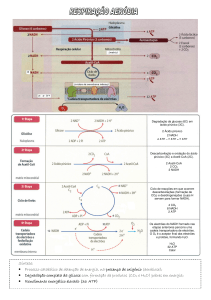
METABOLISMO LIPÍDICO
O metabolismo lipídico resumido:
triacilgliceróis
lípidos de membrana
ácidos gordos
β-oxidação
NADPH
FADH2
ATP
NADH
síntese de ácidos gordos
colesterol
acetil-CoA
ciclo de
Krebs
corpos cetónicos
fosforilação oxidativa
NADH
ATP
FADH2
GTP
Catabolismo de lípidos
1. Hidrólise de triacilgliceróis
2. Activação dos ácidos gordos
(membrana mitocondrial externa)
Transporte dos ácidos gordos activados (acis-CoA)
para o interior da mitocôndria
carnitina
carnitina
Acil-CoA
Acil-CoA
CoA
Acil-carnitina
Citosol
CoA
Acil-carnitina
Mitocôndria
membrana mitocondrial
interna
Resultados 1 - 10 de cerca de 448.000 para
carnitina. (0,15 segundos)
…e cerca de 3.880.000 para carnitine!!!
Vendida como “queimador de gorduras”
…mas não há provas científicas que o apoiem
… há até blogs dedicados á carnitina!!!!
http://grupo11a-lcarnitina.blogspot.com/
Há uma doença de origem genética (autossómica recessiva), em que os
transportadores de carnitina para as células não funcionam adequadamente →
o doente não tem capacidade de utilizar os ácidos gordos (e portanto os
lípidos) como fonte de energia.
Cromossoma 5
Gene SLC22A5
3. β-oxidação dos
ácidos gordos
β-oxidação do ácido palmítico
(16 átomos de C)
“contabilidade” da β-oxidação do ácido palmítico
(16 átomos de C):
ganho
β-oxidação 7 NADH
7 FADH2
ciclo de Krebs
(acetil CoA)
(21ATP)
(14ATP)
gasto
1 ATP (activação)
8x3NADH (72ATP)
8x1FADH2 (16ATP)
8x1GTP
(8ATP)
131ATP
Global: 130 ATP
Glucose (6C)
Ácido capróico (6C)
2 ATP (glicólise)
2 NADH (glicólise)
2 NADH (piruvato→Acetil-CoA)
6 NADH
ciclo de Krebs
2 FADH2
2 GTP
36-38 ATP/molécula
β-oxidação 2 NADH
2 FADH2
6 ATP
4 ATP
ciclo de 9 NADH
27 ATP
Krebs 3 FADH2
6 ATP
3 GTP
3 ATP
46 ATP
-1 ATP (activação)
45 ATP/molécula
M.M.glucose=180.16g/mole
1g glucose ⇔ 5.55 moles
M.M.ác.capróico=116.16g/mole
1g ác. capróico ⇔ 8.61 moles
210.19 mmoles ATP/g
387.45 mmoles ATP/g
387.45
= 1.84!!!
210.9
∴Lípidos → modo mais
eficiente de armazenar
energia
-se houver excesso de Acetil-CoA
(lípidos como fonte principal de energia, p.ex. em jejum prolongado ou
diabetes):
no fígado (matriz mitocondial):
corpos cetónicos
utilização de
corpos
cetónicos
como fonte
de energia
músculo cardíaco e esquelético
cérebro, em jejum prolongado
Casos especiais na β-oxidação:
- ácidos gordos com nº ímpar de átomos de Carbono:
n CH3-C-S-CoA
+
CH-CH2-C-S-CoA
O
propionil-CoA
O
acetil-CoA
succinil-CoA
ciclo de Krebs
- ácidos gordos insaturados (igual, excepto):
H H
=
O
CH3(CH2)7 C=C CH2(CH2)6C S CoA
oleoil-CoA
3 ciclos da β-oxidação
acetil-CoA 3 CH3-C-S-CoA
O
O
=
H H
CH3(CH2)7 C=C CH2C S CoA
∆3-cis-dodecenoil-CoA
H
enoil-CoA isomerase
=
O
CH3(CH2)7CH2 C=C CH2C S CoA
∆3-trans-dodecenoil-CoAH
H2O
O
=
OH
CH3(CH2)7CH2 C CH2C S CoA
H
intermediário da β-oxidação!!!
