FETUS-CENTRO DE ESTUDOS
PÓS-GRADUAÇÃO EM MEDICINA FETAL
SISTEMA NERVOSO CENTRAL
ANORMALIDADES DE ESTRUTURA DA LINHA MÉDIA
-AGENESIA DE CORPO CALOSO
-HOLOPROSENCEFALIA
CRISTINA LOPES RIBEIRO
SÃO PAULO,2011
CRISTINA LOPES RIBEIRO
SISTEMA NERVOSO CENTRAL
ANOMALIAS ESTRUTUTAIS DE LINHA MÉDIA
-AGENESIA DE CORPO CALOSO
-HOLOPROSENCEFALIA
MONOGRAFIA APRESENTADA PARA POSGRADUAÇÃO EM MEDICINA FETAL DO
CENTRO DE ESTUDOS –FETUS, COMO
\
REQUISITO PARCIAL PARA CONCLUSÃO
DO CURSO.
ORIENTADOR: Dr EDUARDO V. ISFER
SÃO PAULO, 2011
DEDICATÓRIA
Dedico esta monografia, primeiramente, a
Deus, pelo dom que recebi e pela nova chance
de dedicar-me a minha profissão, a meus pais
que me proporcionaram a oportunidade de
estar aqui, pelo incentivo de lutar pelos meus
ideais e principalmente por sempre
acreditarem e confiarem em mim. Ao meu
marido que mesmo distante, sempre esteve
presente em todos os momentos, aos meus
filhos pelo amor incondicional e compreensão
pelos períodos de ausência.
RESUMO
As anomalias de linha média, aqui representadas pela Agenesia de Corpo Caloso e a
Holoprosencefalia, ocorrem devido a uma falha de clivagem durante o período
embrionário.
Atualmente com o advento da ultra-sonografia, principalmente na melhora significativa
na qualidade dos equipamentos com conseqüência da imagem fetal, tornou-se
rotineiro e fundamental o diagnóstico pré-natal.
Este diagnóstico possibilita a equipe médica e multidisciplinar escolher a melhor
conduta e orientar os pais e familiares sobre o prognóstico, possíveis tratamentos
neonatais e aconselhamento genético visando gestações futuras.
PALAVRAS CHAVES:
AGENESIA DO CORPO CALOSO, HOLOPROSENCEFALIA E ULTRA-SOM.
1
SUMÁRIO
I. OBJETIVO...............................................................................03
II. INTRODUÇÃO........................................................................04
III. CONCEITO............................................................................06
IV. AGENESIA DO CORPO CALOSO
- INCIDÊNCIA...........................................................................14
- ETIOPATOGÊNIA..................................................................14
- CLASSIFICAÇÃO...................................................................15
- ANOMALIAS ASSOCIADAS..................................................17
- DIAGNÓSTICO PRÉ-NATAL.................................................18
- DIAGNÓSTICO DIFERENCIAL.............................................21
- CONDUTA PRÉ-NATAL........................................................22
- CONDUTA OBSTÉTRICA.....................................................23
- ASSISTÊNCIA NEONATAL...................................................23
- PROGNÓSTICO....................................................................24
V. HOLOPROSENCEFALIA
- INCIDÊNCIA..........................................................................26
- ETIOPATOGÊNIA..................................................................26
- CLASSIFICAÇÃO...................................................................28
- ANOMALIAS ASSOCIADAS..................................................32
2
- DIAGNÓSTICO PRÉ-NATAL.................................................36
- DIAGNÓSTICO DIFERENCIAL.............................................37
- CONDUTA PRÉ-NATAL........................................................38
- CONDUTA OBSTÉTRICA.....................................................38
- ASSISTÊNCIA NEONATAL...................................................39
- PROGNÓSTICO....................................................................40
VI. CONCLUSÃO..........................................................................42
VII.REFERÊNCIAS BIBLIOGRÁFICAS........................................43
3
OBJETIVO
Este estudo tem como objetivo a realização de revisão bibliográfica sobre as
anomalias estruturais da linha média, em especial a Agenesia do Corpo Caloso
e a Holoprosencefalia.
O conhecimento dos médicos em geral, principalmente dos obstetras, fetólogos
e pediatras é de fundamental importância, pois permite que estes profissionais
possam fazer o diagnóstico adequado, classificar estas patologias, para
posteriormente escolher a melhor conduta e principalmente orientar os pais e
familiares.
Todos sabemos que no advento de uma gestação, os pais sempre idealizam
a criança perfeita, sendo assim possuem uma certa ansiedade em relação ao
desenvolvimento motor e intelectual da mesma. Este fato se agrava quando é
realizado qualquer diagnóstico de alteração do sistema nervoso central.
Assim sendo o diagnóstico preciso da anormalidade e o conhecimento dos
fatores que implicam estas patologias, possibilitam um atendimento
diferenciado a estas gestantes e uma abordagem mais detalhada, bem como
acompanhamento por uma equipe multidisciplinar, sem esquecer da
necessidade de aconselhamento genético visando futuras gestações.
4
INTRODUÇÃO
A ultra-sonografia vem sendo utilizada para estudo fetal desde a década de 60,
onde foi largamente utilizada por obstetras e radiologistas. ( Isfer, 1996 e
Mauad, 2006 ).
Sabe-se que nas últimas duas décadas houve um grande aprimoramento da
ultra-sonografia obstétrica, devido a melhora dos equipamentos ( Doppler,
ultra-som tridimensional e melhora na metodologia ), aumento no treinamento
e experiência dos profissionais que realizam estes exames.
Atualmente o ultra-som tornou-se um exame de rotina durante o pré-natal,
inclusive na rede pública de saúde, onde o acesso das gestante a este
método aumentou significativamente juntamente com a criação de protocolos
de atendimento à gestantes. Hoje o ultra-som é o único método não invasivo e
direto, onde o feto pode ser avaliado, fazendo com que o obstetra deixe de
ter uma conduta contemplativa e passe a avaliar e a buscar possíveis
anormalidades e/ou alterações nos compartimentos fetais, maternos e
placentários.
