ANOTAÇÕES EM FARMACOLOGIA E FARMÁCIA CLÍNICA
2) medicamentos que atuam no sistema nervoso autônomo - adrenérgicos
Síntese, armazenamento, liberação e metabolismo da
noradrenalina
Receptores adrenérgicos
A principal classificação farmacológica divide os
receptores adrenérgicos em alfa e beta. Por sua vez, existem
os subtipos alfa-1 e alfa-2, beta1, beta2 e beta3, todos
pertencentes à super-família dos receptores acoplados à
proteína G.
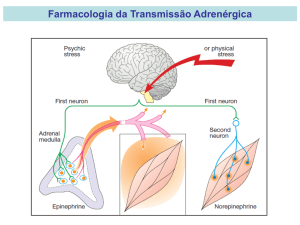
A noradrenalina é sintetizada através da conversão da
L-tirosina em DOPA pela tirosina hidroxilase nos
neurônios catecolaminérgicos. Por sua vez, a DOPA é
convertida em dopamina pela descarboxilase. Em seguida,
a dopamina é convertida em noradrenalina pela dopaminabeta-hidroxilase, localizada nas vesículas sinápticas. Na
medula supra-renal, a noradrenalina é convertida em
adrenalina pela feniletanolamina-N-metil transferase.
Os receptores alfa-1 ativam a fosfolipase C,
produzindo assim, IP3 e DAG como segundos mensageiros;
os receptores alfa-2 inibem a adenilato ciclase e, portanto,
diminuem a formação de AMPc; todos os tipos de
receptores beta estimulam a adenilato ciclase.
A noradrenalina é armazenada em vesículas sinápticas,
juntamente com ATP e cromogranina, e seu transporte para
o interior das vesículas é realizado por transportador
(VMAT – transportador de monoamina vesicular) que pode
ser bloqueado pela reserpina. Atualmente a reserpina não
possui utilidade clínica devido ao seu efeito irreversível
sobre o VMAT e de sua associação com depressão
psicótica.
A liberação do neurotransmissor ocorre normalmente
por exocitose mediada por Ca2+ (geração de um potencial
de ação – despolarização da membrana – abertura dos
canais de cálcio – entrada de cálcio – fusão da vesícula e
descarga por exocitose), e é controlada pela
retroalimentação auto-inibitória, mediada pelos receptores
alfa-2. Essa liberação de catecolaminas é iniciada por sinais
que se originam em um conjunto de áreas de processamento
no SNC, particularmente no sistema límbico. Esses
neurônios do SNC projetam axônios que fazem sinapse em
neurônios pré-ganglionares simpáticos nas colunas
intermédio-laterais da medula espinhal. Os axônios préganglionares projetam-se para os gânglios simpáticos, onde
liberam acetilcolina. Esse neurotransmissor inicia
potenciais pós-sinápticos excitatórios nos neurônios pósganglionares, ativando os receptores nicotínicos de
acetilcolina. Os axônios pós-ganglionares simpáticos
formam varicosidades ou sinapses nos órgãos-alvos ou
sobre eles. A chegada de um potencial de ação nessas
terminações abre os canais de Ca+ regulados por voltagem,
e o consequente influxo de cálcio deflagra o processo de
exocitose das vesículas sinápticas contendo catecolaminas.
A noradrenalina sofre rápida difusão da varicosidade présináptica e regula localmente as respostas teciduais através
da ativação dos receptores adrenérgicos pós-sinápticos.
A noradrenalina modula numerosas funções vitais,
incluindo a frequência e a força da contração cardíaca, a
resistência dos vasos sanguíneos e bronquíolos, a liberação
de insulina e a degradação da gordura.
A ação desse neurotransmissor é interrompida
principalmente por recaptação pelas terminações nervosas,
através do transportador de noradrenalina (NET). Essa
captação é bloqueada por antidepressivos tricíclicos, pela
fenoxbenzamina, cocaína e anfetaminas. Além disso, as
catecolaminas (noradrenalina, dopamina e serotonina) são
metabolizadas pelas enzimas MAO e pela COMT.
Os principais efeitos da ativação dos receptores
adrenérgicos estão relacionados na tabela 1.1, em anexo.
