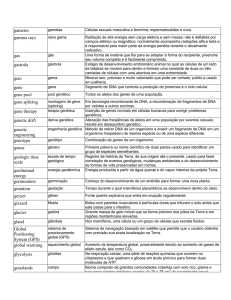
Genética
e
Melhoramento de Plantas
Universidade de Évora
2005
Estrutura dos ácidos nucleicos
Ácido Desoxirribonucleico (DNA)
Ácido Ribonucleico (RNA)
Dos genes às proteínas (eucariotas): transcrição e tradução
Engenharia Genética de Plantas
Identificação de genes de interesse
Extracção de RNA total e purificação de mRNA
Isolamento e clonagem de genes de interesse
Desenho de primers degenerados
Desenho de primers específicos para isolamento das
extremidades (5’ e 3’ de um gene)
Construção de Vectores de Clonagem
Avaliação de colónias recombinantes com enzimas de restrição
DNA: Ácido DesoxirriboNucleico
Constituído por uma sequência de
apenas quatro nucleótidos.
Os diferentes nucleótidos
apresentam uma estrutura comum:
à Um grupo fosfato ligado por uma
ligação fosfoester a uma pentose
(desoxirribose), a qual se liga a uma
base azotada resultando num
desoxirribonucleótido.
à As bases azotadas podem ser purinas
(adenina e guanina) ou pirimidinas
(citosina e timina).
à Os desoxirrinucleótidos ligam-se entre
si por ligações fosfodiester originando
uma cadeia simples.
Duas cadeias simples ligam-se
entre si devido à
complementaridade das suas
bases azotadas (A=T, G=C),
através de ligações por pontes de
hidrogénio, originando uma
cadeia dupla.
James Watson and Francis Crick
(1953):
Estrutura em dupla hélice
com orientação das duas cadeias
antiparalela.
RNA: Ácido RiboNucleico
Constituído por uma sequência de
apenas quatro nucleótidos.
Os diferentes nucleótidos
apresentam uma estrutura comum:
à Um grupo fosfato ligado por uma
ligação fosfoester a uma pentose
(ribose), a qual se liga a uma base
azotada resultando num
desoxirribonucleótido.
à As bases azotadas podem ser purinas
(adenina e guanina) ou pirimidinas
(citosina e uracilo).
Apresenta uma estrutura em cadeia
simples.
à Os ribonucleótidos ligam-se entre si por
ligações fosfodiester originando uma
cadeia simples.
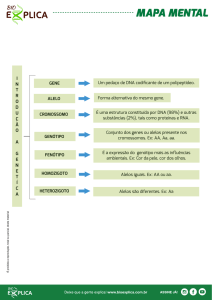
Estrutura de um gene:
Promotor: Região do DNA à qual se liga a enzima RNA polymerase antes de iniciar a
transcrição do DNA em RNA.
Determina: região de início da transcrição; a cadeia de DNA que servirá como template; número de
transcritos; frequência da transcrição.
Gene: Segmento de DNA constituído por um conjunto de intrões e exões que será transcrito em
mRNA, codificando os exões a síntese proteica.
Terminador: Região terminal que bloqueia a enzima RNA polimerase e induz a sua dissociação.
5’ – Promoter
UTR Exon1
Intron1
Exon2 UTR Terminator – 3’
GENE
Transcription
Poly A
Transcrição (decorre no núcleo):
síntese de uma cadeia de RNA mensageiro (mRNA) pela enzima RNA
polimerase que decorre de 5’ para 3’ tendo como modelo apenas uma
das cadeias de DNA. A nova cadeia resultante será complementar à
cadeia de DNA que lhe serviu de molde apresentando uracilo em vez
de timina:
Modificações pós-transcrição:
5’ capping: ligação de uma molécula de 7-metilguanilato à
extremidade 5’ do mRNA durante a transcrição, tendo como
objectivo a protecção da nova cadeia.
Clivagem da extremidade 3’ e adição de uma cauda Poly (A)
Ainda no núcleo o mRNA
transcrito primário sofre a
clivagem da extremidade 3’ e
posterior adição de uma cauda
poly(A).
A dimensão da cauda poly(A)
varia entre 100 a 250
adeninas.
No citoplasma a dimensão da
cauda diminui.
A funçãon da cauda poly(A)
não é ainda bem conhecida.
