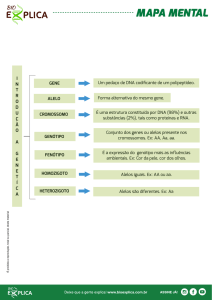
Células
PESQUISA
Tronco-Embrionárias
Células tronco-embrionárias e a geração de modelos animais para doenças genéticas humanas
Alexandre Kerkis
Professor visitante do Departamento de Biologia do Instituto de Biociências USP.
Marina Soukoian
Professora visitante do Departamento de Biologia do Instituto de Biociências USP.
Irina Kerkis
Professora visitante do Departamento de Biologia do Instituto de Biociências USP.
Christian Merkel
Aluno de Mestrado do Programa
Interunidades em Biotecnologia IPEM
Butantã ICB - USP.
Marco R. B. Mello
Aluno de Doutorado do Departamento de
Reprodução Animal, Faculdade de Veterinária e Zootecnia - USP.
Lygia da Veiga Pereira
Professora Assistente do Departamento de
Biologia do Instituto de Biociências USP.
[email protected]
Fotos cedidas pelos autores
20
mantidas em seu estado não diferenciado por múltiplas divisões celulares.
Por outro lado, essas células podem
ser induzidas a iniciar um programa
de diferenciação in vitro. Por exemplo, quando cultivadas em suspensão,
as células ES formam espontaneamente agregados de células diferenciadas chamados corpos embrióides
(Ebs - do inglês, embryoid bodies),
que simulam o desenvolvimento de
um embrião pré-implantado. Através
de análises morfológicas, imuno-histoquímicas e moleculares, encontrase uma grande variedade de linhagens embrionárias dentro dos EBs
hematopoética, neuronal, endotelial,
cardíaca e muscular (Ling e Neben,
1997) (Figura 1c, d).
Essas propriedades das células ES
levaram ao seu uso como modelo in
vitro de desenvolvimento embrionário precoce. Nelas, podem ser estudados os mecanisFigura 1: Células ES.
mos de diferenciação celular, o
(A) Blastócisto de
processo de iniciação da inaticamundongo. O asterisvação do cromossomo X, e os
co indica o botão
efeitos de substâncias tóxicas e
embrionário. (B) Células
biologicamente ativas no deES em cultura. As setas
senvolvimento embrionário in
indicam grumos contenvitro.
do dezenas de células
ES. (C) Corpo
Modelos animais para
embrióide. (D) Análise
doenças genéticas
histológica de corpos
humanas:
embrióides contendo
diferentes estruturas e
Nos últimos anos, células ES
tipos celulares: cavidade
vêem
sendo utilizadas para procística (cc); adipócitos
duzir
camundongos transgêni(ad); músculo (m); fibras
cos
e
modelos animais de dode colágeno (cl);
enças
genéticas humanas. Escondrócitos (cd); precurses
modelos
animais são imsores sanguíneos (os)
portantes ferramentas para o
inhagens de células tronco
embrionárias (células ES do inglês embryonic stem)
são células não diferenciadas, derivadas do botão embrionário de blastócistos, que têm
como característica principal pluripotência (Evans e Kaufman, 1981;
Martin, 1981) (Figura 1a, b). Ou seja,
quando reintroduzidas em um blastocisto, as células ES possuem capacidade de retomar o desenvolvimento normal, colonizando diferentes
tecidos do embrião, incluída a linhagem germinativa. Somente aquelas
linhagens de células ES capazes de
colonizar a linhagem germinativa de
um embrião são consideradas altamente pluripotentes.
Quando cultivadas em condições
específicas, as células ES podem ser
Biotecnologia Ciência & Desenvolvimento - nº 20 - maio/junho 2001
pela versão mutada do mesestudo das patologias associmo gene, construída no laadas com uma doença especíboratório (Figura 2). Dessa
fica, além de servirem como
forma, são obtidas linhagens
um sistema no qual podem
de células ES geneticamente
ser testadas, novas terapias. O
modificadas. Essas são agrerecente desenvolvimento de
gadas a mórulas de camuntécnicas de manipulação do
dongos in vitro e, assim, ingenoma de camundongos faz
corporadas ao embrião (Fido camundongo transgênico
gura 3). Os camundongos
o melhor sistema disponível
resultantes serão quimeras
para o estudo de doenças geformadas de células do emnéticas humanas (Capecchi,
brião recipiente e das células
1989).