continuação
da β -oxidação
6 CH3-C-S-CoA
O
- degradação de lípidos sujeita a sinais hormonais:
interacção
hormona-receptor:
adrenalina,
glicagina
membrana
plasmática
Adipócito
adenilato
ciclase
Adipócito
Proteína cinase
(inactiva)
Proteína cinase
(activa)
Triacilglicerol
lipase (inactiva)
Triacilglicerol
Ác. gordo
Diacilglicerol
Triacilglicerol
do
gor
lipase (activa)
Ác .
o
Monoacilglicerol rd
o
.g
Ác
Glicerol
- Ácidos gordos livres: exportados para a corrente sanguínea, complexados com albumina
- nos outros tecidos (fígado, músculo), β−oxidação estimulada pelo aumento da concentração de
ácidos gordos
Síntese de ácidos gordos
(fígado, tecido adiposo e glândula mamária)
* Processo eminentemente CITOSSÓLICO ( até ácido palmítico)
. Alongamento da cadeia
MIT (AG cadeia média e curta)
e RE
. Dessaturação
RE
* Precursor
acetil-CoA (formado sobretudo na mitocôndria)
* Processo redutor
Passagem
para o citossol
NADPH = agente redutor
Fontes
de NADPH
. Via das pentoses
. Acção do enzima málico
. Acção do isocitrato desidrogenase extramitocondrial
• a primeira reacção, de carboxilação do acetil-CoA, é
catalisada pela acetil CoA carboxilase, que tem como
cofactor a biotina:
Enzima-biotina
HCO3 + ATP
1
ADP + Pi
Enzima-biotina-CO2
O
ll
CH3-C-SCoA
acetil-CoA
2
Enzima-biotina
O
-
ll
O2C-CH2-C-SCoA
malonil -CoA
• à carboxilação da biotina (1), dependente de ATP, seguese a transferência do carboxilo para o Acetil-CoA (2)
Enzima-biotina
HCO3 + ATP
1
ADP + Pi
Enzima-biotina-CO2
O
ll
CH3-C-SCoA
acetil-CoA
2
Enzima-biotina
O
-
ll
O2C-CH2-C-SCoA
malonil -CoA
Reacção global:
HCO3− + ATP + acetil-CoA ADP + Pi + malonil-CoA
• o malonil-CoA, este liga-se ao enzima ACP, “Acyl Carrier
Protein”
• uma molécula de acetil-CoA liga-se a outro ACP
• o acetil-ACP e o malonil-ACP reagem para formar o
acetoacetil-ACP, numa reacção catalisada por uma enzima
condensadora, acetil-malonil-ACP
•o
acetoacetilACP,
numa
sequência de 3
reacções (idênticas
às da β-oxidação
mas em sentido
inverso),
origina
butiril-ACP
• o butiril-ACP reage com uma nova molécula de malonil-ACP
• depois de mais uma sequência como antes, já são 6 átomos
de C
• o alongamento dá-se repetindo esta sequência de reacções,
em geral até ao palmitoil-CoA, com 16 átomos de C
• este é libertado sob a forma de ácido palmítico
• o ácido palmítico
endoplásmico
pode
ser
modificado
no
retículo
• nos mamíferos, estes enzimas estão todos englobados num
complexo chamado Ácido gordo sintetase
A Acetil-CoA Carboxilase, é o passo mais importante
na regulação da síntese de ácidos gordos.
Nos mamíferos, o enzima é regulado por
fosforilação
alosteria
Alterações conformacionais associadas à
regulação:
multímero
activo
na conformação activa, a Acetil-CoA
carboxilase associa-se em complexos
multiméricos
nPi
nPi
a transição para a forma inactiva
envolve
uma
dissociação
dos
monómeros
monómeros
inactivos
Regulação da
Acetil-CoA
Carboxilase por
metabolitos
(regulação
alostérica):
Protómero fosforilado da Acetil- Carboxilase (inactiva)
CoA
Citrato
Palmitoil-CoA
.
