.
O volume de distribuição fornece uma estimativa de extensão da distribuição da droga,
sendo o volume aparente o qual se reporta a totalidade da dose do medicamento para que a
sua concentração plasmática seja a que é medida no sangue. Assim, para que se mantenha
uma concentração plasmática desejada tem de se ajustar a dose ao volume de distribuição, ou
seja, se o volume de distribuição aumentar, também a dose tem de aumentar: VD=dose/Cp.
A biodisponibilidade é a velocidade e extensão em que a droga administrada, em
determinada forma farmacêutica, penetra intacta na circulação sistémica, o que vai afectar o
efeito da droga.
O conhecimento da depuração renal de uma droga é fundamental para determinar a
dosagem – dose por unidade de tempo, necessária para que ocorra uma certa concentração
média de estado estacionário. Se o clearence de uma droga for muito elevado, em de se
administrar mais vezes a mesma dose da droga para se manter a concentração plasmática
estável.
27. Descrever o papel dos segundos mensageiros como receptores fisiológicos.
Os 2º mensageiros são mediadores intracelulares que funcionam como amplificadores do
sinal transmitido por união do ligando com o receptor. Por cada receptor estimulado liberta-se
grande quantidade de 2ºmensageiros, ou seja, é necessário um ligando para produzir uma
resposta eficaz.
2ª Frequência
1. Caracterize o mecanismo de acção, toxicidade e principais vias de aplicação dos
piretróides e rotenona.
São ambos ectoparasiticidas.
Os piretróides são compostos de síntese que substituem as piretrinas (de origem vegetal).
Exercem a sua acção por modelação cinética da abertura dos canais de sódio dos nervos. Esta
acção resulta em repetidas descargas ou desporalização da membrana e consequente morte
do parasita. Os insecticidas piretróides suprimem os receptores GABA, glutamato e os canais
de cálcio activados por voltagem. Estão entre os ectoparasiticidas de menor toxicidade, os
quais se manifestam de forma análoga ao que sucede aos ectoparasitas, surgindo como sinais
clínicos de toxicidade desordens no sistema nervoso e muscular. Em cães e gatos
moderadamente afectados surge hipersalivação, vómito, diarreia e tremura moderada.
Quando severamente afectados surge hipertermia ou hipotermia, desorientação e apoplexia.
A rotenona tem origem vegetal e exerce o seu efeito inibindo especificamente o sistema
respiratório dos ectoparasitas alvo, a nível da oxidação do NADH, e subsequente obtenção de
energia (ATP). É mais tóxica nos mamíferos do que os piretróides, mas ainda é considerada
segura para uso no cão e gato. É tóxica para suínos, peixes e caprinos, pelo que não deve ser
usada. É o componente activo de pós, sprays e banhos para controlo de pulgas, carraças e
ácaros em cães e gatos.
2. O tartarato de pirantel é efectivo contra um largo espectro de parasitas de ruminantes
e suínos. A que grupos fármaco-terapêutico e químico pertence este composto? E
quais as contra-indicações deste produto?
É um nematocida semelhante à nicotina, com acção a nível da paralização do verme,
causando o bloqueio neuromuscular-desporalizante, semelhante ao levamisol. A sua absorção
intestinal é excelente após administração oral. É rapidamente metabolizado e excretado,
principalmente nas fezes mas também na urina.
É eficaz contra Strongylus, Ascaris e Oxiuros. Tem baixa toxicidade, embora possam
ocorrer vómitos. Como é semelhante ao levamisol não podem ser administrados em conjunto.
3. A ivermectina é um macrólido com larga aplicação terapêutica em veterinária. Qual o
interesse farmacológico da sua utilização? Em que espécies animais se emprega mais
correctamente?
Trata-se de um anti-helmíntico que actua por inibição da motilidade. Aumenta a libertação
de GABA nas sinapses do SN, o que hiperpolariza o potencial de repouso normal das células
pós-sinápticas, tornando mais difícil a neurotransmissão de estímulos para os músculos, pelo
que estes não se contraem. Os vermes ficam paralizados e consequentemente são expelidos.
Tem actividade contra céstodos e tremátodos, sendo necessária uma quantidade
extremamente pequena para actividade anti-helmíntica, tanto por via oral como parentérica.