ES recombinantes. Se essas
A propagação pela linhaúltimas colonizarem a linhagem germinativa de DNA migem germinativa dos animais
croinjetado no pronúcleo de
quiméricos, a mutação será
ovos fertilizados de camunentão transmitida às novas
dongos foi descrita, pela priFigura 2: Recombinação homóloga. Exons
gerações de camundongos
meira vez, em 1980 (Gordon
estão representados por retângulos, íntrons
(Figura 4).
et al., 1980). Essa metodolopor linhas azuis. O vetor de recombinação
A capacidade de modifigia faz com que várias cópias
homóloga possui duas regiões de homologia
car
regiões específicas do
do transgene injetado se inteao gene alvo (homol. 5 e 3). O pareamento
genoma
do camundongo por
grem em tandem em um sítio
dessas regiões com as regiões homólogas do
recombinação
homóloga peraleatório no genoma e sejam
lócus endógeno permite que duas
mite
a
criação
em potencial
transmitidas de forma Menderecombinações recíprocas levem à substituide
camundongos
com qualliana. Desde então, a microinção de um exon (cinza) pela cassete de
quer
genótipo
desejado.
O
jeção pronuclear tem sido utiexpressão neo. A detecção do evento de
vetor
mais
comumente
usalizada geração de modelos
recombinação homóloga é feita por Southern
do para recombinação hoanimais para várias doenças
blot de DNA genômico das colônias de
móloga é o chamado vetor
genéticas dominantes, inclucélulas ES. Uma sonda externa à região de
de substituição de seqüênindo osteogenesis imperfecta
homologia (barra verde) é utilizada em DNA
cia, que contém o gene de
(Bonadio et al., 1990), através
digerido com a enzima de restrição EcoRI
resistência à neomicina (neo)
da inserção de um alelo muta(E). A substituição do exon por neo introduz
flanqueado por seqüências
do - o transgene - no genoma
um sítio de EcoRI no locus, gerando uma
homólogas ao gene alvo (Cado camundongo.
banda menor referente ao alelo mutado.
pecchi, 1989) (Figura 2). AtraNo entanto, esse método
Uma colônia recombinante é detectada (*)
vés de dois eventos de reapresenta algumas limitações.
combinação recíprocos, esse
Por causa do sítio aleatório de
tipo de vetor media a substiintegração do transgene, este
tuição das seqüências endópoderá não estar sob o congenas de DNA pelas seqüêntrole de todos os elementos
cias exógenas contidas no vetor (Fiem cis que controlam a expressão do veis a efeitos de dosagem gênica.
gura 2). Assim, são criadas mutações
gene endógeno. Assim, a expressão
em loci específicos do genoma do
temporal e espacial do transgene não
Manipulação do genoma
camundongo.
seguirá o padrão de expressão do
do camundongo através
gene endógeno. Além disso, a introde células ES:
Mutação condicional dução de um terceiro alelo - o transsistema CRE-loxP:
gene mutante - cria uma situação
Recentemente, a combinação do
artificial no que diz respeito à pro- cultivo de células ES e da recombinaOutro tipo de vetor desenvolvido
porção entre os transcritos normais e ção homóloga resultou na criação de
os mutantes. Enquanto uma pessoa um método alternativo e mais preci- ainda mais recentemente utiliza o
com uma doença genética dominan- so para manipular o genoma do sistema de recombinação em bactérias CRE-loxP (Chambers, 1994).
te possui um alelo normal e um camundongo.