• palmitoil-CoA
(produto final da
Ácido gordo
sintetase): inibe
Protómero desfosforilado da
Acetil-CoA Carboxilase (activa)
Regulação da Acetil-CoA Carboxilase
• citrato (nível
elevado indica
que há acetil-CoA
suficiente a entrar
no ciclo de
Krebs): activa
Glucose-6-P
Gluconeogénese
Glucose
Glicólise
Piruvato
Ácidos gordos
• o excesso de
citrato, via
malonil-CoA, é
convertido em
ácidos gordos
para armazenar
Acetil CoA
citrato
oxaloacetato
activada por
desfosforilação
dependente de
insulina
citrato
Oxidação de Ácidos Gordos
Mitocôndria
ciclo de
Krebs
acetil-CoA
carboxilase
R.E.liso
Malonil-CoA
ácido gordo
sintetase
Palmitato
Acetil-CoA
Acil-CoA
Triacilglicerol
Hepatócito (fígado)
citrato
Acil-CoA
Transporte de
Ácidos Gordos
Complexo
ácido gordo/albumina
Ácido gordo
na
teí
ro se
p
po pa
Li li
Lipoproteínas
de baixa densidade
(VLDL)
Armazenamento
de gorduras
inibição
corpos
cetónicos
Palmitato
outros
A.G.
Libertação de
Ácidos Gordos
para o sangue
Ácidos gordos
livres
lipase sensível
às hormonas
activação
sujeito a regulação
a longo prazo
Regulação do
metabolismo dos
Ácidos Gordos
corpos
cetónicos
Acetil-CoA
Transporte de
Triacilgliceróis
Acetil CoA
Ciclo de Krebs
Biossíntese de Ácidos Gordos
inibida por
fosforilação
dependente
de AMPc
Corpos cetónicos
Colesterol
Triacilgliceróis
Adipócito
inibida pela
insulina
activada por
fosforilação
dependente
de AMPc
Insulina +
Insulina +
Insulina +
Insulina +
Armazenamento
de lípidos
Insulina -
+ Adrenalina
+ Noradrenalina
?+ Glicagina
Mobilização
de lípidos
Lipoproteínas do plasma: transporte de lípidos entre os tecidos
éster de colesterol
triacilglicerol
apolipoproteína
colesterol
livre
fosfolípido
Tipo
Composição (%)
QM
VLDL
LDL
VHDL
HDL
Proteína
2
9
21
33
57
Fosfoacilgliceróis
7
18
22
29
21
Colesterol
2
7
8
7
3
Ésteres do colesterol
6
15
38
23
14
83
50
10
1
1
Triacilgliceróis
Ácidos gordos
QM: QuiloMicra; VLDL: Very Low Density Lipoprotein;
LDL: Low Density Lipoprotein; HDL: High Density Lipoprotein;
VHDL: Very High Density Lipoprotein
- e o colesterol?
dieta alimentar
(ex: ovos)
síntese de novo
no organismo
(fígado e intestino)
reserva de colesterol
do organismo
vitamina D
membranas
celulares
sais biliares
síntese de
hormonas
(estrogénios,
testosterona)
excreção
- num indivíduo saudável, o
nível de colesterol está bem
regulado:
↑↑ colesterol na dieta ⇒ ↓↓ síntese de colesterol
↓↓ colesterol na dieta ⇒ ↑↑ síntese de colesterol
- síntese de colesterol:
2 NADPH
+ 2H+
Acetil-CoA
Acetoacetil-CoA
2 NADP+
Ác.mevalónico + CoA
HMG-CoA
HMG-CoA
redutase
Acetil-CoA
colesterol
HMG-CoA: hidroximetilglutaril-CoA
colesterol
da dieta
alimentar
QM
síntese de colesterol
(fígado,
mucosa intestinal)
LDL
tecidos do
organismo
HDL
acumulação/
utilização
reciclagem
no fígado
excreção na bílis
(colesterol livre,
ácidos biliares)
QM
- problemas no metabolismo do colesterol:
excesso de colesterol nas
LDL (“mau” colesterol)
⇒
acumulação de ésteres de
colesterol na parede das
artérias (aterosclerose)
⇒
⇒
tromboses, infartos
de miocárdio,
acidentes vasculares