Tem uma ampla faixa de eficácia em bovinos e ovinos, tanto para endoparaditas como
alguns ectoparasitas, sobre os quais diminuem o potencial reprodutivo, pois interrompem a
alimentação, fecundação e produção de ovos, apesar de não provocar morte ou destacamento
imediato.
Também muito utilizado em suínos e equinos. Em cães é útil na prevenção de Dirofilariose
e no tratamento de sarnas.
Tem grande margem de segurança em todas as espécies.
4. O que se entende por antissépticos? Dar três exemplos de produtos utilizados para
este fim e comparar em termos de eficácia e toxicidade.
Os antissépticos são germicidas que se aplicam em tecidos vivos. São agentes químicos
que diminuem a população microbiana da pele e outros tecidos vivos. Na maior parte dos
casos actuam de forma não-específica, destruindo as membranas celulares ou enzimas pelo
que devem ser cautelosamente utilizados, para não causar danos nos tecidos do animal.
Os derivados de iodo, como o soluto alcoólico de iodo, combinações de iodo com a
polivinilpirrolidona, são usados como antissépticos da pele e feridas, em ginecologia, na
prevenção contra mamites e em cirurgia.
A água oxigenada ou peróxido de hidrogénio, é um agente oxidante muito eficaz na
antissepsia de feridas sujas de terra, particularmente de microorganismos anaeróbios, sendo
também um hemostático potente. Não danifica instrumentos metálicos nem material de
penso. É inactivada pelo permanganato de potássio.
O etanol existe como álcool absoluto, 95° e 70°, sendo este último o que tem maior poder
antisséptico externo, porque a desnaturação de proteínas microbianas necessita da presença
de água. Muito eficaz em uso externo, sendo tóxico quando ingerido.
5. Em que se distinguem os antissépticos dos desinfectantes?
Os antissépticos são substâncias químicas germicidas que se aplicam nos tecidos vivos,
enquanto que os desinfectantes são aplicados nos objectos inanimados.
Os primeiros podem-se tornar inactivos em objectos e os segundos podem ser perigosos
para os tecidos vivos. Por vezes o mesmo princípio activo pode apresentar-se em diferentes
formulações consoante o fim a que se destina.
6. Caracterize o mecanismo de acção, as características cinéticas e as principais
aplicações veterinárias das sulfonamidas.
As sulfonamidas são activas sobre microorganismos que sintetizam o seu próprio ácido
fólico, pois inibem a síntese deste composto, interferindo com a produção normal de RNA,
síntese de proteínas e mecanismos de replicação microbiana. Exercem esta acção de inibição
do metabolismo intermediário, interferindo com a produção de ácido fólico, enquanto que as
diaminopirimidinas (trimetropim), interferem mais tarde nesta via por bloqueio da produção
de ácido tetrahidrofólico. Por isso quando utilizadas isoladamente as sulfonamidas são
bacterioestáticas.
Têm um largo espectro, actuam sobre Gram (+) e (-) e muitos protozoários. São usadas em
infecções do SNC, tracto respiratório, gastrointestinal e urinário.
Quanto à susceptibilidade a estes compostos, os microorganismos são considerados em:
Muito susceptíveis – Actinomycetes spp, Bacillus spp, Brucella spp, Streptococcus spp,
Chlamydia spp, Cryptosporidium spp, várias coccídeas.
Moderadamente susceptíveis – Gram(+) aeróbios como estafilococus e enterococus;
Gram(-) aeróbios como Enterobacter spp, E.coli, Klebsiella spp, Proteus spp,
Actinobacillus spp, Hemophilus spp, Pasteurella spp, Pseudomonas spp, entre outros.
Resistentes – Mycobacterium spp, Mycoplasma spp, Rickettsia spp, P.aeruginosa,
Espiroquetas.
São geralmente absorvidos rapidamente no tracto gastrointestinal quando administrados
por via oral. A administração I.M. ou S.C. requer que a solução seja previamente tamponada,
para evitar reacções perivasculares e de sensibilização devido às características alcalinas
destes compostos. Também são bem absorvidas aquando de administração I.P.