Splicing:
clivagem dos intrões no inteior da
cadeia de mRNA e simultânea
ligação dos exões,
correspondendo à última fase do
processamento das moléculas de
mRNA no núcleo.
Genoma
Total conjunto de genes
de um organismo
Transcritoma
Conjunto de sequências
transcritas (mRNA)
Proteoma
Conjunto de proteinas
codificado pelo genoma
Código genético:
envolve o reconhecimento de tripletos
(conjunto de três nucleótidos) do
mRNA denominados codões pelos
complementares anticodões do tRNA.
Codão de iniciação: AUG (Met)
Codões stop: UAA; UAG; UGA
O código genético é universal e
degenerado (61 codões codificam para
a síntese de apenas 20 aa):
vários codões = 1 aa
1 codão ≠ vários aa
Início da tradução:
ão
reconhecimento do
codão de iniciação
(mRNA) AUG pelo
anticodão (tRNA)
UAC transportando
o aminoácido inicial
Meteonina.
O ribossoma
(rRNA+proteina)
movendo-se ao
longo da cadeia de
mRNA (de três em
três nucleotidos)
cataliza a síntese
proteica.
Final da tradução:
ão
reconhecimento do
codão de terminação
(mRNA) UAG pelo
anticodão (tRNA)
AUC com a
consequente
separação das duas
subunidades que
constituem o
ribossoma.
Melhoramento genético:
Consiste na adição ou subtracção de um ou mais genes de um
organismo, introduzindo ou silenciado uma determinada característica.
Vantagem:
Ultrapassar a barreira da espécie: um gene que codifica uma
característica numa determinada espécie pode ser introduzido numa
outra espécie mesmo que filogenéticamente muito afastada recorrendo a
técnicas de biologia molecular.
Interesse:
Criar plantas tolerantes ou resistentes a determinadas condições de stress
biótico e/ou abiótico.
Exemplo:
Introdução de Resistência ao Fungo Uncinula necator (Oídio) em
Videira (Vitis vinifera)
O fungo em questão possui parede de quitina pelo que as proteínas com
capacidade de a hidrolizar – quitinases – podem conferir à planta
potencialidade para resistir a esse stress biótico.
1º PASSO:
Pesquisar na bibliografia a existência de espécies vegetais resistentes a este fungo.
Ex.: Vitis rupestris
2º PASSO:
Pesquisar na base de dados se o gene está descrito para a espécie classificada como
resistente. Caso não esteja, será objectivo isolar este gene na espécie
resistente para introduzir na espécie sensível (http://www.ncbi.nlm.nih.gov/).
3º PASSO: Isolamento de RNA total do organismo do qual se pretende isolar o gene
(espécie resistente – V. rupestris) e purificação de mRNA.
mRNA
4º PASSO: Transcrição Reversa
5º PASSO: Desenho de primers degenerados permitindo-nos amplificar uma
sequência correspondente ao gene que se pretende isolar. Para tal
seleccionam-se na base de dados as sequências ORF (Open Reading
Frame) de várias espécies, ou seja, a região estritamente necessária para a
codificação da síntese da proteína. A ORF inicia-se com a sequência
Kozak (CCGCCATGGG) e finaliza com um dos codões stop,
correspondendo sempre à maior sequência encontrada;
6º PASSO: Alinhamento das diferentes sequencias de um mesmo gene codificando a
mesma proteína em diferentes espécies e desenho de um primer
degenerado (sequencia de 20 a 30 oligonucleótidos) forward
(extremidade 5’) e outro reverse (extremidade 3’);
7º PASSO: Amplificação de uma sequência de cDNA por PCR utilizando os primers
degenerados.
Extracção de RNA total (Protocolo do “Hot Borate”)
A integridade do RNA é avaliada em gel de agarose onde
devem ser visíveis duas bandas mais fortes correspondentes
ao RNA ribossomal (28S e 18S);
Pode ocorrer contaminação com DNA;
O trabalho com RNA requer mais cuidados, uma vez que este
é mais instável que o DNA;
A grande desvantagem de trabalhar com o RNA será a de não
existir forma de o amplificar.