1
Marcelo A. Cabral
ANOTAÇÕES EM FARMACOLOGIA E FARMÁCIA CLÍNICA
Efeitos dos fármacos simpaticomiméticos sobre sistemas e
órgãos
g) trato geniturinário: o útero humano possui receptores alfa
e beta-2. a ativação dos receptores beta-2 do útero produz
relaxamento. A ativação dos receptores alfa da bexiga, uretra e
próstata promovem a continência urinária.
a) vasos sanguíneos: O tônus do músculo liso vascular é
regulado por receptores adrenérgicos. Os receptores alfa
aumentam a resistência arterial, enquanto os receptores beta-2
promovem o relaxamento do músculo liso. Os vasos cutâneos
apresentam receptores alfa e sofrem contração na presença de
adrenalina e noradrenalina. Os vasos no músculo esquelético
podem contrair-se ou dilatar-se, dependendo da ativação dos
receptores alfa ou beta. Portanto, os efeitos globais de um
agente simpaticomimético sobre os vasos sanguíneos dependem
das atividades relativas das drogas nos receptores alfa e beta.
h) glândulas exócrinas: as glândulas salivares contêm
receptores adrenérgicos, e sua ativação causa redução da
produção da saliva (boca seca). Por outro lado, as glândulas
sudoríparas que se localizam na palma das mãos são
estimuladas pelos simpaticomiméticos. Essas glândulas estão
associadas ao estresse psicológico.
e) efeitos metabólicos: a ativação dos receptores beta
adrenérgicos nas células adiposas resulta em aumento da
lipólise, com liberação aumentada de ácidos graxos livres e
glicerol no sangue. Por outro lado, a ativação dos receptores
alfa-2 dos lipócitos inibe a lipólise. Os receptores alfa e beta
adrenérgicos expressos nas ilhotas pancreáticas tendem a
aumentar e a diminuir a secreção de insulina, respectivamente,
embora o principal regulador da liberação de insulina seja a
concentração plasmática de glicose.
b) coração: os efeitos diretos sobre o coração são
determinados, em grande parte, pelos receptores beta-1,
embora estejam envolvidos os receptores beta-2 e, em menor
grau, os receptores alfa. A ativação dos receptores beta resulta
em aumento do influxo de cálcio nas células cardíacas, com
consequências tanto elétricas quanto mecânicas: aumento da
frequência e força de contração cardíacas.
c) pressão arterial: os efeitos das drogas simpaticomiméticas
sobre a pressão arterial podem ser explicados com base nos
seus efeitos sobre o coração, a resistência vascular periférica e
o retorno venoso. Um agonista alfa puro (fenilefrina) aumenta
a resistência arterial periférica e diminui a capacitância
venosa, além de poder exercer uma ação inotrópica positiva
moderada. Por outro lado, a resposta da pressão arterial a um
agonista puro dos receptores beta aumenta o débito cardíaco
(ativação dos receptores beta-1), além de reduzir a resistência
periférica ao ativar os receptores beta-2, produzindo
vasodilatação em certos leitos vasculares. O efeito final
consiste em manter ou elevar levemente a pressão sistólica,
permitindo, ao mesmo tempo, uma queda da pressão
diastólica.
h) função endócrina e leucocitose: a secreção de renina é
estimulada pelos receptores beta-1 e inibida pelos receptores
alfa-2. a adrenalina e agentes relacionados em altas
concentrações causam leucocitose, em parte, ao promover a
desmarginação dos leucócitos sequestrados da circulação geral.
i) efeitos sobre o sistema nervoso central: a ação dos agentes
simpaticomiméticos sobre o sistema nervoso central varia
acentuadamente, dependendo de sua capacidade de atravessar a
barreira hematoencefálica. As catecolaminas são quase
totalmente excluídas por essa barreira. Por outro lado, as não
catecolaminas de ação indireta, como as anfetaminas, que
penetram facilmente no SNC a partir da circulação, produzem
desde leve estado de alerta, elevação do humor, insônia, euforia,
anorexia e até um comportamento psicótico. Esses efeitos não
são facilmente atribuídos a ações mediadas pelos receptores alfa
e beta e podem representar uma intensificação dos processos
mediados pela dopamina ou outros efeitos dessas drogas no
sistema nervoso central.
d) olho: a ativação dos receptores alfa do músculo dilatador
da pupila da íris (fenilefrina) provoca midríase. Os
estimulantes alfa e beta adrenérgicos exercem efeitos
importantes sobre a pressão intra-ocular. Os agonistas alfa
aumentam o efluxo de humor aquoso do olho, enquanto os
antagonistas beta diminuem a produção de humor aquoso.