Quanto à rapidez de acção podem ser de curta duração, acção intermédia ou de longa
duração, de acordo com o seu perfil de concentração plasmática em função do tempo.
Distribuem-se largamente por todos os tecidos. De um modo geral ligam-se às proteínas
do plasma, aumentando o tempo de meia-vida. Por serem ácidos fracos não atingem grandes
concentrações no leite, atingindo-o por difusão passiva, no entanto os valores não são
suficientes para que possam ser utilizadas em mastites.
A principal via de metabolização é a acetilação, a qual ocorre no fígado e pulmões, sendo
evidente em bovinos, caprinos e suínos, mas ausente nos carnívoros. No entanto a conjugação
com o ácido glucorónico e a hidroxilação aromática são também vias seguidas na
biotransformação destes fármacos.
São excretadas principalmente pelos rins, quer sob a forma de produtos inalterados ou
dos seus metabolitos, pela via de filtração glomerular, transporte activo ou absorção passiva
da droga não ionizada do fluido tubular distal. Podem ser também excretadas pelas lágrimas,
fezes, bílis, suor ou leite. O pH urinário baixo favorece a reabsorção tubular, prolongando o
tempo de meia-vida destes compostos.
A nível de toxicidade podem surgir situações de cristalúria renal, ceratoconjuntivite seca
nos cães, anemia, trombocitopénia e carcinogenese.
7. Caracterize quimicamente as penicilinas e cefalosporinas e descreva as principais
razões para desenvolvimento de resistências a estas substâncias. Descrever os
mecanismo de acção destes antibióticos.
Quer as penicilinas quer as cefalosporinas são antibióticos β-lactâmicos, tal como os
inibidores da β-lactamase (ex:ácido clavulânico). O anel β-lactâmico é a estrutura comum a
estes antibióticos. O grupo inclui ainda antibióticos similares cuja estrutura não inclui a fusão
dos dois anéis, mas cuja actividade é semelhante ao nível da β-lactamase.
Estes antibióticos inibem a síntese da parede celular bacteriana, promovendo a destruição
da mesma. Estes compostos ligam-se a diversos enzimas envolvidos nas etapas finais da
biossíntese da parede celular bacteriana, por apresentarem analogia estrutural com a ponte
D-alanina
D-alanina, que faz as ligações cruzadas das cadeias de peptidoglicano que
compõem a referida parede celular – substituem as ligações normais , enfraquecendo a
estrutura da parede celular. As diferentes afinidades destes antibióticos para se ligarem às
referidas proteínas, explica as diferenças observadas nos seus espectros de actividade que não
são causados pela presença ou ausência de β-lactamase.
Existem 3 factores independentes que determinam a susceptibilidade a estes antibióticos:
A produção de β-lactamase;
A permeabilidade da parede celular;
A sensibilidade das proteínas a que se ligam estes antibióticos.
A produção de β-lactamases, que são enzimas que inactivam o antibiótico por hidrólise do
anel β-lactâmico, é o principal mecanismo de defesa das bactérias a estes antibióticos.
Diferentes bactérias produzem β-lactamases que diferem nas propriedades físicas,
químicas e funcionais. Algumas β-lactamases são específicas para as penicilinas (penicilases),
outras específicas de cefalosporinas (cefalosporinases), e outras ainda são activas contra estes
dois tipos de antibióticos.
Contra estes dois tipos de antibióticos conhecem-se dois mecanismos de produção de βlactamases: cromossomal (desenvolve-se face a um grupo β-lactâmico) e plasmídio (que
podem ser transferidas entre bactérias, aumentando o nº das que são resistentes a um
determinado antibiótico.) Nas Gram(+) são de origem cromossomal (cefalosporinases),
enquanto que nas Gram (-) são também de origem cromossomal (cefalosporinases) ou
mediadas por plasmídeos (antibióticos β-lactâmicos de largo espectro).
Penicilinas
Existem 4 grupos de penicilinas:
I.
Penicilinas naturais - penicilina G e penicilina V.
II.
Aminopenicilinas – amoxilina e ampicilina.
III.
Penicilinas resistentes à penicilase – cloxacilina, dicloxacilina e oxacilina.
IV.