O primeiro diagnóstico de malformação fetal foi realizado por Sudem em 1964,
e se tratava de uma anomalia do sistema nervoso central, a anencefalia. Desde
então entre os diagnósticos mais importantes de avaliação fetal estão as
anomalias neurológicas, pois são responsáveis por aproximadamente por 50%
das anomalias estruturais diagnosticada no período pré-natal, e possuem um
índice de 90-95% dos casos de diagnóstico de malformação neurológica
diagnosticado no exame ultra-sonográfico de rotina. ( Isfer, 1996) .
A sensibilidade diagnóstica ultra-sonográfica na detecção de holoprosencefalia
5
segundo Mauad, 2006, é de 89%.
A acurácia da ultra-sonografia no diagnóstico de malformações fetais depende
da população estudada; assim, a sensibilidade no diagnóstico das anomalias
fetais em gestações de baixo risco varia de 14 a 85% e a especificidade, de 93
a 99%. Por outro lado, quando a avaliação é feita em gestantes de alto risco, a
sensibilidade do método varia de 27 a 99%, com especificidade de 96 a 100%
( Park, 2001 ).
A sistematização do exame morfológico fetal deve obedecer a uma seqüência
metodológica, de forma a permitir uma completa avaliação do feto, sugere-se
iniciar o exame pela avaliação da cabeça e da face, em seqüência descreverei
a metodologia adequada para a avaliação e detecção de anomalias do sistema
nervoso central e da face :
- cabeça/face : diâmetro biparietal , diâmetro occipto-frontal, circunferência
craniana, diâmetro interorbitário, ventrículo lateral, plexo coróide, fossa
posterior, cerebelo, vérmis, corpo caloso, pálpebras, narinas, maxilares,
germes dentários, palato, língua, orofaringe, orelhas e perfil ( Costa, 2005 ).
Devendo seguir com o exame detalhado de todo o feto.
A ultra-sonografia é o método ideal para orientar o clínico em relação a idade
gestacional, bem-estar e anatomia fetal, destaca-se pela inocuidade, baixo
custo e alta capacidade diagnóstica, portanto é um método propedêutico que
inegavelmente, trouxe vários benefícios para a obstetrícia moderna, sendo um
dos principais métodos de avaliação fetal o que possibilitou a avaliação
detalhada das estruturas fetais, possibilitando o diagnóstico precoce de
anomalias fetais e possibilitando o tratamento do feto como um paciente.
Por estes fatos a ultra-sonografia tornou-se obrigatória no acompanhamento de
rotina pré-natal, possibilitando o diagnóstico de malformações do sistema
nervoso central, neste caso da holoprosencefalia, da agenesia do corpo
6
caloso e de outras malformações associadas.
È inegável que após avaliação detalhada do feto malformado o prognóstico
poderá ser traçado de maneira mais adequada, favorecendo além dos pais e
familiares, todos os profissionais envolvidos no acompanhamento do caso,
como obstetras, fetólogos, neonatologistas, cirurgião pediátrico, cirurgião
plástico, fonoaudiólogo, ortodontista, neurologista, psicólogo, etc).
CONCEITO
Para iniciarmos o estudo sobre as anormalidades da linha média precisamos
relembrar sobre o desenvolvimento embrionário do sistema nervoso central .
Sabemos que a Agenesia do Corpo Caloso é uma anomalia congênita,
motivada por um defeito migratório telencefálico que pode ser parcial ou
completo ( Maranhão, 2010) e que a Holoprosencefalia é uma malformação
complexa e rara que acomete o sistema nervoso central na sua embriogênese,
no qual durante o desenvolvimento cerebral ocorre comprometimento dos seus
estágios iniciais de diferenciação ( Massonetto, 2003 ).
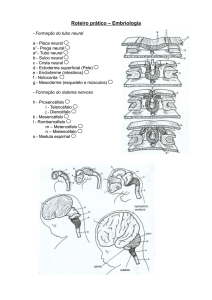
EMBRIOLOGIA, DIVISÕES e ORGANIZAÇÃO GERAL do SISTEMA NERVOSO
O estudo do desenvolvimento embrionário do sistema nervoso é muito
importante pois nos permite entender muitos aspectos da sua anatomia. Muitos
termos largamente utilizados para demonstrar partes do encéfalo adulto
baseiam-se na embriologia.
7
.
Fig 1: Fonte goianoycolombia.blogspot.com
Origem do Sistema Nervoso
Dos três folhetos embrionários é o ectoderma que está em contato com o meio
externo ( sabe-se que durante a evolução, os primeiros neurônios surgiram na
superfície externa dos organismos, fato este significativo, tendo em vista a
função primordial do sistema nervoso de relacionar o animal com o ambiente )
e é deste folheto que se origina o sistema nervoso.
O primeiro indício de formação do sistema nervoso consiste em um
espessamento do ectoderma, situado acima da notocorda, formando a
chamada placa neural.
Sabe-se que para a formação desta placa e a subseqüente formação e
desenvolvimento do tubo neural, tem importante papel, a ação indutora da
notocorda e do mesoderma.
A placa neural cresce progressivamente, torna-se mais espessa e adquire um
sulco longitudinal denominado sulco neural, que se aprofunda para formar a
8
goteira neural, desenvolvendo as células que formam de cada lado uma lâmina
longitudinal denominada crista neural, situada dorsalmente ao tubo neural .
O tubo neural dá origem a elementos do sistema nervoso central, enquanto a
crista neural dá origem a elementos do sistema nervoso periférico.
Tubo Neural
O fechamento da goteira neural e, concomitantemente, a fusão do ectoderma
não diferenciado é um processo que se inicia no meio da goteira neural e é
mais lento nas suas extremidades. Assim, em uma determinada idade, temos
um tubo neural no meio do embrião e a goteira nas extremidades.
Mesmo em fases mais adiantadas, permanecem nas extremidades cranial e
caudal do embrião dois pequenos orifícios , que são denominados,
respectivamente, neuróporo rostral e neuróporo caudal. Estas são as últimas
partes do sistema nervosa à se fecharem.
9
Fig 2: Fonte /www.forp.usp.br/mef/embriologia/geral.htm
Dilatações do Tubo Neural
Desde o início da sua formação, o calibre do tubo neural não é uniforme. A
parte cranial, que dá origem ao encéfalo adulto, torna-se dilatado e constitui o
encéfalo primitivo, ou arquencéfalo, a parte caudal dá origem à medula do
adulto, permanece com calibre uniforme e constitui a medula primitiva do
embrião.