Essas classes de fármacos são usadas no tratamento do
glaucoma (brimonidina + timolol).
1) Medicamentos adrenérgicos:
Os medicamentos adrenérgicos são também
denominados simpaticomiméticos em virtude de sua capacidade
de produzir efeitos semelhantes aos produzidos pelo sistema
nervoso simpático.
e) trato respiratório: o músculo liso brônquico contém
receptores beta-2 que causam relaxamento. A ativação desses
receptores resulta em broncodilatação. Os vasos sanguíneos da
mucosa das vias respiratórias superiores contêm receptores
alfa. A ação dos descongestionantes dos receptores
adrenérgicos é clinicamente útil.
A capacidade dos agonistas dos receptores
adrenérgicos de iniciar uma sinalização distal é proporcional ao
número de receptores ativados. Por conseguinte, a ocorrência de
mudanças na densidade dos receptores existentes sobre a
superfície celular irá alterar a eficácia aparente de um agonista.
Assim, as alterações tanto em curto prazo (dessensibilização)
quanto em longo prazo (infra-regulação) no número de
receptores adrenérgicos funcionais são importantes na regulação
da resposta do tecido. Quando um agonista ativa o receptor
adrenérgico, a dissociação das proteínas G heterotriméricas leva
a uma sinalização distal, bem como a um mecanismo de
retroalimentação negativa que limita as respostas dos tecidos. O
f) trato gastrintestinal: é possível produzir relaxamento do
músculo liso gastrintestinal com agentes alfa e beta
estimulantes. Os receptores beta, que parecem estar
localizados diretamente nas células musculares lisas, medeiam
o relaxamento através de hiperpolarização e diminuição da
atividade em espícula nessas células. Os agonistas alfa-2
diminuem a atividade muscular indiretamente através da
redução pré-sináptica da liberação de acteilcolina e de outros
estimulantes do sistema nervoso entérico.
2
Marcelo A. Cabral
ANOTAÇÕES EM FARMACOLOGIA E FARMÁCIA CLÍNICA
acúmulo das subunidades BY na membrana recruta uma cinase
do receptor acoplado à proteína G (GRK), que fosforila o
receptor nos resíduos da extremidade C-terminal, que atuam
como importantes alvos de proteínas inativadoras
(dessensibilização). Alternativamente, a proteinocinase A e a
proteinocinase C podem fosforilar as proteínas G. o estado
fosforilado de uma proteína G pode ligar-se a outra proteína
denominada B-arrestina, que inibe estericamente a interação
receptpr-proteína G, silenciando efetivamente a sinalização do
receptor. Em uma escala temporal maior, o complexo receptorb-arrestina é seuqestrado, através de um mecanismo dpendente
de clatrina, em um compartimento endocítico para
internalização, um processo denominado infra-regulação. Cada
um desses processos é importante na regulação da
responsividade do tecido em curto ou em longo prazo.
A noradrenalina e a adrenalina apresentam efeitos
semelhantes sobre os receptores beta-1 do coração, com
potência também semelhante nos receptores alfa.
A
noradrenalina tem pouco efeito sobre os receptores beta-2. Por
conseguinte, a noradrenalina aumenta a resistência periférica e a
pressão arterial tanto sistólica quanto diastólica.
A noradrenalina também aumenta a freqüência cardíaca,
porém esse efeito é tipicamente superado pela atividade vagal
reflexa em resposta à elevação da pressão arterial. Ela é
utilizada com frequência no tratamento de emergência do
choque distributivo (caracterizado pela queda do tônus
vasomotor por vasodilatação e hipovolemia relativa – comum
acontecer em choque séptico, depressão do SNC, traumatismos
graves, etc).
Os simpaticomiméticos ditos indiretos promovem
a liberação do neurotransmissor para a fenda sináptica, por
deslocá-lo de vesículas de armazenamento. Já os diretos
acoplam-se a receptores simpáticos pós-sinápticos. Os
medicamentos adrenérgicos produzem seus esfeitos ao estimular
os receptores alfa e/ou beta adrenérgicos. Eles são classificados
em dois grupos segundo as suas estruturas químicas: as
catecolaminas e as não-catecolaminas.