28S
18S
Purificação do mRNA
DNA
mRNA
Permite isolar genes que se estejam a expressar
(constitutivos ou de expressão diferencial, que só se expressem
sob uma determinada condição de stress);
A incapacidade de o amplificar levou ao desenvolvimento de
técnicas (Transcrição Reversa)
Reversa que permitiram converter este
novamente em DNA (DNA complementar ou cDNA).
cDNA
Transcrição Reversa
Enzimas denominadas Transcriptases Reversas, descobertas por Temin e
Baltimore (1960) em retrovírus, catalizam a síntese de cDNA a partir de
RNA.O cDNA é mais estável e apresenta a vantagem de poder ser
amplificado por técnica de PCR.
Transcrição Reversa
mRNA template
AAAAAAA (…) Poly(A)
VTTTTTTTTTTTTTTTTCAGCTGTAGCTATGCGCACCAG (V=A,C ou G) VIAL 8
AAAAAAA (…) Poly(A)
VTTTTTTTTTTTTTTTTCAGCTGTAGCTATGCGCACCAG (V=A,C ou G)
AAAAAAA (…) Poly(A)
Degradação da cadeia
de mRNA com RNase
VTTTTTTTTTTTTTTTTCAGCTGTAGCTATGCGCACCAG (V=A,C ou G)
VTTTTTTTTTTTTTTTTCAGCTGTAGCTATGCGCACCAG (V=A,C ou G)
Amplificação por PCR
GTCGACATCGATACGCGTGGTC
VIAL 9
VTTTTTTTTTTTTTTTTCAGCTGTAGCTATGCGCACCAG (V=A,C ou G)
Pefw
PeRev
AAAAAAAAAAAAAAAGTCGACATCGATACGCGTGGTC
A transcrição (reversa) do cDNA tem a vantagem de permitir posterior amplificação de um gene de interesse
limpo de intrões. O conhecimento da sequência das extremidades do cDNA permite a posterior amplificação
das extremidades de um gene. Para tal deverá utilizar-se um primer específico que emparelhe com uma das
extremidades e um outro primer que emparelhe com uma zona específica da nossa sequência.
Primer: Sequência de oligonucleótidos (16-24 nucleótidos) complementar a uma
região do DNA que ao emparelhar permite o funcionamento da enzima Taq
polimerase e consequente formação de uma nova cadeia de DNA
complementar à existente.
Forward (5’)
Reverse (3’)
5’
AGGCTA(…)
AGGCTA(…)
GGCAAT(…)
CCGTTA(…)
Fw
Rv
5’ AGGCTA(…)
3’
5’ (…)ATTGCC
Reverse complement
Os primers devem ser desenhados tendo em conta os seguintes pontos:
a) primer Forward – próximo da extremidade 5’ com a sequência igual à da
cadeia modelo; Reverse – próximo da extremidade 3’ com a sequência complementar à
da cadeia modelo, sendo a sua sequência colocada em reverse devido a efeitos de
encomenda;
b) cada primer não deverá exceder os 25 nucleótidos;
c) o número de nucleótidos de um primer depende da sua temperatura de
emparelhamento, determinada da seguinte forma: [2x(A+T)]+[4x(G+C)];
d) a extremidade 3’ de cada primer deverá ser mais rica em G e/ou C.
Polymerase chain reaction (PCR)
Amplificação exponencial de uma sequência específica de DNA, determinada
pelo emparelhamento dos primers.
Condições necessárias para a reacção:
a) sequência de DNA que servirá de template
b) um par de primers (forward e reverse)
c) DNA polimerase (Taq DNA polimerase: Thermus aquaticus)
d) dNTPs (C, T, G, A)
c) tampão de PCR com MgCl2
Programa de PCR:
1º: Desnaturação: temperatura de 90-95°C
2º: Emparelhamento: temperatura varia com primers
3º: Temperatura de síntese: temperatura 60-72°C
Mix de PCR:
1. cDNA …………………..… 1µl
2. PCR Buffer (10x) ……….... 5µl
3. MgCl (50mM) …………..… 2µl
4. dNTPs (10mM) …………... 1µl
5. Primer forward (10mM) ….. 1µl
6. Primer reverse (10mM) ……1µl
7. H2O Milli-Q estéril ………. 38.5µl
8. Taq DNA polimerase …….. 0.5µl
50µl
Programa de PCR:
∞
35 ciclos
330 pb
5’
800 pb
Rv
5’
3’
MM
5’
3’
± 1000 pb
5’
Fw
PCR 1
-
± 500 pb
A utilização de primers degenerados permitenos amplificar fragmentos específicos da
espécie em estudo (± 500pb);
1500pb
600pb
500pb
100pb
+
Por vezes ocorre a amplificação de outros
fragmentos denominados inespecíficos dado o
emparelhamento inespecífico do primer (se
existir uma pequena sequência na extremidade
3’ do primer, sobretudo G ou C, que
emparelhem com alguma zona complementar
no templete, ocorrerá igualmente síntese de
DNA).