10
Dilatação do tubo neural:
Fig3. Fonte:cienciasmorfologicas2.blogspot.com
No arquencéfalo distinguem-se inicialmente três dilatações, que são as
vesículas encefálicas primordiais denominadas : prosencéfalo, mesencéfalo e
rombencéfalo.
Com o subseqüente desenvolvimento do embrião, o prosencéfalo dá origem a
duas vesículas , telencéfalo e diencéfalo. O mesencéfalo não se modifica, e o
rombencéfalo origina o metencéfalo e o mielencéfalo.
11
Fig 4: Encefalon. Fonte: pt.wikipedia.org
Fig: 5- Embrião 4 semanas: Fonte Google imagens.
12
- telencéfalo
- prosencéfalo
Dilatações
do tubo
neural
-arquiencéfalo
- mesencéfalo
-medula
primitiva
- rombencéfalo
- diencéfalo
- metencéfalo
- mielencéfalo
Fig. 6: Embriologia e divisão do sistema nervoso central- Neuroanatomia Funcional,
2 edição- Ângelo Machado.
O telencéfalo compreende uma parte mediana, da qual se invaginam duas
porções laterais, as vesículas telencefálicas laterais. A parte mediana é
fechada anteriormente por uma lâmina que constitui uma porção mais cranial
do sistema nervoso e se denomina lâmina terminal. As vesículas telencefálicas
laterais crescem muito para formar os hemisférios cerebrais e escondem
quase completamente a parte mediana e o diencéfalo.
O diencéfalo apresenta quatro pequenos divertículos: dois laterais, as vesículas
ópticas, que formam a retina ; um dorsal, que forma a glândula pienal; e um
ventral, o unfundíbulo que forma a neuro-hipófise.
Cavidade do Tubo Neural
A luz do tubo neural permanece no sistema nervoso do adulto, sofrendo em
algumas partes, vária modificações. A cavidade dilatada do rombencéfalo
forma o IV ventrículo. A cavidade do diencéfalo e a parte mediana do
diencéfalo formam o III ventrículo. A luz do mesencéfalo permanece estreita e
13
constitui o arqueoduto de Sylvius, que une o III ao IV ventrículo.
A luz das vesículas telencefálicas laterais formam, de cada lado, os ventrículos
laterais, unidos ao III ventrículo pelos dois forames interventriculares ( ou de
Monro ).
O corpo caloso se desenvolve entre a 12ª e a 22ª semana de gestação. A
ruptura ou falha na formação vascular pode ocasionar a agenesia completa ou
parcial.
Entre a 4ª e a 8ª semana de desenvolvimento embrionário ocorre a
segmentação normal dos hemisférios cerebrais e do diencéfalo, também
chamados de diverticulação. Quando existe falha da separação do prosencéfalo
e do telencéfalo que formam os hemisférios cerebrais, e do diencéfalo, que
forma o tálamo e o hipotálamo, podemos observar o formato final da vesívcula
prosencefálica, que não apresentou seu desenvolvimento normal,
segmentanto os hemisférios cerebrais., conhecido como Holoprosencefalia.
14
ANORMALIDADES DE ESTRUTURA DA LINHA MÉDIA
AGENESIA DO CORPO CALOSO
INCIDÊNCIA
A prevalência da agenesia do corpo caloso estimada é de 1% da população
( Sanders et al, 1999 ) e ( Santos et al, 2008 ).
Sabe-se que a agenesia de corpo caloso é uma malformação relativamente
freqüente, atingindo taxa de 0,7% dos casos examinados de por
pneumoencefalografia. Sua freqüência real ainda é desconhecida, pois existem
casos assintomáticos. Os achados anatomopatológicos mostram incidência
maior em relação aos diagnósticos tomográficos realizados na infância e na
vida adulta ( Isfer, 1996 ).
ETIOPATOGÊNIA
O corpo caloso, a maior das comissuras inter-hemisféricas, é formado por um
grande número de fibras mielínicas que cruzam o plano sagital mediano e
penetram em cada lado no centro branco medular do cérebro, unindo áreas
simétricas do córtex cerebral de cada hemisfério. Emergindo abaixo do
esplênio do corpo caloso e arqueando-se em direção da comissura anterior,
esta o fórnix, feixe complexo de fibras. Entre o corpo caloso e o fórnix estendese até o septo pelúcido, constituindo-se por duas delgadas lâminas de tecido
nervoso que delimitam o seu cavum ( cavidade do septo pelúcido). O septo
15
pelúcido separa os dois ventrículos laterais.
O corpo caloso é uma estrutura encefálica mediana cujas as fibras nervosas
interconectam os hemisféricos cerebrais, permitindo troca de informações e a
divisão do aprendizado e da memória entre os membros, portanto responsável
pela função de aprendizado e memória.. Desenvolve-se entre a 12ª a 18ª
semana de gestação.
A agenesia completa se dá quando o fator desencadeante, freqüentemente
eventos hipóxico-isquêmicos, ocorre precocemente na gestação, no período
embrionário. A agenesia parcial ocorre tardiamente no desenvolvimento e pode
afetar a porção anterior ou posterior do corpo caloso. A ausência parcial
anterior é provavelmente causada por lesões destrutivas, inflamatória e
anóxicas, enquanto a ausência posterior é resultado de agenesia focal ( Mauad,
2006).
A ruptura ou falha na formação da vascular pode ocasionar agenesia completa
ou parcial ( Sanders, 1999).
CLASSIFICAÇÃO:
A Agenesia do Corpo Caloso pode ser classificada em três grupos:
1) Sem envolvimento de outras áreas do cérebro;
2) Associada a outras disgenesias telencefálicas;
3) Fazendo parte de uma síndrome.
A etiologia é heterogênea e inclui:
1) Anormalidades citogenéticas:
2) Metabólicas (erros inatos do metabolismo) ;
3) Síndromes genéticas;
16
4) síndrome alcoólica fetal.
TERATOGÊNICOS
Não há relados de nenhum teratogênico conhecido.