O isoproterenol é um agonista muito potente dos
receptores beta, que exerce pouco efeito sobre os receptores
alfa. A droga possui ações cronotrópica e inotrópica positivas. O
isoproterenol é um potente vasodilatador, pois atua
exclusivamente sobre os receptores betas.
A dopamina, precursor metabólico imediato da
noradrenalina, ativa os receptores D1 em vários leitos
vasculares, resultando em vasodilatação.
O fenoldopam
também é um agonista dos receptores D1, sendo indicada sua
administração intravenosa para o tratamento da hipertensão
grave. A dopamina é administrada em baixas doses para
melhorar o fluxo sanguíneo dos rins, uma vez que faz dilatar os
vasos sanguíneos renais. Neste caso, os receptores
dopaminérgicos D1 ativam a adenilil ciclase nas células
musculares lisas vasculares, resultando em aumento dos níveis
de AMPc e em vasodilatação. Em concentrações
suprafisiológicas, a dopamina também pode atuar como
agonista nos receptores alfa-1(vasoconstrição) e beta-1
(inotropismo positivo). Em face disso, a dopamina é utilizada no
tratamento do choque, particularmente nos estados de choque
causados por baixo débito cardíaco e acompanhados de
comprometimento da função renal, resultando em oligúria
(volume da urina excretada menor que o necessário para
eliminação de catabólitos). Apesar de a dopamina ser um
neurotransmissor proeminente do SNC, a sua administração
sistêmica tem poucos efeitos sobre o SNC, visto que ela não
atravessa facilmente a barreira hematoencefálica.
a) Catecolaminas: são os simpaticomiméticos com
núcleo catecólico (o-diidroxibenzeno). As catecolaminas mais
comuns são a dobutamina, dopamina, adrenalina,
noradrenalina, cloridrato e sulfato de isoproterenol.
As catecolaminas não podem ser administradas por via
oral, visto que são destruídas pelas enzimas digestivas, por outro
lado, são absorvidas rapidamente quando administradas por via
sublingual. A absorção por via SC é lenta, pois esses fármacos
provocam constrição dos vasos sanguíneos ao redor do local de
aplicação. A absorção IM é mais rápida devido a menor
constrição dos vasos sanguíneos locais.
A adrenalina em baixas concentrações possui efeitos
predominantemente beta-adrenérgicos, ao passo que, em altas
concentrações, predominam os efeitos alfa. É um vasoconstritor
e estimulante cardíaco muito potente. Promove a elevação da
pressão arterial sistólica devido suas ações inotrópica e
cronotrópica positivas (ativação dos receptores beta-1) e pela
vasoconstrição induzida em muitos leitos vasculares (ativação
receptores alfa). A adrenalina também ativa os receptores beta-2
existentes em alguns vasos (vasos sanguíneos do músculo
esquelético), resultando em sua dilatação. Por conseguinte, a
resistência periférica total pode diminuir, explicando a queda da
presão diastólica após injeção de adrenalina. A ativação dos
receptores beta-2 relaxa a musculatura lisa brônquica, aumenta
as concentrações de glicose e de ácidos graxos livres no sangue.
As catecolaminas que estimulam os receptores alfa são
utilizadas no tratamento da hipotensão. As catecolaminas que
estimulam os receptores B1 são utilizadas no tratamento da
bradicardia, do bloqueio cardíaco e no tratamento da taquicardia
nodal ou atrial paroxística noturna (surto de frequência cardíaca
rápida).
A adrenalina é utilizada no tratamento da crise asmática
aguda e anafilaxia. Aplicada localmente em altas doses provoca
vasoconstrição e prolonga a ação dos anestésicos locais. Ela
possui rápido início e breve duração de ação, sendo ineficaz por
via oral. O aumento da excitabilidade cardíaca induzido pela
adrenalina pode levar a arritmias cardíacas, e a acentuada
elevação da pressão arterial pode provocar hemorragia cerebral.
As drogas B1 adrenérgicas (isoproterenol e adrenalina)
também são usadas no tratamento da fibrilação ventricular, na
assistolia e na parada cardíaca. Já as drogas que possuem
atividade B2 (isoproterenol e dobutamina) são utilizadas no
tratamento da asma brônquica, enfisema, bronquite e nas
reações de hipersensibilidade aguda às drogas.