Sequenciação do fragmento amplificado
à Na reacção de sequenciação (PCR com apenas 1 primer) o fragmento vai ser
amplificado com nucleótidos marcados que o sequenciador vai reconhecer, sendo o
resultado da leitura dado por uma cor diferente para cada nucleótido;
à A leitura do fragmento de DNA amplificado permite determinar se este
corresponde de facto a um fragmento do gene que se pretende isolar.
Isolamento das extremidades 5’ e 3’ do gene
à Desenho de primers específicos
5’………………………………(330pb)GGGATACTGCTACCTCAGGGAACAAGGCAGCCCCGGAGCTTACTGTGTTCC
CAGCCCCGGAGCTTACTGTGTTCC
TAGTGCACAGTGGCCTTGTGCCGCTGGTAGGAAATACTATGGCCGAGGCCCCATACAGATTTCCTACAACTACAACT
ATGGGCAAGCTGGGAAAGCCATAGGGGTAGACCTGGTAAACAACCCTGATCTAGTAGCAACAGATGCAGTCATATC
ATTCAAGACAGCCTTCTGGTTCTGGATGACACCCCAGTCACCCAAGCCTTCCTGCC
CCCCAGTCACCCAAGCCTTCCTGCCATAATGTCATCACAGGAGG
ATGGACCCCATCAGGTGCAGATAGGTCAGCAGGGCGGCTTCCCGGTTTTGGTGTTATCACAAACATCATCAATGGAG
GTGTTGAATGTGGGAAAGGGGTAGTTCCTCAGGTCCAGGACCGCATAGGTTTCTATAAGAGGTACTGTGATATACTT
AGGGTTAGCTATGGCAATAACCTGGACTGCAACAACCAAAGGCCTTTCGGGTCTGGCC(800pb)……………………...3’
Ta=(4x15)+(2x9)-10=78-10= 68
Ta=(4x17)+(2x8)-10=84-10= 74
± 500pb
cDNA
5’
3’
GGG
5’PCR primer
CCC
Rv
Fw
± 700pb
Poly(A)
AAAAAA
Poly(T)
5’
3’
Clonagem do Gene Completo
A enzima Taq DNA polimerase possui a capacidade adicional de terminal
transferase (para além de DNA polimerase), ou seja, adiciona ao final da
cadeia amplificada uma base de adenina (A).
Esta capacidade permite a ligação do fragmento amplificado a um vector de
clonagem linearizado possuindo extremidades de timina (T).
5’AATGGGGTTGTGGGCATTGGTAGCTTTCTGTCTGTTGTCATTAATACTGGTTGGCTCAGCAGAGCAATGTGGAGGGCAAGCTGG
GGGTAGAGTTTGCCCAGGGGGGGCATGCTGCAGCAAGTTTGGTTGGTGTGGCAACACTGCTGATTACTGTGGCAGTGGCTGCCA
AAGCCAGTGCAGTTCCACTGGTGACATTGGCCAGCTTATTACCAGGTCCATGTTCAATGATATGCTTAAGCATAGAAATGAGGGG
AGTTGCCCTGGCAAGGGCTTCTACACCTATGACGCTTTCATAGCTGCTGCTAAGGCCTTTCCTGGCTTTGGAACAACTGGTGATAC
CACTACTCGTAAAAGGGAAATCGCAGCCTTCTTGGCTCAAACTTCTCATGAAACCACTGGGGGGTGGGCTAGTGCACCTGATGGC
CCATACGCTTGGGGATACTGCTACCTCAGGGAACAAGGCAGCCCCGGAGCTTACTGTGTTCCTAGTGCACAGTGGCCTTGTGCCG
CTGGTAGGAAATACTATGGCCGAGGCCCCATACAGATTTCCTACAACTACAACTATGGGCAAGCTGGGAAAGCCATAGGGGTAG
ACCTGGTAAACAACCCTGATCTAGTAGCAACAGATGCAGTCATATCATTCAAGACAGCCTTCTGGTTCTGGATGACACCCCAGTC
ACCCAAGCCTTCCTGCCATAATGTCATCACAGGAGGATGGACCCCATCAGGTGCAGATAGGTCAGCAGGGCGGCTTCCCGGTTTT
GGTGTTATCACAAACATCATCAATGGAGGTGTTGAATGTGGGAAAGGGGTAGTTCCTCAGGTCCAGGACCGCATAGGTTTCTATA
AGAGGTACTGTGATATACTTAGGGTTAGCTATGGCAATAACCTGGACTGCAACAACCAAAGGCCTTTCGGGTCTGGCCTCCTGCT
GGACACCATCTAAA3’
Vector de Clonagem
Gene LacZ
Ampicilina
Permite a selecção de
bactérias transformadas.