Mas há relatos de que crianças expostas ao álcool intra-útero, apresentam
alterações do neurodesenvolvimento do sistema nervoso central, que se
manifestam como alterações estruturais do cérebro, incluindo a Agenesia
do Corpo Caloso, fazendo com que esta seja uma das anomalias mais
freqüentes na Síndrome Alcoólica Fetal, podendo acometer cerca de 6%
destas crianças
Por razões éticas, não é possível a realização de estudos experimentais para
avaliar a ação do álcool em mulheres grávidas. Estudos em aves e em animais
foram realizados para avaliar os efeitos teratogênicos do álcool e também mais
detalhadamente , in vitro, por meio de culturas de determinados tecidos e
células.
Em diferentes fases da gravidez, o álcool retardou a maturação de áreas
craniofaciais de embriões de camundongos, devido ao dano celular ao longo da
borda da prega neural anterior ( Mesquita, 2005 ).
Existem, ainda relatos associando a agenesia de corpo caloso fetal com
infecção congênita ( toxoplasmose e rubéola ), esclerose tuberosa e
mucopolissacoridose.
17
CARACTERÍSTICAS HEREDITÁRIAS
A maioria dos defeitos isolados é esporádica. As síndromes autossômicas
dominantes, recessivas e ligadas ao X têm sido descritas.
A agenesia de corpo caloso pode estar associada a síndromes cromossômicas
como as trissomias 13 e 18 ( como parte integrante da seqüência da
holoprosencefalia ) e translocações ( Sanders, 1999 ) e ( Isfer, 1996 ).
ANOMALIAS ASSOCIADAS
A agenesia de corpo caloso é rara, e consiste em um defeito do
desenvolvimento da comissura inter-hemisférica podendo ser total ou parcial,
manifestando-se como uma forma particular de hidrocefalia . Pode ocorrer
isolada ou associada a outras anomalias.
Entre estas incluem-se:
- Dismorfismo facial em 30% a 45% dos casos ( microftalmia, hipertelorismo,
fenda palatina );
- Outras malformações em 45% dos casos, tais como anomalias vertebrais
( vértebras em bloco, hemivértebras ) e vísceras ( genito-urinárias e
cardíacas ).
- Lipomas de linha média;
- Cistos inter-hemisféricos;
- Malformação de Dandy-Walker, a investigação da fossa posterior deve também ser
detalhada, pois a associação com esta síndrome não é rara.
- Malformação de Arnold-Chiari tipo II;
- Heterotopias;
- Porencefalias;
18
- Anomalias cardíacas;
- Anomalias genitourinárias;
Mais de oitenta síndromes cromossômica, genéticas e esporádicas tem sido
descritas com agenesia do corpo caloso, incluindo as trissomias do 13 e do 18.
DIAGNÓSTICO PRÉ-NATAL
O diagnóstico no ultra-som é geralmente tardio. Antes da 26ª semana a
agenesia de corpo caloso total pode ser suspeitada frente a sinais indiretos,
são eles :
- a não identificação do cavum do septo pelúcido;
- a ausência ou interrupção do eco médio, isolada ou associada a hidrocefalia;
- presença de malformações faciais ou vertebrais.
A partir da 26ª semana de gestação, a distenção moderada do III ventrículo,
em corte transverso ( diâmetro biparietal , e o afastamento dos ventrículos
laterais obrigam o ultra-sonografista a praticar outros cortes ( coronal anterior e
sagital mediano ) , os quais podem revelar as seguintes alterações:
- expansão ascendente do III ventrículo, interposto entre os ventrículos laterais;
-afastamento anormal dos cornos frontais e dos corpos dos ventrículos laterais,
cuja parede interna torna-se côncava em sua região superior (aspecto de
cabeça de touro);
-ausência de visibilidade total ou parcial do corpo caloso;
- disposição radial e espessamento característico dos sulcos da face interna
dos hemisférios em torno dos ventrículos.
Após a 34ª semana, a dilatação moderada dos cruzamentos ventriculares e o
alargamento dos cornos occipitais contrastam com os cornos frontais
19
adelgaçados.
Esses sinais indiretos podem ser os únicos a alertar sobre a existência de uma
possível agenesia de corpo caloso parcial.
O diagnostico de ACC requer exame de imagem, sabe-se o fato que o exame
de ultra-sonografia reconhece a condição já a partir de vigésima semana de
gestação e a deixar de salientar que a ressonância magnética (RM) é o padrão
ouro, revelando aspectos considerados clássicos, especialmente na fase
neonatal, mas atualmente sendo usada para diagnóstico pré-natal através da
avaliação fetal.
O corpo caloso geralmente não é visualizado através dos cortes habituais da
ultra-sonografia, assim o seu diagnóstico deve ser considerado ante a
presença de outros sinais ecográficos indiretos, como a elevação do terceiro
ventrículo , a septação dos ventrículos laterais, a configuração em lágrima dos
ventrículos laterais ( causada pelo aumento do diâmetro dos cornos posteriores
do mesmo ) e a ausência do cavum do septo pelúcido. Em cortes sagitais , o
uso do mapeamento Doppler para avaliação da artéria pericalosa pode auxiliar
no seu diagnóstico.
20
Fig 07: RNM Crânio-Agenesia do Corpo Caloso.
Fonte: Neurosanjuan.com.ar
Fig 08: RNM Agenesia Corpo Caloso, Fonte: Enscer.com.br
21
Fig 09 : Ultra-som, corte sagital, ausência de corpo caloso.
Fonte: Centrus.com.br
DIAGNÓSTICO DIFERENCIAL
- Holprosencefalia lobar pois os ventrículos laterais e o terceiro ventrículo
aparentemente estão unidos. A foice não é visualizada, pois não está presente
na linha mediana e os ventrículos laterais estão separados;
- Cisto Aracnóide, apresentando-se de forma irregular e dificilmente visualizado
na linha média, em geral não observa-se dilatação ventricular;
- Ventrículomegalia Lateral Moderada, onde todo o ventrículo está dilatado e o
terceiro ventrículo está moderadamente dilatado e na posição normal.
Atenção:
- A síndrome de Dandy-Walker é a associação mais comum conforme já foi
22
relatado, nesta situação o vérmis cerebelar encontra-se menor que o esperado
e estará circundado por um quarto cisto ventricular;
- Na encefalocele e na mielomeningocele a cisterna magna não será visível e o
cerebelo estará “achatado”, descrito como o sinal da “banana”;
- A Síndrome de Aicardi ocorre apenas em fetos do sexo masculino e pode
estar associado a anomalias vertebrais;
- Quando houver suspeita de anomalias cromossômicas, como trissomias do 8,
13 e 18, procurar os estigmas próprios destas anomalias;
-deve-se sempre pesquisar se há existência de hérnias diafragmáticas,
anomalias cardíacas, agenesia ou displasia do pulmão, agenesia renal ou rim
displásico.