mutado, o camundongo transgênico
As células ES podem ser modifi- Nesse microorganismo, duas seqüênpossuirá dois alelos endógenos nor- cadas geneticamente, em cultura, atra- cias específicas de 38 pb, denominamais e diversas cópias do alelo mu- vés da recombinação homóloga, pro- das loxP, são reconhecidas pela protante (transgene). Essa proporção cesso que promove a substituição do teína CRE do bacteriófago P1. Por
pode ser crítica em doenças suscetí- alelo normal de um gene específico meio da recombinação entre duas
Biotecnologia Ciência & Desenvolvimento - nº 20 - maio/junho 2001
21
caras e podem ser usadas
sequências loxP, a protedurante um tempo limitaína CRE promove a remodo, por já estarem em culção da seqüência de DNA
tivo há a um período reladentre estas sequências,
tivamente longo. Assim,
resultando em uma delepara a produção a longo
ção.
prazo de animais transgêO sistema CRE-loxP
nicos é fundamental a
pode ser usado junto com
obtenção de novas linhaa recombinação homólogens de células ES.
ga para se criarem mutaNós estabelecemos três
ções no genoma do canovas linhagens de célumundongo (Chambers,
las ES a partir do botão
1994). Nesse caso, o veembrionário de blastócistor de recombinação contos de camundongos 129/
tém, além da cassete de
Sv (pelagem agouti): USPexpressão neo, duas se1, USP-2 e USP-3. Sua pluqüências loxP delimitanripotência foi avaliada in
do a região a ser deletada
vitro através de testes con(Figura 5). O primeiro
vencionais. Eles incluíram
evento de recombinação
a cariotipagem das céluhomóloga introduzirá as
Figura 3: Sistema de recombinação CRElas, a atividade de fosfataseqüências loxP nos sítiloxP. Seqüências loxP estão representadas por
se alcalina e a capacidade
os específicos do gene
triângulos verdes. O primeiro evento de
das células de formarem
alvo. Um segundo evento
recombinação homóloga introduz os sítios loxP
EBs complexos. Essas três
de recombinação, mediaentre o exon a ser deletado. Em um segundo
linhagens apresentaram
do pela proteína CRE, leevento, a expressão da proteína CRE irá mediar
cariótipo 40, XY normal,
vará à deleção a região do
a recombinação entre os dois sítios loxP,
alta atividade de fosfatase
alelo recombinante flandeletando a seqüência entre eles. A expressão
alcalina, e formaram EBs
queada pelas seqüências
de CRE pode ser controlada de forma que
complexos, com diversos
loxP.
induza a recombinação somente em tecidos ou
tipos celulares e cardiomiA recombinação meestágios de desenvolvimento específicos do
ócitos pulsantes, demonsdiada por CRE pode ser
camundongo
trando um alto grau de
feita ainda nas células ES
diferenciação (Figuras 1 e
em cultura, através da
6). Estas características inexpressão transiente de
CRE que há nelas. Uma vantagem quando presentes desde a concep- dicam um alto nível de pluripotência
desse sistema é que, com ele, o gene ção ou em todos os tecidos do ani- das novas linhagens de células ES
USP-1, USP-2 e USP-3.
neo não fica inserido no gene alvo. A mal.
Foi avaliada a capacidade da lipresença de neo em íntrons de genes
A combinação dessas estratégias
pode introduzir seqüências que atra- de manipulação do genoma do ca- nhagem USP-1 de popular a linhapalhem o splicing correto do gene mundongo, quando realizadas em gem germinativa de animais quiméalvo, ou que, de alguma ou outra células ES, permitem a criação de ricos. Através da agregação e do
cocultivo das células USP-1 com móforma, diminuam o nível de expres- linhagens de animais mutantes.
rulas de animais CD-1 (pelagem bransão do alelo mutado ou de genes na
ca), foram gerados animais quimérisua proximidade. Esse tipo de efeito
Estabelecimento de novas
cos (Figura 3). O nível de quimerisjá foi descrito em alguns genes (Pelinhagens de células ES:
mo, estimado a partir da coloração
reira et al., 1997; Pereira et al., 1999).