Apenas as bactérias
transformadas, ou seja,
que possuam o vector
plasmídico no seu interior
apresentam resistência a
este antibiótico.
MCS
Origem de replicação
Responsável pela iniciação da
síntese proteica.
Responsável pela síntese do
fragmento ω da enzima βgalactosidase que por
complementação com o fragmento
α sintetizado pelo cromossoma
bacteriano produz actividade da
enzima.
No meio da sequência deste gene é
conhecida uma zona com vários
locais de restrição identificados
(Multiple Cloning Site).
Qual a importância desta região?
Vector de Clonagem: Qual a importância da MCS?
à Os locais de restrição identificados nesta região são únicos no vector, o que
significa que as enzimas de restrição específicas deste locais apenas cortarão o
vector uma única vez.
à As extremidades que se geram no vector após digestão com uma determinada
enzima de restrição são compatíveis com as extremidades geradas num fragmento
digerido com a mesma enzima, permitindo assim a integração desse fragmento no
vector.
à Estando a região MCS inserida no meio da sequência do gene LacZ (responsável
pela síntese da enzima β-galactosidase e consequente expressão azul das colónias
bacterianas) a interrupção do gene por inserção de um fragmento, leva à inibição da
expressão do gene. As bactérias que possuam plasmídio com um fragmento
inserido na região MCS (transformadas recombinantes) possuem coloração branca.
Transformação de bactérias
Introdução de um vector plasmídico, integrando o gene de interesse,
em bactérias desprovidas de qualquer DNA desta natureza (bactérias
competentes).
O objectivo desta técnica consiste obter milhares de cópias do vector
introduzido na bactéria e assim do gene inserido neste.
Screening de bactérias recombinantes com enzimas de restrição
O screening de bactérias recombinantes portadoras do fragmento de interesse pode ser efectuados de
duas formas:
1. Por PCR utilizando os primers específicos do gene ou do vector;
2. Por digestão com enzimas de restrição que flanqueiem o local de inserção do gene.
Neste caso é necessário a incubação prévia das colónias seleccionadas em LB líquido para posterior
extracção do DNA plasmídico (protocolo da aula). Somente após esta extracção é que se poderá
efectuar a digestão do DNA utilizando enzimas de restrição específicas da zona MCS (podendo ser
apenas uma enzima com dois locais de restrição, um na zona 5’ do fragmento e outro na zona 3’ ou
duas enzimas diferentes).
Mix Digestão:
1. DNA plasmídico extraido ………. 10µl
2. Buffer (10x) …………………….. 2µl
3. Enzima 5’ ………………………. 0.3µl
4. Enzima 3’ …………………….… 0.3µl
5. H2O Milli-Q estéril ………….…. 7.4µl
20µl
Incubar 2-3h a xºC (a temperatura óptima
varia com as enzimas, sendo na maior parte dos
caso 37ºC)
Screening de bactérias recombinantes com enzimas de restrição
1.
MM
A reacção de digestão não foi completa,
pois observam-se no gel bandas
correspondentes a formas circulares e
superenroladas;
2.
Apenas as colónias 2 e 4 apresentam o
fragmento de interesse (1000 pb);
3.
A colónia 1 apresenta um fragmento
clonado de peso inferior ao desejado;
4.