cerebrais
estreitamento
sanguíneos
dos
vasos
Progressão e Repercussões da
Doença Cardiovascular
Consequências Clínicas:
Angina
Enfarte do
Miocárdio
Hipertrofia
Ventricular
Esquerda
Insuficiência
Cardíaca
AVC
Insuficiência
Renal
Doença
Vascular
Periférica
METABOLISMO DOS AMINOÁCIDOS
E PROTEÍNAS
enzimas
componentes estruturais
das células
Proteínas/aminoácidos
músculo (animais)
a longo prazo,
podem ser
reserva de
glucose e
energia
Utilização dos aminoácidos
síntese proteica
sínteses várias
(aminas, carnitina,
purinas, pirimidinas,
porfirinas, etc)
excedente
grupo α-amina
esqueleto
carbonado
ureia
ciclo de Krebs
gluconeogénese
Plantas → sintetizam todos os aminoácidos
Microorganismos → variável
Vertebrados → só sintetizam cerca de 50% dos
aminoácidos (os outros têm de vir da
alimentação)
- nos humanos:
Não essenciais
Essenciais
alanina
asparagina
ácido aspártico
cisteína*
ácido glutâmico
glutamina
glicina
prolina
serina
tirosina#
arginina+
histidina
isoleucina
leucina
lisina
metionina
fenilalanina
treonina
triptofano
valina
*cisteína produzida só a partir do
aminoácido essencial metionina;
#tirosina produzida só a partir do
aminoácido essencial fenilalanina;
+arginina só necessária nas fases
de crescimento
• Valor biológico duma proteína alimentar:
medida da capacidade que essa proteína tem
de
fornecer
os
aminoácidos
essenciais
necessários para a manutenç
manutenção dos tecidos do
nosso organismo
Fonte
Valor
bioló
biológico
Proteíínas animais
Prote
Ovos
100
Carne
100
Peixe
87
Leite
85
Proteíínas vegetais
Prote
Soja
67
Batatas
67
Pão de trigo integral
30
as proteínas de
fontes vegetais têm
geralmente um valor
biológico mais baixo
que as animais; mas
podem-se combinar
de modo a dar um
resultado nutricional
equivalente à proteína
animal
- Outras boas combinaç
combinações de vegetais:
feijão (rico em Lys, pobre em Met)
+ arroz (pobre em Lys, rico em Met)
Chenopodium quinoa Willd. (“cereal mãe” no idioma
dos índios quechua)
- Foi o alimento básico dos Incas,
unido à sua religião e cultura
Com
a
chegada
dos
conquistadores espanhóis,
foi
substituída pelo milho e batata e
praticamente desapareceu.
AMINOÁCIDOS
QUINOA
TRIGO
LEITE
Histidina *
4.6
1.7
1.7
Isoleucina *
7.0
3.3
4.8
Leucina *
7.3
5.8
7.3
Lisina *
8.4
2.2
5.6
Metionina *
5.5
2.1
2.1
Fenilalanina *
5.3
4.2
3.7
Treonina *
5.7
2.7
3.1
Triptofano *
1.2
1.0
1.0
Valina *
7.6
3.6
4.7
Acido Aspártico
8.6
--
--
Acido Glutâmico
16.2
--
--
Cisteina
7.0
--
--
Serina
4.8
--
--
Tirosina
6.7
--
--
Arginina *
7.4
3.6
2.8
Prolina
3.5
--
--
Alanina
4.7
3.7
3.3
Glicina
5.2
3.9
2.0
*Aminoácidos
essenciais
apesar de terem uma grande diversidade de vias
anabólicas e catabólicas, algumas reacções
de
transferência ou de eliminação do grupo amina são comuns
a muitos aminoácidos
permitem obter alguns aminoácidos a partir doutros
ou simplesmente eliminar o grupo NH3+ e inserir o
esqueleto carbonado do aminoácido noutras vias, como o
catabolismo para se obter ATP ou, se houver em excesso,
sintetizar lípidos para reserva
Proteínas na
alimentação