Alternativamente, o segundo
Um parâmetro crítico para uso de da pelagem dos animais, variou de 0
evento de recombinação pode ser células ES na geração de modelos a 100%. Os animais mais compostos
realizado in vivo. Dessa forma, con- animais é a sua pluripotência, que de células derivadas das células USPtrolando-se o padrão temporal e o pode ser perdida durante o cultivo 1, de pelagem predominantemente
espacial da expressão de CRE, pode- prolongado. O uso de linhagens en- agouti, foram cruzados com fêmeas
se controlar o período de desenvol- velhecidas dessas células pode levar CD-1. Como a pelagem agouti é
vimento e os tecidos onde a mutação a uma baixa freqüência de recombi- dominante sobre a pelagem branca
será induzida, o que chamamos de nação homóloga e à não colonização dos animais CD-1, a pelagem agouti
mutação condicional. Esse sistema é da linhagem germinativa de animais da ninhada resultante demonstrou a
utilizado quando se deseja estudar o quiméricos (Nagy e Rossant, 1993). colonização da linhagem germinatiefeito de mutações que são letais Células ES murinas comerciais são va dos animais quiméricos pela li22
Biotecnologia Ciência & Desenvolvimento - nº 20 - maio/junho 2001
nhagem de células ES USP-1 (Figura
4). Essa linhagem está, no momento,
sendo utilizada para gerar um modelo animal para a síndrome de Marfan.
Geração de um modelo animal
para a síndrome de Marfan:
A síndrome de Marfan (SMF) é a
mais comum das doenças genéticas
do tecido conjuntivo. Herdada de
forma autossômica dominante, essa
síndrome tem uma incidência de,
aproximadamente, 1 em 10.000 indivíduos (McKusick, 1955). O fenótipo
da SMF é 100% penetrante, e suas
manifestações clínicas afetam primariamente três áreas: esquelética (crescimento excessivo dos membros, hiperextensibilidade articular, escoliose e deformidades na região anterior
do tórax); ocular (ectopia lentis, miopia e deslocamento da retina); e
cardiovascular (dilatação da raíz da
aorta, dissecção da aorta, prolapso
da válvula mitral e regurgitação mitral).
A sobrevida de pacientes com a
SMF é de, aproximadamente, dois
terços do normal - 40 anos na média.
Em 90% dos casos, a morte é causada
por falha cardiovascular (McKusick,
1955). O diagnóstico precoce seguido de intervenção médica leva a um
prolongamento da sobrevida.
A SMF é causada por mutações no
gene FBN1. Esse gene codifica a
fibrilina-1, componente estrutural
mais abundante das microfibras na
matriz extracelular. As microfibras
estão largamente distribuídas pelo
organismo e se encontram associadas à elastina nas fibras elásticas.
Acredita-se que mutações no gene
FBN1 geram o fenótipo da SMF através do modelo dominante-negativo
(Dietz et al., 1993). Segundo esse
modelo, proteínas mutantes interagem com as fibrilinas normais, incorporando-se às microfibras e perturbando, assim, sua organização e integridade. Ainda de acordo com esse
modelo, os indivíduos portadores de
um alelo mutante com uma expressão de fibrilina-1 mais baixa podem
apresentar um fenótipo menos severo da doença devido à prevalência de
fibrilinas normais produzidas a partir
do alelo normal (Pereira et al., 1997).
A identificação de uma proteína
altamente homóloga à fibrilina-1, denominada fibrilina-2, levou à classificação destas como uma nova família
de proteínas extracelulares: fibrilina1, produto do gene FBN1; e fibrilina2, produto do gene FBN2. Estudos
dos padrões de expressão desses
dois genes durante a embriogenia
mostram que estes são expressos de
maneiras diferentes, tanto em termos
de distribuição em tecidos quanto de
estágios de desenvolvimento (Zhang
et al., 1995). Em geral, mRNAs da
fibrilina-2 aparecem mais cedo e se
acumulam por um período mais curto de tempo do que os da fibrilina-1.
Síntese de fibrilina-1 está relacionada
com o período mais tardio da morfogenese e com o aparecimento de
estruturas mais definidas.