A colónia 3 apesar de apresentar uma
banda correspondente a um fragmento
de 1000 pb, apresenta uma outra banda
correspondente a um fragmento de peso
inferior a 100 pb. Como o vector não
possui mais locais de restrição para as
enzimas utilizadas, as duas bandas
corresponderão a um fragmento de peso
total de 1050pb que possui um local de
restrição para uma das enzimas
utilizadas a 50 pb de uma extremidade.
cl.1
cl.2
cl.3
cl.4
Polissacáridos
DNA circular com nicks
DNA linear
1500 pb
DNA circular superenrolado
600 pb
Fragmento linear digerido
100 pb
SacI
5’
(0pb)
KpnI
3’
(1000pb)
Alguns Mecanismos de Transferência de genes de
interesse
Construção de Vectores de Expressão
Agrobacterium
Bombardeamento de Partículas
Mecanismos de selecção de transgénicos
Melhoramento Clássico vs. Biotecnologia
Melhoramento genético: potencialidades, benefícios e
riscos
Construção de Vector de Expressão
Um vector de expressão corresponde a um vector plasmídico integrando uma sequência
denominada “cassete de expressão”- sequência de DNA linear que inclui para além dos genes,
um promotor e um terminador que controlam a expressão de cada gene em particular. Na
“cassete de expressão” podem existir:
a) apenas o gene de interesse (ex.: chitinase), não existindo forma de selecção;
b) o gene de interesse e o gene de selecção (resistência a antibiótico ou herbicida);
c) o gene de interesse, o gene de selecção e um gene repórter:
- β-glucuronidase (GUS);
- Green Fluorescent Proteins (GFP).
CHITINASE
Vitis rupestris
GUS
pHKTL1
(12.020 Kb)
Agrobacterium tumefaciens
É uma bactéria que existe naturalmente
no solo infectando um grande número
de dicotiledóneas.
Penetra nas raízes através de feridas
provocando uma reacção no tecido que
se traduz numa multiplicação anormal
das células dando origem a tumores.
Os tumores resultam da transferência, integração e expressão nas células da planta de
um segmento específico do DNA bacteriano designado de T-DNA (DNA transferido).
Esta capacidade faz da bactéria A. tumefaciens um instrumento natural de engenharia
genética.
O T-DNA constitui parte do plasmídio Ti (tumour inducing) que ocorre naturalmente nas
células de A. tumefaciens e é responsável pela capacidade infecciosa da bactéria.
A forma de explorar a capacidade do A.
tumefaciens consiste em:
alterar a zona de DNA que é
transferida (T-DNA), eliminando os genes
tumorais e o gene para a opina (aminoácido
modificado utilizado como fonte de carbono e
azoto pela bactéria), substituindo-os pelo gene ou
genes que se pretendem introduzir;
as sequências dos limites são os
únicos elementos que é necessário manter (LE e
LD) permitindo a transferência da sequência do
T-DNA para as células do hospedeiro.
Mapa genético do plasmídio Ti.
Devido às grandes dificuldades em manipular grandes moléculas de DNA como o
plasmídeo Ti, foram desenvolvidos vectores em que toda a manipulação é feita
recorrendo à Escherichia coli.
Esquema do plasmídeo Ti utilizado para transformação de
células vegetais mediada por A. tumefaciens.
Etapas da Transformação Genética mediada por Agrobacterium:
1. O vector plasmídico Ti retirado da
bactéria e manipulado por alteração a
região T-DNA. Esta manipulação
envolve enzimas de restrição específicas
que permitem a digestão do vector em
zonas conhecidas;
3. Transformação da estirpe de bactéria
Agrobacterium com o vector Ti
recombinante, originando bactérias
recombinantes;
Genomicc DNA
Genomic DNA
Ti plasmid
2. A digestão do vector permite a
inserção nessas zonas dos genes de
interesse (flanqueados por um promotor
e um terminador): vector recombinante;
Vitis
rupestris
cells
Vitis
rupestris
cells
Plant
cell
Agrobacterium
(carries the gene of
interest)
interest
Restriction
enzyme A
Restriction
enzyme A
+
Empty
plasmid
Gene of
interest : chitinase
Ti plasmid with the gene of interest
4. Infecção (co-cultura que decorre em média
2 dias) do hospedeiro com transferência do
vector Ti para as células desse (o material
vegetal utilizado depende do sistema de
regeneração seguido);
5. Transferência da região T-DNA (suporta os
genes de interesse) do vector Ti da bactéria
para o genoma do hospedeiro;
6. Integração do T-DNA no genoma da planta:
células transformadas;
Agrobacteriumtumefaciens
Ti plasmid with the newgene
cell’s
DNA
+
Agrobacterium
Plant cell
The new
gene
7. Multiplicação da região T-DNA integrada
no genoma da planta originando uma nova
planta integrando esse(s) gene(s): planta
transgénica.