As reações adversas às catecolaminas podem incluir:
inquietação, ansiedade tonteira, cefaléia, palpitações, arritmias
3
Marcelo A. Cabral
ANOTAÇÕES EM FARMACOLOGIA E FARMÁCIA CLÍNICA
cardíacas, hipotensão, hipertensão, AVC, angina e aumento da
glicemia.
As reações adversas às não-catecolaminas são:
cefaléia, inquietação, ansiedade, irritação, tremor, sonolência,
insônia, aturdimento, convulsão, hipertensão, hipotensão,
bradicardia, taquicardia, parada cardíaca, etc.
b) Não-catecolaminas: Os principais representantes
são: mefentermina, metaraminol, metoxamina, fenilefrina,
albuterol, salbutamol, efedrina, terbutalina e metaproterenol.
2) Medicamentos bloqueadores adrenérgicos:
A fenilefrina é um agonista alfa relativamente puro. Como
não se trata de um derivado catecólico, a fenilefrina não é
inativada pela COMT e apresenta duração de ação muito mais
prolongada que as catecolaminas. Trata-se de um midriático e
descongestionante eficaz, que pode ser utilizado para elevar a
pressão arterial.
Esses fármacos atuam ao bloquear a transmissão dos
impulsos nos neurônios adrenérgicos ou nos receptores
adrenérgicos.
De acordo com seu local de
medicamentos bloqueadores são classificados em:
A efedrina é encontrada em várias plantas e vem sendo
utilizada na China há mais de 2.000 anos. Como se trata de uma
fenilpropanolamina não-catecólica, a efedrina possui alta
biodisponibilidade e duração de ação relativamente longa.
Devido a seu acesso ao SNC, atua como estimulante leve. A
pseudo-efedrina, um dos quatro enantiômeros da efedrina, é
disponível sem prescrição médica como componente de muitas
misturas descongestionantes.
ação,
os
- bloqueadores alfa-adrenérgicos; e
- bloqueadores beta-adrenérgicos.
Efeitos dos antagonistas alfa-adrenérgicos
Como o tônus das arteríolas e das veias é determinado,
em grande parte, pelos receptores alfa no músculo liso
vascular, as drogas antagonistas dos receptores alfa produzem
redução da resistência vascular periférica e da pressão arterial.
Os antagonistas dos receptores alfa podem causar hipotensão
postural e taquicardia reflexa. A hipotensão postural é devida
ao antagonismo da estimulação dos receptores alfa-1 pelo
sistema nervoso simpático no músculo liso venoso. A
taquicardia pode ser mais pronunciada com agentes que
bloqueiam os receptores alfa-2 pré-sinápticos no coração.
O metaraminol provoca vasoconstrição e é utilizado
no tratamento da hipotensão em casos de choque grave
(ativação dos receptores alfa-1). A ritodrina e a terbutalina são
administradas para interromper o trabalho de parto pré-termo
(ativação dos receptores beta-2).
O salbutamol, salmeterol, ritodrina e a terbutalina são
agonistas seletivos dos receptores beta-2 adrenérgicos, isentos,
nas doses usuais, de efeitos estimulantes cardíacos. Por efeito de
relaxamento das musculaturas brônquica e uterina, são
utilizados clinicamente para alívio da crise de asma, e
administrados intravenosamente no trabalho de parto prematuro.
Com aumento da dose ocorrem efeitos beta-1, e a taquicardia,
tremores e nervosismo são os principais efeitos adversos.
Os efeitos de menor importância que indicam bloqueio
dos receptores alfa em outros tecidos incluem miose e
congestão nasal. O bloqueio dos receptores alfa-1 da base da
bexiga e da próstata está associado a uma redução da
resistência ao fluxo de urina.
Os medicamentos adrenérgicos não catecolaminas são
utilizados para:
Fármacos antagonistas alfa-adrenérgicos
Os principais fármacos antagonistas alfa-adrenérgicos
são a fentolamina, a tolazozina, a fenoxbenzamina, prazosina,
terazosina e doxazosina.
- produzir a contração local ou sistêmica dos vasos
sanguíneos (mefentermina, metaraminol, metoxamina e
fenilefrina) (receptores alfa-1);
A fentolamina, derivado imidazólico, é um potente
antagonista competitivo no nível dos receptores tanto alfa-1,
quanto alfa-2. A fentolamina produz redução da resistência
periférica através do bloqueio dos receptores alfa-1 e,
possivelmente, dos receptores alfa-2 no músculo liso vascular.