Por outro lado, a síntese da fibrilina-2 coincide com etapas iniciais da
morfogenese, em particular com o
começo da elastogenese. Logo, as
microfibras morfologicamente homogêneas são, na verdade, heterogêneas, no que diz respeito à sua composição por diferentes fibrilinas. Com
base nesses resultados, foi proposto
que as fibrilinas têm funções distintas, porém relacionadas, na fisiologia
das microfibras. Segundo essa teoria,
a fibrilina-1 provê principalmente
suporte e força, enquanto a fibrilina2 tem uma função principal na regulação do processo inicial da formação da fibra elástica (Zhang et al.,
1995).
Para estudarmos a função específica de cada fibrilina na formação da
fibra elástica e na manutenção da
integridade de diversos tecidos, e os
mecanismos de patogênese envolvidos na SMF, criamos camundongos
com diferentes mutações no gene
Fbn1, homólogo ao gene FBN1 humano, através da recombinação homóloga em células ES (Figura 7).
D:
Linhagem mgD
Figura 4: Geração de animais
quiméricos. Células ES
derivadas de um animal agouti
são cocultivadas com mórulas
de um animal de pelagem
branca. As células se integram
ao embrião, e os animais
resultantes são quimeras,
compostas de células derivadas
do embrião receptor e de
células derivadas das células ES
A primeira linhagem (mgDD) possui uma substituição dos exons 19-24
do gene Fbn1 por um cassete de
expressão do gene da neomicina
(neo) (Pereira et al., 1997) (Figura 7).
Esta mutação levou à produção de
monômeros de fibrilina-1 com uma
deleção interna, porém, devido à
presença de neo no íntron 18, com
um nível de expressão 10 vezes menor do que o do alelo normal. Assim,
a mutação mgD representa uma combinação de uma mutação estrutural e
hipomórfica.
Em acordo com o modelo dominante-negativo, animais heterozigotos não apresentam nenhum fenótipo, provavelmente devido ao excesso de fibrilinas normais em relação à
forma mutante. Por outro lado, animais homozigotos mgD/mgD morrem de complicações cardiovasculares durante as duas primeiras semanas de vida pós-natal. A histopatologia da parede vascular desses animais é extremamente similar àquela
de pacientes com SMF, com a mesma
Biotecnologia Ciência & Desenvolvimento - nº 20 - maio/junho 2001
23
Figura 5: População da
linhagem germinativa das
quimeras pelas células ES.
Animais quiméricos cruzados
com fêmeas CD-1 (pelagem
branca) deram origem a uma
ninhada de animais de
pelagem agouti. Isso indica
que a linhagem germinativa
das quimeras foi populada
pelas células ES e, assim, o
genótipo dessas células foi
transmitido à prole
fragmentação das fibras elásticas. Mesmo assim, em tecidos não afetados,
as fibras elásticas são morfologicamente normais.
A conclusão mais importante desse estudo foi a de que, mesmo na
ausência de fibrilina-1 normal, há
formação da fibra elástica (Pereira et
al., 1997). Assim, confirmamos a hipótese de que na verdade, a fibrilina2 expressa mais cedo, durante o
desenvolvimento embrionário, dirige o depósito de elastina na formação da fibra elástica, e que a fibrilina
1 teria um papel posterior na manutenção da integridade e no suporte
dessas fibras.
Linhagem mgR:
O segundo alelo Fbn1 mutante
(mgR) foi criado através da inserção
do gene neo entre os exons 18 e 19
(Pereira et al., 1999) (Figura 7). Apesar dessa inserção não causar nenhuma perda de seqüência do gene
Fbn1, ela leva a uma redução de 5
vezes na expressão do alelo mutado.
Figura 7: Mutações no gene Fbn1 geradas por recombinação
homóloga em células ES. A proteína fibrilina está esquematizada.