CONDUTA PRÉ-NATAL
A conduta pré-natal deve ser realizada de forma adequada, de maneira
individualizada e sem esquecer de realizar uma orientação adequada à
gestante e aos familiares, sem esquecer de solicitar um cariótipo fetal, pois é
essencial que seja feito o estudo cromossômico. Também devemos fazer uma
busca minuciosa da morfologia fetal para diagnosticar anomalias associadas,
para tanto é importante associar a avaliação morfológica fetal e a ecocardiografia
fetal está indicada .
Como as infecções congênitas podem ser as causadoras desta anomalia, a
sorologia para toxoplasmose, rubéola, citomegalovírus e do vírus da herpes
simples devem ser realizados através do sangue materno.
As consultas adicionais ao pré-natal de baixo risco vão depender das
alterações e/ou patologias associadas.
23
CONDUTA OBSTÉTRICA
Não existe indicação de alteração da conduta obstétrica quando a Agenesia do
Corpo Caloso cursa de forma isolada, não devendo ocorrer complicações nesta
situação.
Sempre que houver associação com outras alterações deverá haver mudança
na conduta .
Preferencialmente o parto deverá ocorrer em um centro de atenção terciária,
com amplos recursos para diagnóstico e controle dos recém-nascidos, em
especial aqueles que possuem malformações múltiplas.
ASSISTÊNCIA NEONATAL
Geralmente, quando ha necessidade de ressuscitação, não tem sido relatado
nenhum problema específico. Mas em casos onde existam defeitos associados,
principalmente do sistema nervoso central, esta anomalia pode se tornar um
fator dominante e de piora do quadro clínico, sendo assim, as decisões
deverão ser tomadas em decorrência destas alterações.
Como já relatado o nascimento deverá ocorrer em um centro de atenção
terciário, pois o transporte não é indicado nestas situações.
Após o nascimento está indicado a confirmação do defeito e avaliação de
possíveis alterações associadas, tendo como padrão ouro a ressonância
magnética nuclear. Também é importante realizar um rastreamento dos
distúrbios metabólicos hereditários.
No berçário o desenvolvimento e o controle subseqüente são determinados
24
pela(s) lesões e/ou patologias associadas.
PROGNÓSTICO
O corpo caloso é uma estrutura filogeneticamente recente, assim sendo, sua
ausência não é essencial para as funções vitais ( Isfer, 1996 ).
A agenesia isolada pode ser compatível com bom desenvolvimento mental,
mesmo em casos onde pode estar associada a cisto inter-hemisférico.
Também pode apresentar de maneira assintomática, sendo descoberta apenas
em exame neurológico pela pesquisa de déficits sensitivos ( dificuldade para
diferenciação de formas de objetos, temperatura e peso ).
Por outro lado em caso de malformações cerebrais associadas, o prognóstico
neurológico torna-se sensivelmente comprometido.
Do ponto de vista clínico e prognóstico, pode-se descrevê-la da seguinte maneira:
1 - Macrocefalia com ou sem sinais neurológicos e/ou retardo mental;
2 - Síndrome de Aicardi ( convulsões, corioretinite lacunar, retardo mental,
microcefalia e anomalias vertebrais ). Acomete exclusivamente o sexo
feminino, sendo letal para o masculino. Esta síndrome é classificada como
autossômica dominante ligada ao sexo.
3- Normo ou microcefalia com sinais neurológicos de retardo mental.
As duas últimas formas possuem um péssimo prognóstico. Entretanto, a
primeira é a mais freqüente, cursando, de modo geral com prognóstico
reservado. Porém, existem casos descritos de intelecto normal.
Em vista destes aspectos, torna-se difícil propor uma decisão objetiva nos
casos de diagnóstico intra-útero de agenesia de corpo caloso, principalmente
quando se referem aos casos do tipo “1” ( descrito acima ) ou das formas
assintomáticas.
25
Em resumo, a importância do diagnóstico antenatal da agenesia do corpo caloso,
serve para diferenciar se esta é ou não decorrente de outras malformações e avaliar
anomalias associadas. Em caso de hidrocefalia, se for decorrente da agenesia de
corpo caloso isolada, à conduta obstétrica não difere da habitual.
Pacientes com agenesia do corpo caloso isolada são freqüentemente
assintomáticos, sendo assim, o seu prognóstico favorável,quando isolada e
assintomática, em até 80% dos pacientes, ou apresentar discreto déficit
neurológico. Os casos mais graves podem apresentar graus variados de
psicose, epilepsia ou retardo mental.
As anomalias associadas incluem hidrocefalia, microcefalia, paquigiria e
lissencefalia.
Os achados clínicos relatados são, principalmente, dificuldade no aprendizado
e epilepsia (> 40%). Mais recentemente vem sendo associada a várias
desordens neuropsiquiátricas, incluindo déficit de atenção, hiperatividade e
esquizofrenia.
26
HOLOPROSENCEFALIA
INCIDÊNCIA
Na Holoprosencefalia a prevalência ao nascimento é de 0,06% a 0,02/1.000,
sendo estimada em 4/1.000 nos embriões, também à relatos de 1/16.000
nascidos vivos e 1/200 abortos espontâneos, com maior predominância no
sexo feminino ( 3:1 ) ( Isfer, 1996 ). Podendo ser descrito mais detalhadamente
como para cada feto do sexo masculino ocorrem três do sexo feminino na
holoprosencefalia alobar e de maneira igual na forma lobar.
Apresenta-se de forma freqüente nos embriões humanos provenientes de
abortamento espontâneo, atingindo a proporção de 1 caso para cada 250
abortos espontâneos ( Castro, 2000 ) e podendo estar presente em até 4% dos
casos ( Isfer, 1996 ), sendo esta incidência 60 vezes maior que a encontrada
em neonatos.