A estimulação cardíaca induzida pela fentolamina é devida à
ativação da estimulação simpática do coração em resposta a
mecanismos barorreflexos. O antagonismo dos receptores
alfa-2 pré-sinápticos pode provocar aumento da liberação de
noradrenalina dos nervos simpáticos (inibição da
retroalimentação).
- descongestão nasal e ocular, e dilatação dos bronquíolos
(salbutamol, efedrina, isoetarina, metaproterenol e terbutalina)
(receptores beta-2);
- relaxamento do músculo liso (ritodrina e terbutalina)
(receptores alfa-2 e beta-2).
É importante lembrar que os agonistas alfa-2
adrenérgicos seletivos têm importante capacidade de reduzir a
pressão arterial através de ações no sistema nervoso central,
embora sua aplicação local direta a um vaso sanguíneo possa
causar constrição. Como exemplo desses fármacos pode-se citar
a clonidina, a metildopa, a guanfacina e o guanabenz.
Os principais efeitos adversos da fentolamina estão
relacionados à estimulação cardíaca, que pode causar
taquicardia intensa, arritmias e isquemia do miocárdio,
particularmente após administração intravenosa.
Os agentes alcalizantes da urina, como a
acetazolamida e o bicarbonato de sódio, retardam a excreção
dos medicamentos não-catecolaminas, prolongando sua ação.
Este fármaco tem sido utilizado no tratamento do
feocromocitoma (intra-operatório) bem como da disfunção
4
Marcelo A. Cabral
ANOTAÇÕES EM FARMACOLOGIA E FARMÁCIA CLÍNICA
erétil masculina através de injeção intra-cavernosa e
administração oral.
doses convencionais, geralmente não causam hipotensão em
indivíduos sadios com pressão arterial normal.
A fenoxibenzamina liga-se de forma covalente ao
receptor alfa, causando bloqueio irreversível de longa duração.
A droga também inibe a recaptação da noradrenalina liberada
pelas terminações nervosas adrenérgicas pré-sinápticas. A
fenoxibenzamina bloqueia os receptores histamínicos H1, de
acetilcolina e de serotonina, bem como os receptores alfa. O
aspecto mais importante do seu uso consiste na capacidade da
fenoxibenzamina de atenuar a vasoconstrição induzida pelas
catecolaminas. O seu principal uso é encontrado no tratamento
do feocromocitoma. Os efeitos adversos mais comuns do seu
uso são a hipotensão postural, taquicardia, fadiga, sedação e
náusea.
Os antagonistas dos receptores beta execem efeitos
proeminentes sobre o coração. Os efeitos inotrópicos e
cronotrópicos negativos são previsíveis, com base no papel
desempenhado pelos receptores adrenérgicos na regulação
dessas funções. No sistema vascular, o bloqueio dos receptores
beta opõe-se à vasodilatação mediada por beta-2. As drogas
beta-bloqueadoras antagonizam a liberação da renina induzida
pelo sistema nervoso simpático. De qualquer modo, enquanto os
efeitos agudos dessas drogas podem incluir um aumento da
resistência periférica, a sua administração crônica resulta em
queda da resistência periférica em pacientes com hipertensão.
A prazosina é eficaz no tratamento da hipertensão. É
altamente seletiva para os receptores alfa-1. Isso pode explicar a
ausência relativa de taquicardia observada com o uso da
prazosina, em comparação com a relatada com a fentolamina e a
fenoxibenzamina. A prazosina provoca relaxamento do músculo
liso tanto arterial quanto venoso, devido ao bloqueio dos
receptores alfa-1.
b) efeitos sobre o trato respiratório:
O bloqueio dos receptores beta-2 no músculo liso
brônquico pode determinar um aumento da resistência das vias
aéreas, particularmente em pacientes com asma. Os antagonistas
dos receptores beta-1, como o metoprolol ou o atenolol, podem
ter alguma vantagem sobre os antagonistas beta não-seletivos
quando se deseja um bloqueio dos receptores beta-1 no
coração, enquanto o bloqueio dos receptores beta-2 não é
desejável.
A terazosina é outro antagonista alfa-1 seletivo e
reversível que se mostra eficaz na hipertensão. Foi também
aprovada para uso em homens com sintomas urinários causados
por hiperplasia prostática benigna (HPB).
c) efeitos sobre o olho:
Vários agentes beta-bloqueadores reduzem a pressão
intra-ocular, particularmente em olhos com glaucoma. Em geral,
o mecanismo relatado consiste em diminuição da produção de
humor aquoso.