Regiões em cinza correspondem àquelas modificadas nos alelos
mutantes. Exons 1, 17 a 28 do gene selvagem (Fbn1wt) estão representados. O alelo mgD consiste na substituição dos exons 18 a 24 pela
cassete de expressão neo, o que leva à produção de monômeros de
fibrilina com uma deleção interna, porém em baixa quantidade. O alelo
mgR é uma inserção dessa cassete no íntron 18 do gene Fbn1, resultando na baixa expressão de fibrilina normal a partir desse alelo. O alelo
Fbn10 é um alelo nulo do gene Fbn1. Nele, parte do exon 1, incluído o
códon de início de tradução (ATG) e a seqüência codificadora do
peptídeo sinal, foi substituída pelo gene repórter da fosfatase alcalina
humana (hFA) e a cassete de expressão neo. O gene hFA fica colocado
sob controle do promotor do gene Fbn1 (seta)
24
Biotecnologia Ciência & Desenvolvimento - nº 20 - maio/junho 2001
Assim, o alelo mgR consiste em
uma mutação hipomórfica que produz 20% de moléculas de fibrilina1 normais.
Como esperado, animais heterzigotos mgR/+ são normais. Em
contraste, camundongos homozigotos mgR/mgR desenvolvem cifose severa e morrem abruptamente
de falha do sistema cardiovascular
até os três meses de idade. Esses
animais mutantes também apresentam hérnia de diafragma e ossos
alongados, similar a pacientes com
SMF. A análise histopatológica dos
animais mgR/mgR revelou uma
participação importante da resposta inflamatória na progressão das
manifestações vasculares da mutação (Pereira et al., 1999). Assim,
esses estudos sugerem que a inibição dessa resposta em indivíduos
com a SMF pode retardar as manifestações da doença no sistema
cardiovascular.
Fbn10:
Finalmente, uma terceira mutação no gene Fbn1 foi gerada em
nosso laboratório nas células ES
USP-1: um alelo nulo (Fbn10) (Figura 7). Para isso, o vetor de recombinação homóloga foi construído de
maneira que promovesse uma substituição de parte do exon 1 do gene
Fbn1 pelo gene repórter da fosfatase alcalina e pela cassete de expressão neo. Espera-se que a deleção
do códon de iniciação de tradução
e da seqüência codificante do peptídeo sinal, presentes no exon 1, irá
inviabilizar a tradução correta da
fibrilina-1. Além disso, a presença
do gene repórter da fosfatase alcalina sob controle do promotor endógeno do gene Fbn1 facilitará a
análise da expressão temporal e
espacial desse gene em animais
heterozigotos.
Perspectivas:
Nos últimos 10 anos, células ES
de camundongo tornaram-se um
poderoso instrumento de pesquisa.
Nelas são estudados os mecanismos de diferenciação celular e os
Figura 6: Caracterização da linhagem USP-1 de células ES. (A)
Cariótipo. (B) Análise de atividade de fosfatase alcalina (FA). Grumos
de células escuras indicam alta atividade de FA
eventos iniciais do desenvolvimento embrionário, reproduzidos por
essas células diferenciadas in vitro.
As linhagens estabelecidas em nosso laboratório estão sendo utilizadas também para o estudo in vitro
do efeito de novas substâncias nesse estágio de desenvolvimento.
Além disso, a capacidade de
células ES de se diferenciarem em
qualquer tipo de tecido representa
um enorme potencial de aplicação
médica. Sob condições específicas,
induzidas a se diferenciarem in
vitro, em um tipo celular determinado, elas poderão, no futuro, ser
uma fonte ilimitada de tecidos para
transplante no tratamento de doenças. Um passo importante nessa
direção foi o estabelecimento de
linhagens de células ES humanas
(Thomson et al., 1998). Agora, experimentos realizados com células
ES murinas poderão ser repetidos e
adaptados às linhagens humanas.