8. Expressão dos genes introduzidos no
hospedeiro quando sob a condição de stress
que se tentou combater.
Transformation
Transgenic plant
Cell division
Selecção de Plantas Transgénicas
Calli embriogénicos transformados
A análise histoquímica com
X-Gluc permite verificar a
eficiência da transformação.
A área azul corresponde a
zonas onde ocorreu a
integração do gene GUS e
sua expressão com síntese da
proteina β-glucoronidase.
A regeneração de plantas
(embriogénese somática) a partir de
zonas integrando o genes GUS origina
plantas transformadas. A análise
histoquímica realizada em plantas
permite detectar por expressão da
coloração azul as plantas que integram
este gene repórter.
Limitações da Técnica
9 Apesar de ser utilizado com sucesso na transformação de muitas dicotiledóneas,
apresenta como grande limitação: a reduzida capacidade na transformação de
monocotiledóneas.
9 Na tentativa de incrementar esta capacidade de transformação testaram-se diferentes
estirpes de Agrobacterium, condições fisiológicas da bactéria e hospedeiro, bem como
técnicas de co-cultura (Godwin et al., 1992), não se conseguindo ainda assim
resultados favoráveis.
9 A transformação de monocotiledóneas requer um sistema alternativo de
transformação genética, como por exemplo o bombardemanto de particulas ou a fusão
de protoplastos.
9 A integração de genes pelo Agrobacterium dá-se unicamente ao nível do DNA
genómico do hospedeiro. Caso se pretenda introduzir informação noutro qualquer
organelo celular, terá de se recorrer a outras técnicas, como por exemplo o
bombardeamento de partículas.
Bombardeamento de Partículas, “Gen Gun” ou Biolística
Métodologia que permite transformar geneticamente plantas de espécies que o
Agrobacterium não infecta (como por exemplo o arroz, o milho, a cevada, etc.).
A técnica basea-se em:
projectar a uma pressão elevada (variando esta com o material vegetal em causa, para
calli de videira é utilizada uma pressão entre 1100 e 1550 psi) partículas de ouro ou
tungsténio de diâmetro variável (1.00µm) com o DNA (vector plasmídico com a
cassete de expressão ou apenas a cassete de expressão) agregado.
A força mecânica gerada por uma velocidade elevada permite que sejam quebradas barreiras
biológicas, podendo ocorrer integração do DNA estranho no genoma das células
bombardeadas (DNA nuclear). Para além deste, esta técnica possibilita a integração ao
nível do genoma dos cloroplastos (Ye et al., 1990; Daniell, 1993) e das mitocondrias.
A aplicação desta técnica requer:
- uma preparação prévia muito cuidada;
- DNA circular ou linear de boa qualidade;
- existência de um protocolo de regeneração optimizado.
O aparelho utilizado:
Câmara de Bombardeamento
Suporte do disco de ruptura (1)
Estrutura onde se colocam os
macrocarriers, respectivos
suportes e stopping screen (2)
Suporte do material Vegetal (3)
O disco de ruptura determina a pressão à qual serão projectadas as
partículas com o DNA agregado.
Existem diferentes discos de acordo com a pressão que suportam: um
disco de 1550psi significa que suporta pressões inferiores a esta e que
ao atingir este valor rompe, permitindo que as partículas sejam
projectadas sob o material vegetal.
(1)
Disco de ruptura
Suporte do disco de ruptura
(2)
Suportes do Macrocarrier
Tampa
Macrocarrier
Stopping Screen
(3)
O material vegetal utilizado no bombardeamento de partículas depende do sistema de
regeneração utilizado para a espécie vegetal que se pretende transformar, podendo ser
calli, embriões, discos foliares, etc.
Análise histoquímica 2 dias após o bombardeamento permite determinar o sucesso da
transformação. O número de pontos azuis corresponde aos pontos em que ocorreu integração do
gene GUS e sua expressão.