Penicilina de largo espectro – carbenicilina.
A via mais comum é a I.M. A penicilina é degradada pelo suco gástrico, pelo que a
administração oral requer doses elevadas. A difusão da penicilina para os tecidos e fluidos
ocorre quando a concentração do fármaco não ligado no plasma é maior que a dos tecidos e
fluidos. Obtêm-se elevadas concentrações nos rins, fígado e pulmões. Não penetram no SNC
em grande extensão. Difundem-se através da placenta por circulação fetal.
A metabolização não se dá em grande escala, mas passa pela hidrólise do anel β-lactâmico
e os seus metabolitos são inactivos. São excretados pela urina juntamente com o fármaco
intacto, por excreção tubular. Também é excretada pelo leite.
São seguras e com poucos efeitos adversos.
Cefalosporinas
Consideram-se 4 grupos de cefalosporinas:
I. C. de 1ª geração – cefadroxil, cefazolina, cefalexina, cefalotina, cefapirina, cefradina. Têm
um espectro semelhante ao das aminopenicilinas, mas são mais activas contra
Staphylococcus ssp.
II. C. de 2ª geração – cefaclor, cefamandol, cefmetazole, cefonicid, ceforamida, cefotetan,
cefoxitina, cefprozil, cefuroxima e loracarbef. Têm actividade semelhante às anteriores mas
são mais activas contra Gram(-).
III. C. de 3ª geração – cefixima, cefoperazona, cefotaxima, ceftazidima, ceftiofur, ceftizoxima,
ceftriaxone e moxalactam. Têm menor actividade contra Staphylococcus spp, mas são ainda
mais activas contra Gram(-). Apresentam alguma actividade contra bactérias anaeróbias.
IV. C. de 4ª geração – cefepime (maxipime) e cefpirome. Cefepime é mais resistente a
algumas β-lactamases como as produzidas por Enterobacter, e penetra bem no fluido
raquidiano.
As cefalosporinas são absorvidas rapidamente após administração I.V. ou S.C. A
biodisponibilidade varia com o fármaco e com a espécie. A afinidade para as proteínas
plasmáticas também é variável, assim como a taxa de eliminação renal, dando lugar a
diferentes perfis farmacocinéticos das diferentes cefalosporinas.
Estes compostos são amplamente distribuídos pelo corpo, obtendo-se elevadas
concentrações no sangue, urina, bílis, fluidos pleural, pericardial e sinovial, e no córtex ósseo.
A maior parte destes antibióticos não passa a barreira cefalo-raquidiana. São pouco
penetrantes no tecido prostático e nos humores vítreo e aquoso.
A maior parte não é metabolizada, mas algumas são desacetiladas.
A principal via de excreção é a filtração renal, mas também pode sofrer eliminação biliar.
Podem provocar principalmente reacções locais: dor, abcesso estéril após administração
I.M. e tromboflebite após administração I.V. A administração oral pode originar vómitos e
diarreias.
8. Caracterize o mecanismo de acção, as características cinéticas e as principais
aplicações veterinárias das tetraciclinas. Discuta quais os factores a ter em conta na
escolha da via de administração das tetraciclinas.
As tetraciclinas constituem um grupo de antibióticos cujas moléculas apresentam um
conjunto de 4 anéis com carácter anfotérico e que se distinguem entre si pelos substituintes
químicos específicos que se encontram em diferentes posições nos anéis.
São bacteriostáticos de largo espectro nas concentrações terapêuticas. Actuam sobre as
bactérias ligando-se à subunidade ribossomal 30S, afectando a ligação aminoacil-RNAt ao
RNAm e, consequentemente, interferindo com a síntese proteica bacteriana no crescimento
ou multiplicação dos microorganismos. Têm muito menor afinidade para os ribossomas dos
mamíferos, mas ocorre sempre alguma inibição quando se administra estes antibióticos, o que
se traduz por um efeito catabólico.