ETIOPATOGENIA
O termo holoprosencefalia deriva do aspecto final apresentado pela condição,
o qual nos remete ao formato final da vesícula prosencefálica, que não
apresentou o seu desenvolvimento normal segmentando-se nos hemisféricos
cerebrais e diencéfalo, também chamados de distúrbio de diverticulação. Nesta
situação podemos afirmar que consiste em uma malformação da linha média,
caracterizada pela falha da separação entre o prosencéfalo eo telencéfalo, que
formam os hemisférios cerebrais, e o diencéfalo, que forma o tálamo e o
hipotálamo.
27
Este evento ocorre entre a 4ª e a 8ª semana de desenvolvimento embrionário.
A gravidade da lesão depende do grau de separação dos hemisférios
cerebrais.
A sua etiopatogenia se refere à ausência de clivagem da vesícula
prosencefálica. Tal anomalia relaciona-se às seguintes condições:
- aneuploidias ( mais frequentemente trissomia do 13 );
- triploidias;
- monossomias;
-mosaicisnmos;
- síndrome de Meckel;
- pacientes diabéticos tipo I.
- teratogênicos como o alcalóide da planta Veratum californium, o etanol,
salicilatos, fenitoína e ácido retinóico;
- tabagismo também tem sido estudado como um possível agente teratogênico;
- infecções congênitas como o citalomegalovírus, rubéola, toxoplasmase;
Sabe-se que até o momento nenhum mecanismo fisiopatológico foi definido em
relação ao mecanismo de ação dos agentes infecciosos.
- CARACTERÍSTICAS HEREDITÁRIAS
O fator familiar também tem sido implicado na sua gênese, mas com marcada
heterogênicidade genética. Pode apresentar herança dominante, recessiva ou
ligada ao sexo. Esta anomalia tem sido descrita como parte integrante em
diversas síndromes, como as síndromes ,de Aicardi, de Andermann,
acrocalosal, da fenda facial mediana, entre outras.
28
Raras famílias apresentam herança autossômica dominante e autossômica
recessiva. Mas existem relatos de síndromes de malformações múltiplas,
incluindo a Síndrome de Meckel ( autossômica recessiva ), a Síndrome de
Aicardi ( dominante ligada ao x ), a Síndrome de Fryns e a Síndrome de
Hidroletalus ( autossômica recessiva ), são descritas com holoprosencefalia
( Sanders, 1999 ).
CLASSIFICAÇÃO
Pode ser classificada devido ao grau de diferenciação da vesícula
prosencefálica e se associa com o prognóstico e o número de anomalias
associadas, concomitantes em cada tipo , sendo mais grave e com
apresentação fenotípica mais extrema no tipo alobar, portanto a
holoprosencefalia pode ser classificada em três tipos:
Fig. 10: Fonte www.fetalmed.net
1 – Alobar: na holoprosencefalia alobar, a forma mais grave, há completa
ausência de diverticulação. Pacientes tem defeitos característicos de linha
média, como ciclopia ( uma órbita com prosbócide e nariz ausente ),
29
hipertelorismo, fenda labial e micrognatia.
No exame ultra-sonográfico há cavidade ventricular única na linha mediana, tálamos
ecogênicos fundidos na linha mediana e um manto cortical indiferenciado
cobrindo a cavidade monoventricular.
O terceiro ventrículo, a foice e a fissura inter-hemisférica estão ausentes. O
cerebelo e o tronco cerebral permanecem, todavia, relativamente normais.
Fig 11: Ultra-som Holoprosencefalia Alobar.
Fonte: Goldbamboo.com
30
Fig 12 : RNM Holoprosencefalia Alobar.
Fonte: Theneurostation.com
2 – Semilobar: na holoprosencefalia semilobar , uma forma intermediária, uma
separação parcial dos hemisférios cerebrais pode ocorrer. Alterações faciais
moderadas podem ocorrer, incluindo hipotelorismo, lábio leporino e palato
fendido.
Os achados ultra-sonográficos, incluem um corpo ventricular único com os tálamos
fundidos, rudimentos da fissura inter-hemisférica. O corpo caloso pode estar
ausente ou incompleto. O terceiro ventrículo está normal ou ausente.
O quarto ventrículo e o cerebelo freqüentemente estão normais.
31
Fig 13: Ultra-som Holoprosencefalia semi-lobar com presença de hemisférios
cerebrais.
Fonte: Drmimeneuroanatomia.blogspot.
3 - Lobar : a holoprosencefalia lobar é a forma menos grave. Anomalias faciais
não se encontram presentes. Os cornos frontais permanecem fundidos e o
septo pelúcido está ausente ou incompleto.
Na ultra-sonografia os ventrículos são alargados e os cantos angulados. A
fissura inter-hemisférica está presente e alargada.
A porção posterior encontra-se freqüentemente normal.
32
Fig 14: Holoprosencefalia lobar.
Fonte: Scielo.br
ANOMALIA ASSOCIADAS
A Holoprosencefalia pode ocorrer de forma isolada ou associada a diferentes
anormalidades craniofaciais, pois geralmente estão associadas à
malformações da linha média facial, aumentando em 80% dos casos a
gravidade da doença.
As anormalidades faciais mais freqüentes são :
- Clicopia: representa um defeito grave no desenvolvimento inicial da linha
média facial, incompatível com a vida, com prevalência de 1-3 em cada
100.000 nascimentos ( Saldarriaga W, 2007 ) . Associada a holoprosencefalia
alobar , onde observa-se a fusão das órbitas, que pode ser parcial ou
completamente fundidos, formando um olho central, portanto podem
apresentar-se de forma anoftálmica ( ausência congênita dos olhos ),
33
monooftálmica ( um olho único ) ou sinoftálmica ( fusão dos dois olhos ).
Também não há formação de um nariz verdadeiro, com as placas olfativas
estando consolidadas numa prosbóscite única localizada acima da única órbita,
implantação baixa do pavilhão auricular e ausência do filtro do lábio superior.
Os ossos etmoidais e outras estruturas ósseas da linha média estão ausentes.
A trissomia do 13 é a alteração cromossômica mais freqüente associada à esta
anomalia .
Fig 15: Ciclopia.
Fonte: Google imagens.
- Etmocefalia: ausência de nariz, com placas olfativas consolidadas numa
prosbócide única. Ocorre hipotelorismo extremo ( duas órbitas protusas ), com
o prosbócide localizando-se geralmente no mesmo plano que as órbitas.
34
Fig 16 : Feto sinoftalmia.