Finalmente, estamos vivendo o
momento histórico do final do seqüenciamento do genoma humano. A análise da primeira versão
completa do genoma humano identificou, aproximadamente, 30.000
genes (Lander et al., 2001). Desses,
uma grande proporção não tem
função conhecida. O grande desafio da era pós-genoma será determinar a função gênica. Uma estratégia poderosa para o estudo de
função gênica in vivo é a geração de
mutantes. No caso de genes humanos, modelos em camundongos são
utilizados devido à nossa capacidade
de manipulação do genoma desse
animal através da recombinação homóloga em células ES. A implementação desse instrumental de pesquisa
no Brasil permitirá seu uso pelos
diversos grupos envolvidos no estudo de função gênica.
REFERÊNCIAS
Capecchi, M.R. (1989). The new
mouse genetics: Altering the genome
by gene targeting. Trends in Genetics, 5: 70-6.
Chambers, C.A. (1994). TKOed:
Lox, stock and barrel. BioEssays, 16:
865-8.
Dietz, H.C., McIntosh, I., Sakai,
L.Y., Corson, G.M., Chalberg, S.C.,
Pyeritz, R.E., and Francomano, C.A.
(1993). Four novel FBN1 mutations:
significance for mutant transcript level
and EGF-like domain calcium binding
in the pathogenesis of Marfan syndrome. Genomics, 17:468-75.
Evans, M. and Kaufman, M.
(1981). Establishment in culture of
pluripotential cells from mouse embryos. Nature, 292: 154- 6.
Gordon, J.W., Scangos, G.A.,
Plotkin, D.J., Barbosa, J.A., e Ruddle, F.H. (1980). Genetic transformation of mouse embryos by microinjection of purified DNA. Proc. Natl.
Acad. Sci. USA., 77: 7380-4
Lander, E.S., Linton, L.H., Birren, B., et al., (2001). Initial sequencing and analysis of the human genome. Nature, 409:860-921.
Ling, V. and Neben, S. (1997). In
vitro differentiation of embryonic stem
cells: immunophenotypic analysis of
cultured embryoid bodies. J Cell Physiol., 171:104-5.
Martin, G.R. (1981). Isolation of a
pluripotent cell line from early mouse
embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc.
Natl. Acad. Sci. USA., 78: 7634-8.
McKusick, V. (1955). The cardiovascular aspects of Marfans syndrome: a heritable disorders of connective tissue. Circulation, 11:321-42.
Nagy, A. and Rossant, J. (1993).
Production of completely ES cell-derived fetuses. In: Gene Targeting. A
Practical Approach (Joiner, A.L., ed.),
IRL Press, Oxford, pp.147-180.
Pereira, L., Andrikopoulos, K.,
Tian, J., Lee, S.Y., Keene, D.R., Ono,
R., Reinhardt, D.P., Sakai, L.Y., Biery, N.J., Bunton, T., Dietz, H.C.,
Ramirez, F. (1997). Targetting of the
gene encoding fibrillin-1 recapitulates
the vascular aspect of Marfan syndrome. Nat. Genet., 17:218-22.
Pereira, L., Lee, S.Y., Gayraud,
B., Andrikopoulos, K., Shapiro,
S.D., Bunton, T., Biery, N.J., Dietz,
H.C. Sakai, L.Y., and Ramirez, F.
(1999). Pathogenetic sequence for
aneurysm revealed in mice underexpressing fibrillin-1. Pros.Natl.Acad.Sci.
USA, 96:3819-23.
Thomson, J.A., Itskovitz-Eldor,
J., Shapiro,S.S., Waknitz, M.A., Swiegiel, J.J., Marshall, V.S., Jones, J.M.
(1998). Embryonic stem cell lines derived from human blastocysts. Science, 282:1145-7.
Zhang, H., Apfelroth, S.D., Hu,
W., Davis, E.C., Sanguineti, C., Bonadio, J., Mechan, R.P., and Ramirez, F. (1994). Structure and expression of fibrillin-2, a novel microfibrillar
component preferentially located in
elastic matrices. J. Cell Biol., 124: 85563.
Zhang, H., Hu, W., and Ramirez,
F. (1995) Developmental expression
of fibrillin genes suggests heterogeneity of extracellular microfibrils. J.
Cell Biol., 123:1165-76.
Biotecnologia Ciência & Desenvolvimento - nº 20 - maio/junho 2001
25