As primeiras células do material vegetal a serem bombardeadas são normalmente destruídas, no
entanto, junto a essas existe uma área de células envolvente em que as partículas penetraram sem
as destruir. Algumas destas células vão sobreviver à agressão provocada pela ferida das partículas
e incorporar no genoma o DNA transportado. Após a incorporação do(s) gene(s) no genoma da
planta, as células podem expressá-lo com a consequente produção da proteína por ele codificada.
Vantagens relativamente ao Agrobacterium:
9 Possível utilização em dicotiledóneas e monocotiledóneas;
9 Inexistência de falsos positivos, correspondentes a plantas não
transformadas mas que possuindo a bactéria utilizada como vector da
transformação, manifestam a presença do gene;
9 Necessidade de apenas um antibiótico para selecção das plantas
transformadas. A utilização de A. tumefaciens requer a utilização de
dois antibióticos, um para eliminar a bactéria e um outro para a
selecção das plantas transformadas.
Limitações da técnica:
9 Elevada destruição celular: as camadas celulares mais externas são denominadas de
“zona de morte” por serem destruídas devido ao impacto ou fragmentação das
partículas ou à rajada de ar produzida (Birch and Franks, 1991);
9 Baixa estabilidade dos transformante: apesar de ocorrer integração de um gene no
genoma de uma célula que poderá regenerar uma planta, esta integração não é estável,
apenas 1-5% das células expressam integração estável (Finer and McMullen, 1990);
9 Limitado na dimensão das construções genéticas: quanto maior o vector de expressão
menor a possibilidade de sucesso;
9 Permite repetições: existe a possibilidade de integrar várias vezes o mesmo gene no
genoma da célula vegetal, dando origem a repetições e consequente expressão
diferencial do mesmo gene;
9 Desconhecimento do efeito que a velocidade das partículas pode causar, bem como
da composição das mesmas, partículas de tungsténio podem oxidar resultando tóxicas
para as células.
Selecção de tecidos geneticamente transformados
Após transformação, mediada por Agrobacterium ou Bombardeamento de Partículas,
o material vegetal será transferido para meio de cultura suplementado com antibiótico ou
herbicida (depende do marcador de selecção utilizado. Ex.: se na cassete de expressão se
integrou o gene que confere resistência ao antibiótico higromicina, o agente de selecção
adicionado ao meio de cultura será este antibiótico).
Apenas as células expressando o gene de selecção conseguem sobreviver ao agente
utilizado originando plantas resistentes a esse. Assume-se que estas plantas, por possuírem o
gene de selecção possuem também o gene de interesse (ambos os genes foram integrados na
cassete de expressão utilizada).
A integração dos genes no genoma das
plantas requer a extracção de DNA
cromossomal das plantas
putativamente transgénicas e sua
análise por PCR.
Na reacção de amplificação são utilizados
primers específicos que permitem amplificar
um fragmento do gene de interesse, ou
qualquer zona presente na “cassete de
expressão” (zona dos promotores,
terminadores, gene de selecção, repórter ou
de interesse).
O resultado do PCR é observado por
electroforese, onde a separação dos
fragmentos de DNA ocorre de acordo com o
seu peso molecular, através de uma corrente
eléctrica, migrando do pólo negativo para o
positivo.
Hygromycin
(HPTII)
β-glucuronidase
(GUS)
A obtenção de resistência a determinadas condições de stress biótico ou abiótico é
possível utilizando métodos tradicionais ou convencionais.
cruzamento entre espécies sensíveis (Vitis vinifera) e
espécies resistentes a esse stress (V. rupestris) originando híbridos, alguns dos quais
possuindo a característica desejada. No entanto, este método é muito moroso e não está
isento de forte controvérsia, já que os híbridos assim obtidos podem colocar em causa a
tipicidade do produto final.
A biotecnologia, através dos seus métodos de propagação e transformação genética,
apresenta grande potencial para o melhoramento de diversas espécies vegetais.
Esta metodologia apresenta, relativamente aos métodos de melhoramento
convencionais, as seguintes vantagens:
- melhoramento de grande precisão, em que apenas o gene pretendido é
introduzido no genoma da planta;
- redução do tempo de obtenção da nova variedade;
- inexistência de barreiras quanto à proveniência do gene responsável pela
característica que se pretende introduzir, podendo ter origem em qualquer outra planta.