Proteínas do
corpo
R-CH2-NH3+-COOaminoácidos
Azoto do
grupo
amina
neurotransmissores,
catecolaminas (ex:
adrenalina, heme,
componentes das
membranas, bases dos
nucleótidos, etc
Amónia
R-CO-COOcetoácidos
Ureia
(Excreção
na urina) aminoácidos
glucogénicos
aminoácidos
cetogénicos
Piruvato ou
componentes
do ciclo de Krebs
Acetil-CoA
Lípidos
CO2 + H2O
Glucose
(glicogénio)
CO2 + H2O
1. Transaminação
O
NH3+
R-CH-COO-
+
-OOC-CH -CH 2
2 C-COO
α-cetoglutarato
aminoácido
Transaminase
(há várias diferentes)
NH3+
O
R-C-COO-
+
α-cetoácido
-OOC-CH -CH 2
2 CH-COO
glutamato
2. Desaminação
NH3+
NAD(P)+ NAD(P)H + H+
NH2+
-OOC-CH -CH -C-COO2
2
-OOC-CH -CH 2
2 C-COO
H
glutamato
Glutamato
desidrogenase
O
-OOC-CH -CH - C-COO2
2
α-cetoglutarato
α-iminoglutarato
(intermediário)
H2O
NH4+
- A transaminação pode ser uma maneira de converter uns
aminoácidos noutros
(as principais incluem o par glutamato ↔α-cetoglutarato):
glutamato: oxaloacetato
transaminase
α-cetoglutarato + aspartato
glutamato + oxaloacetato
glutamato: piruvato
transaminase
α-cetoglutarato + alanina
glutamato + piruvato
glutamato: tirosina
transaminase
glutamato + p-hidroxifenilpiruvato
α-cetoglutarato + tirosina
glutamato: leucina
transaminase
glutamato + α-cetoisocaproato
α-cetoglutarato + leucina
- correspondência entre ceto-ácidos
aminoácidos correspondentes:
Piruvato
Oxaloacetato
α-cetoglutarato
e
os
Alanina
transaminação
Aspartato
Glutamato
O glutamato como
“intermediário” das
desaminações
e transaminações:
arginina
prolina
glutamina
histidina
sequências de reacções diversas
glutamato desidrogenase
(desaminação oxidativa)
ácido glutâmico
ácido α-cetoglutárico
α-cetoácido
transaminase
outros aminoácidos
ácido α-cetoglutárico
Catabolismo de aminoácidos – fígado
Aminoácido
esqueleto carbonado
corpos cetónicos
ureia
- principal via de degradação de aminoácidos no fígado:
α-cetoglutarato
NADH + NH4+
α-cetoácido
glutamato
NAD+ + H2O
O
=
α-aminoácido
H2N-C-NH2
ureia
Degradação dos aminoácidos I -remoção dos aminogrupos
α -Aminoá
Aminoácido
α-Cetoá
Cetoácido
Transaminaçção
Transamina
(Excepções: Lisina, Treonina)
α -Cetoglutarato
L-Glutamato
Desaminaçção oxidativa
Desamina
NH3
CO2
Ureia
Degradação dos aminoácidos II-ciclo da ureia
Passo Reagente
-
Produto
Catalisado por
Localização
1
2ATP + HCO3 +
+
NH4
carbamoil fosfato +
2ADP + Pi
Carbamoil fosfato
sintetase (CPS1)
mitocôndria
2
carbamoil fosfato +
ornitina
citrulina + Pi
Ornitina
transcarbamoilase (OTC)
mitocôndria
3
citrulina + aspartato
+ ATP
arginosuccinato +
AMP + |PPi
Argininosuccinato
sintetase (ASS)
citosol
4
arginosuccinato
Arginina + fumarato
Arginosuccinato liase
(ASL)
citosol
5
Arginina + H2O
ornitina + ureia
Arginase 1 (ARG1)
citosol
CPS1, Carbamoil
fosfato sintetase
(enzima reguladora do
processo)
Equação geral do ciclo da ureia:
NH3+CO2 + Aspartato + 3 ATP + 2 H2O →
→ Ureia + Fumarato + 2 ADP + 4 Pi + AMP
A ureia é libertada para a circulação sanguínea, sendo depois
captada pelo rim e eliminada na urina
O ciclo da ureia está ligado ao ciclo de Krebs – como?