Podem ser administradas via I.V. e I.M. como a oxitetraciclina, mas a via oral é mais preferida
para minimizar os efeitos adversos deste composto, sendo absorvidas no tracto
gastrointestinal. Reagem facilmente com catiões polivalentes dando complexos (quelatos), o
que diminui a sua absorção. Por isso há que ter em atenção a possibilidade de inactivação
destes compostos quando são administrados simultaneamente com o leite e seus produtos,
alimentos ricos em catiões polivalentes (Ca, Mg, Fe e Al). Nas doses terapêuticas não devem
ser administradas a ruminantes ou equinos devido a severos desarranjos na flora intestinal que
podem originar. Contudo podem ser adicionados em concentrações sub-terapêuticas a rações
para vários fins.
Após absorção ligam-se a proteínas do plasma em graus variáveis, dependendo da espécie
considerada. Distribuem-se amplamente através da maior parte dos tecidos após
administração oral ou I.V., acumulando-se no fígado e rins. A distribuição no corpo depende da
solubilidade dos lípidos.
As tetraciclinas não são metabolizadas em grande extensão: cerca de 60% excretada na
urina por filtração glomerular e os restantes 40% são excretados pelas fezes. A resistência a
estes antibióticos ocorre nas bactérias devido a R-plasmídeos.
Como antibióticos de largo espectro as tetraciclinas inibem o crescimento de uma larga
variedade de bactérias, protozoários e organismos intracelulares como micoplasma, riquétsias
e clamídeas. As diferenças observadas no espectro de actividade in vivo das tetraciclinas deriva
intimamente a sua absorção., distribuição, metabolismo, excreção e a concentração do
antibiótico dentro da célula. Quanto maior é a concentração que se obtém no interior da
célula, maior é a actividade antimicrobiana e melhor é a sua eficácia clínica.
O efeito lateral mais comum é a irritação no estômago e duodeno, mas em geral são
consideradas seguras.
9. Quais os usos veterinários de escolha dos aminoglicosidos antibióticos?
Estes antibióticos ligam-se irreversivelmente à subunidade ribossomal 30S dos
microorganismos, mas o mecanismo exacto ainda não está bem clarificado. A passagem
através da membrana celular é um processo que requer oxigénio e é acompanhado por
difusão passiva, pelo que as bactérias aneróbias são resistentes a estas substâncias. Este
transporte está ligado a um sistema transportador de electrões, que carrega negativamente o
citoplasma relativamente ao periplasma e ao meio exterior. Os aminoglicosidos carregados
positivamente ligam-se electrostaticamente ao citoplasma bacteriano. Alguns catiões são
inibidores deste sistema de transporte. São capazes de se difundir através de canais aquosos
formado por proteínas de membrana exterior das bactérias Gram(-).
São utilizados em tratamento de infecções mais sérias causadas por Gram(-).
Não são absorvidos no tracto gastrointestinal por serem altamente polares e de natureza
catiónica. Não são no intestino e são rapidamente eliminados, pelo que têm de ser
administrados via parental para se obterem concentrações plasmáticas com actividade
terapêutica. A sua absorção é praticamente completa por via I.M. ou S.C. e é extremamente
rápida se forem instalados nas cavidades com superfícies serosas por via I.V.
A penetração através das barreiras é muito limitada. Liga-se pouco às proteínas do plasma
(20%) e aos eritrócitos (10%).
São eliminados exclusivamente por filtração glomerular renal sem metabolização, por
todas as espécies animais estudadas.
Apresenta toxicidade e nefrotoxicidade em equinos, ruminantes, cães e gatos.
10. Caracterizar o grupo fármaco-terapêutico a que pertencem a tilosina e a eritromicina e
comparar os mecanismos de acção.
São ambos macrólidos. Estes antibióticos inibem a síntese proteica bacteriana por ligação
à sub-unidade 50S ribossomal, prevenindo a translocação de aminoácidos para a cadeia
peptídica em crescimento.
São bacteriostáticos. A eritromicina impede a ligação do peptidil-RNA, enquanto que a
tilosina impede a ligação do peptidil e aminoacil-RNA.
São amplamente distribuídos por todos os tecidos, excepto o SNC. A eritromicina é
metabolizada no fígado e excretada na urina, enquanto que a tilosina é excretada inalterada
na urina.
A actividade microbiana primária surge contra os Gram(-) aeróbios e anaeróbios e
micoplasmas. Têm poucos efeitos adversos.