Fonte: Scielo.br
-Cebocefalia: possui uma incidência de 1:16.000 nascidos vivos, onde observase características de macaco, apresentando nariz rudimentar, com uma narina
única. As duas órbitas estão presentes mas associadas com hipotelorismo.
Fig 17: Síndrome de Patau.
Fonte: esds1.pt
35
- Fenda labial mediana: também chamada de agenesia pré-maxilar, é a
manifestação da linha média da face envolvendo área do filtro do lábio
superior, na qual este encontra-se ausente.
Diferencia-se do lábio leporino comum, encontrado lateralmente ao filtro, e em
geral sem alterações do sistema nervoso central.
O lábio leporino mediano pode estar associado a hipotelorismo e fenda
palatina.
Fig 18: Síndrome Patau, Fenda Palatina Mediana.
Fonte: Google imagens.
- Outras alterações também podem estar associadas, são elas : polidactilia,
hexadactilia, exoftalmia, cistos renais, displasia renal, agenesia renal,
hidronefrose, hidriopsia fetal, onfalocele, malformações cardiovalculares,
meningomielocele e anormalidades intestinais.
36
DIAGNÓSTICO PRÉ-NATAL
O que chama inicialmente a atenção para o diagnóstico ultra-sonográfico é o
aumento da formação cística intracraniana com ventrículos cerebrais não
segmentados, designado por alguns autores como “ventrículo em ferradura” ou
“ em forma de C “, em cortes coronais.
Neste mesmo plano não se observa o cavo do septo pelúcido, o qual excluiria o
diagnóstico, se presente. A ausência do eco médio e os tálamos fundidos
também são bastante característicos. No diagnóstico de holoprosencefalia o
examinador deve direcionar a investigação de forma detalhada para a face
fetal, já que encontra-se alterada concomitantemente, em até 87% dos casos .
A falha da segmentação do prosencefálo pode ocorrer tanto no plano sagital
( manifestada no aspecto de ventrículo único ) , quanto no plano axial
( manifestada nas alterações dos tratos ópticos e olfativo, por meio de
hipotelorismo, da probócide ou da ciclopia ).
Dentre as anormalidades associadas encontram-se as anormalidades da face ,
incluindo fenda palatina mediana. O diagnóstico diferencial pode ser realizado
com a hidrocefalia, a hidrancencefalia e a agenesia do corpo caloso.
Os tipos menos diferenciados ( alobar e semilobar ) tendem a apresentar
diagnóstico mais reservado no que se refere à sobrevida, assim como no
retardo mental.
A sensibilidade diagnóstica ultra-sonográfica na detecção de hoprosencefalia é
estimada em 89%.
Pode ocorrer polidrâmnia, a placenta geralmente é normal e geralmente
apresenta restrição de crescimento intrauterino.
37
Deve-se salientar que o diagnóstico pode ser realizado precocemente, através
da avaliação transvaginal ao redor da 13ª semana de gestação.
DIAGNÓSTICO DIFERENCIAL
- Hidranencefalia onde pode-se diferenciar por não apresentar os tálamos
fundidos e pode-se observar o terceiro ventrículo;
- Hidrocefalia na sua forma maior, sendo importante a diferenciação;
- A forma alobar pode ser confundida com estenose de aqueduto de, se a fusão
posterior dos ventrículos e a ausência do terceiro ventrículo passarem
despercebidas;
- Esquizencefalia pode envolver ambos os hemisférios cerebrais com seu
defeito cístico, mas geralmente com uma forma assimétrica, e os demais
ventrículos estão com aspecto anormal;
- Cisto de Dandy-Walker maciço pode dar a impressão de um ventrículo
comum dilatado, mas o sistema supratentorial normal estará visível;
- A Agenesia do Corpo Caloso pode simular a forma lobar. O terceiro ventrículo
estará separado dos ventrículos laterais e localizado em um nível superior.
Deve-se prestar atenção se existem estigmas da trissomia do 13, como mãos e
pés anormais, encefalocele e outros problemas associados como, doença
cardíaca congênita ( problemas de saída dupla ), onfalocele e síndrome de
Dandy-Walker.
38
CONDUTA PRÉ-NATAL
Deve ser realizado o estadiamento da holoprosencefalia para tentar prever o
prognóstico.
O diagnóstico de alterações cromossômicas deve ser realizado através do
estudo citogenético, mesmo na gravidez avançada, pois assim poderemos
orientar as futuras gestações.
A ecocardiografia é importante para avaliar se existe malformações cardíacas
associadas.
Deve ser feito uma avaliação cuidadosa da glicemia materna, em busca de
diagnosticar diabetes materna.
Sorologia para para citalomegalovírus, rubéola e toxoplasmose devem ser
solicitados de forma rotineira
Acompanhamento com equipe multidisciplinar é o ideal, e intervenções
terapêuticas intra-útero são contra-indicadas.
CONDUTA OBSTÉTRICA
Não é recomendada intervenções na gravidez como cesariana ou parto
prematuro, pois todas as formas de holoprosencefalia apresentam um péssimo
prognóstico. Muitas vezes a gestação cursa com polidrâmnia, que ocasiona
espontaneamente o parto prematuro.
O pólo cefálico deve ser avaliado para avaliar se há possibilidade de parto
normal.
Na conduta obstétrica é importante o acompanhamento em um centro de
39
referência de medicina fetal. O cariótipo fetal é uma opção na confirmação do
diagnóstico etiológico e no aconselhamento futuro para o casal acometido com
um filho portador da condição. Para casos de transmissão através de
anomalias autossômicas dominantes, o risco de recorrência pode chegar a
18 %, sendo que, para os casos isolados, o risco empírico calculado de
recorrência é de 6 %.
ASSISTÊNCIA NEONATAL
Quando for estabelecido o diagnóstico deve-se ter uma conversa franca com a
família para determinar , antes do parto, sobre as medidas de ressuscitação.
Recomenda-se que o nascimento ocorra em um centro de atenção terciário,
com uma equipe de neuropediatria e exames disponíveis, principalmente se
houver necessidade de confirmação do diagnóstico.
Além dos sinais clínicos recomenda-se realizar tomografia computadorizada ou
ressonância magnética nuclear pós-natal.