Regulação do ciclo da ureia:
Curto prazo:
excesso de arginina ►activação da N-acetilglutamato
sintase ► aumento de N-acetilglutamato
alostérica da carbamoil fosfato sintetase
► activação
Longo prazo:
Dieta com teor de proteína muito elevado ►excesso de
aminoácidos oxidado para produzir ATP e/ou lípidos
►eliminação de NH3 ► aumento ureia
jejum prolongado ►aminoácidos do músculo utilizados na
gluconeogénese ►eliminação de NH3 ► aumento ureia
Aumento da síntese
de enzimas do ciclo
da ureia e de
carbamoil fosfato
sintetase
Fenilcetonúria - doença congénita muito grave:
catabolismo (normal) da fenilalanina:
fenilalanina
hidroxilase
- os doentes têm ausência ou
deficiência
da
fenilalanina
hidroxilase
- há acumulação de fenilalanina
e de fenilpiruvato nos fluidos
corporais
- atraso mental grave ao fim do
1º ano de vida
solução: diagnóstivo precoce
... o “teste do pézinho” pode detectar esta doença!!!
evitar alimentos ricos em fenilalanina desde o nascimento
aspartame – um dos edulcorantes mais
utilizados nos produtos dietéticos
treonina
Ciclo de Krebs no catabolismo
de aminoácidos
s
tran
amin
ase
piruvato
asparagina
alanina
glicina
serina
cisteína
piruvato
aspartato
transaminase
(TA)
fumarato
acetil CoA
asp
TA
acetil CoA
malato
NADH e FADH2
para a produção
aeróbica de ATP
succinato
tirosina
fenilalanina
valina
isoleucina
metionina
treonina
isocitrato
α-cetoglutarato
fumarato
GTP
CITOPLASMA
citrato
oxaloacetato
oxaloacetato
malato
isoleucina
leucina
lisina
fenilalanina
triptofano
tirosina
GDP,Pi
TA
succinil CoA
glu
MITOCÔNDRIA
succinil CoA
α-cetoglutarato
transaminase
(TA)
glutamato
prolina
arginina
histidina
glutamina
lisina
valina
isoleucina
Ciclo de Krebs no anabolismo de aminoácidos
glicólise
hidratos de
carbono
fosfoenolpiruvato
transaminase
piruvato
fenilalanina
triptofano
tirosina
aspartato
r. an
tra
ns
am
in
a
se
OAA
aple
rótic
a
piruvato
PEP
CO2
leucina
lisina
acetil CoA
OAA
acetil CoA
oxaloacetato
alanina
glicina
serina
cisteína
citrato
citrato
TA
malato
aspartato
aspartato
malato
NADH e FADH2
para a produção
aeróbica de ATP
GTP
succinato
GDP,Pi
succinil CoA
MITOCÔNDRIA
CITOPLASMA
isocitrato
NADP
α-cetoglutarato
fumarato
lisina
treonina
metionina
isoleucina
asparagina
isocitrato
TA
glu
α-cetoglutarato
(TA)
glutamato
prolina
arginina
glutamina
Regulação do metabolismo proteico
- depois duma refeição (↓glicagina, ↑insulina) ⇒ síntese proteica
- jejum (↑glicagina, ↓ insulina) ⇒ degradação proteica
- jejum prolongado (↑glicagina, ↓ insulina) ⇒ degradação proteica
intensa
- diabetes (↑glicagina, ↓ insulina) ⇒ degradação proteica
- stress (↑adrenalina) ⇒ degradação proteica
Glucose
cos
éni
c og
glu
AcetilCoA
ce
tog
én
ico
s
piruvato
PEP
gl u
co
s
ico OAA
gé n
aminoácidos
tradução
proteases
Proteínas
citrato
ciclo
de
Krebs
Algumas moléculas sintetizadas a partir de aminoácidos:
Derivados do triptofano
Serotonina
(AA essencial)
Algumas moléculas sintetizadas a partir de aminoácidos:
Derivados do triptofano
Purinas
Algumas moléculas sintetizadas a partir de aminoácidos:
Derivados do triptofano
Heme
Erros congé
congénitos do catabolismo de aminoá
aminoácidos
Os
Os erros
erros congénitos
congénitos podem
podem ser
ser perda
perda total
total ou
ou parcial
parcial da
da actividade
actividade enzimática
enzimática
Na
Na ausência
ausência de
de tratamento
tratamento estas
estas alterações
alterações conduzem
conduzem aa atraso
atraso mental
mental
São
São quase
quase sempre
sempre afecções
afecções pediátricas
pediátricas
Ocorrem
com
uma
frequência
Ocorrem com uma frequência inferior
inferior aa 1/500.000
1/500.000