No berçário a melhor conduta é tentar obter diagnóstico definitivo o mais rápido
possível, e se a analise cromossômica não tenha sido realizado durante o prénatal, é preciso coletar sangue antes de realizar qualquer procedimento
invasivo ou transfusão sanguínea. O apoio respiratório é indicado juntamente
com os cuidados de longo prazo.
40
PROGNÓSTICO
O estadiamento da holoprosencefalia pela classificação De Meyer et al., 1964,
ainda é utilizada até nos dias de hoje, por ser de grande relevância pois ela
permite classificar , estabelecer o prognóstico e também planejar a abordagem
terapêutica que deverá ser proposta por uma equipe multidisciplinar de acordo
com o tipo de holoprosencefalia e das malformações associadas.
Tabela 1 : Classificação de anormalidades holoprosencefálicas
GRUPO
I
CICLOPIA
II
ETMOCEFALIA
III
CEBOCEFALIA
IV A
AGENESIA
PRÉ-MAXILAR
IV B
VA
DISMORFISMO
ATENUADO
VB
MORFOLOGIA
FACIAL
Olho único ou fendido
Arrinia com probóscide
Hipotelorismo severo
Arrinia com probóscide
Hipotelorismo
Narina única
Hipotelorismo
Nariz achatado
Fenda labial mediana
Hipotelorismo
Nariz plano
Fenda labial bilateral
Hipotelorismo
Nariz plano
Fenda palatina
Hipotelorismo
Hipoplasia médio facial
MORFOLOGIA
ENCEFÁLICA
Alobar
Microcefalia
Alobar
Microcefalia
Alobar
Microcefalia
Alobar
Semilobar
Trigonocefalia
Lobar
Lobar
De Meyer et al. 1964
41
- Grupos I a III – não sobrevivem além da infância e ocorrem freqüentemente
em abortos espontâneos e óbito intra-útero.
Pelo prognóstico reservado está totalmente contra-indicada a correção
cirúrgica;
- Grupo IV A - podem sobreviver por anos, mas apresentam grave déficit
neurológico, sendo os benefícios com a correção cirúrgica raros;
- Grupos IV B, V A, V B – são eletivas para a correção cirúrgica, pois
geralmente possuem um quociente de inteligência ( Q.I. ) suficiente para a
independência social, mas podem cursar com algum déficit mental e
psicomotor.
Os pacientes do grupo IV B, por serem mentalmente deficientes, a correção
cirúrgica para correção do palato e do lábio fendido se justifica pela expectativa
de vida aumentada.
Os casos de holoprosencefalia alobar e semilobar geralmente são fatais e
associadas a retardo mental grave, portanto o prognóstico é desfavorável.
42
CONCLUSÃO
Esta revisão permitiu um estudo detalhado, e priorizando a importância do
diagnóstico antenatal da agenesia do corpo caloso e da holoprosencefalia, bem
como as formas de manifestação, associações com outras malformações,
condutas e prognóstico.
Sabe-seque em relação a etiologia muito ainda deve-se estudar para podermos
entender o mecanismo de ação, em especial dos agentes teratógenos, mesmo
assim devemos investigar o máximo possível, possibilitanto um
aconselhamento adequado para as futuras gestações.
O principal método diagnóstico pré-natal é a ultra-sonografia que possui um
alto índice de sensibilidade para o diagnóstico da holoprosencefalia e se baseia
muitas vezes em sinais secundários no caso da agenesia do corpo caloso.
Nestes casos uma abordagem com equipe multiprofissional principalmente
após o estadiamento e avaliação de malformações associadas é fundamental.
43
BIBLIOGRAFIA
1- Castro Jr NP, Granato L, Figueredo MS, Rios OAB. Holoprosencefalia
com prosbócite: caso clínico. Rev Bras de Otorrinolaringologia 2000; 66:273-6
2- Costa AG. Diagnóstico pré-natal de anomalias congênitas por ultrasonografia. Femina 2005, vol 33 nº 11, 847.
3- De Meyer W, Zeman W, Palmer CG. The face predicts the brain: diagnostic
significance of medial facial anomalies for holoprosencephaly. Pediatrics
1964; 34: 256-63.
4- Isfer EV, Sanches RC, Saito M. Sistema Nervoso Central: Anomalias de Linha
Média. In___. Medicina Fetal: Diagnóstico Pré-Natal e Conduta. 1ª Ed. Rio de
Janeiro: Revinter; 1996. P 107.
5- Machado ABM. Embriologia, Divisões e Organização do Sistema Nervoso.
In__Neuroanatomia Funcional. 2ª Ed. São Paulo: Atheneu; 1993. P 07.
6-
Maranhão PA . Agenesia do Corpo Caloso- O Sinal do Candelábro.Rev Bras
Neurol, 46 (2): 51, 2010.
7- Massonetto JC. Holoprosencefalia. Femina, 2003, vol 31 nº 9, 795.
8- Mesquita MA, Segre CAM. Síndrome alcoólica fetal. Rev Bras de Med
Pediatria Moderna, nov/dez; 2005 05 a 41 nº6. Pag 273 a 290.
9- Mauad F. Ultra-sonografia Morfológica Fetal. In___. Ultra-Sonografia na
Prática Obstétrica. 1ª Ed. Rio de Janeiro: Revinter; 2006. P 130.
10-Park YW. Diagnosis of fetal anomalies by sonography. Yonsei Med J 2001;
42:660-8.
44
11- Saldarriaga W, Isaza C, Mastroiacovo P, Castilha EE. Ciclopía em el hospital
universotario Del Valle ( Cali, Colombia ) : reporte de cuatro casos nacidos y
revisión de la literatura. Ver Colombiana de Obstetricia y Ginecología, vol 58,
nº1, 2007.
12-Sanders RC, Blackmon LR, Hogge WA, Wulfsberg EA. O Sistema Nervoso
Central: Agenesia do corpo Caloso e Holoprosencefalia. In___Feto Anomalias
Estruturais Uma Abordagem Completa. 1ª Ed. Rio de Janeiro: Revinter; 1999.
P 14 e 34.
13-Santos LC, Figueredo SR, Souza ASR, Marques M. Anomalias do Sistema
Nervoso Central: Anormalidades de Estrutura da Linha Média. In__Medicina
Fetal-IMIP- Instituto Materno Infantil- Professor Fernando Figueira.1ª Ed. Rio
de Janeiro: Medbook-Editora Científica; 2008. P322.