A doxazosina mostra-se eficaz no tratamento da
hipertensão e da HPB. Difere da prazosina e da terazosina, pela
sua meia-vida mais prolongada, cerca de 22 horas.
As drogas antagonistas alfa-adrenérgicas são utilizadas
no tratamento do feocromocitoma (fenoxibenzamina e
fentolamina), e no tratamento da hipertensão crônica. A
fentolamina tem sido utilizada para reverter a vasoconstrição
local intensa causada por infiltração inadvertida de agonista alfa
no tecido subcutâneo durante a administração intravenosa. O
antagonista alfa é administrado por infiltração local no tecido
isquêmico. Diversos estudos demonstraram a eficácia de vários
antagonistas dos receptores alfa-1 em pacientes com HPB. Os
antagonistas alfa-2 têm relativamente pouca utilidade clínica.
d) efeitos metabólicos e endrócrinos:
Os antagonistas beta, como o propranolol, inibem a
estimulação da lipólise pelo sistema nervoso simpático. A
glicogenólise no fígado humano seja, pelo menos parcialmente,
inibida após bloqueio dos receptores beta-2. Os antagonistas
beta adrenérgicos devem ser usados com muita cautela em
pacientes diabéticos insulino-dependentes, visto que as
catecolaminas podem constituir os principais fatores na
estimulação da liberação da glicose pelo fígado, em resposta à
hipoglicemia. Os antagonistas beta-adrenérgicos são muito mais
seguros em pacientes com diabete tipo 2 que não apresentam
episódios de hipoglicemia.
Efeitos dos antagonistas beta-adrenérgicos
Os efeitos dessas drogas são devidos, em sua maior
parte, à ocupação e ao bloqueio dos receptores beta. Entretanto,
algumas ações podem resultar de outros efeitos, incluindo
atividade de agonista parcial nos receptores beta e ação
anestésica local, que diferem entre os beta-bloqueadores. Os
principais fármacos bloqueadores beta adrenérgicos são o
propranolol, o metropolol, atenolol, nadolol, labetalol,
carvedilol, esmolol e o timolol.
O uso crônico de antagonistas dos receptores betaadrenérgicos tem sido associado a um aumento das
concentrações plasmáticas de colesterol das VLDL e uma
redução das concentrações de colesterol da HDL. Ambas as
alterações são potencialmente desfavoráveis em termo de risco
de doença cardiovascular.
A ação anestésica local, também conhecida como ação
“estabilizadora das membranas”, constitui um efeito
proeminente de vários beta-bloqueadores. Explicar esta merda...
a) efeitos sobre o sistema cardiovascular:
Os fármacos beta-bloqueadores, quando administrados
de forma crônica, reduzem a pressão arterial em pacientes com
hipertensão. Os mecanismos envolvidos podem incluir efeitos
sobre o coração e os vasos sanguíneos, supressão do sistema
renina-angiotensina e, talvez, efeitos sobre o sistema nervoso
central. Em contraste, essas drogas, quando administradas em
5
Marcelo A. Cabral
ANOTAÇÕES EM FARMACOLOGIA E FARMÁCIA CLÍNICA
Fármacos antagonistas beta-adrenérgicos
observados. Essa classe de fármacos também se mostra eficaz
no tratamento das arritmias ventriculares e supra-ventriculares.
O propranolol é o protótipo das drogas betabloqueadoras. Sua biodisponibilidade é baixa e dosedependente, em virtude do extenso metabolismo de primeira
passagem no fígado.
Verificou-se que a administração de beta-bloqueadores
reduz a pressão intra-ocular em pacientes com glaucoma. O
mecanismo parece envolver uma diminuição da produção de
humor aquoso pelo corpo ciliar, que é fisiologicamente ativada
pelo AMPc. O timolol mostra-se apropriado para uso local no
olho, visto que carece de propriedade anestésica local.
O metropolol e o atenolol são membros do grupo
beta-1 seletivo. Esses fármacos podem ser mais seguros para
pacientes que apresentam broncoconstrição em resposta ao
propranolol. Como a sua seletividade beta-1 é bastante limitada,
esses fármacos devem ser utilizados com muita cautela, ou até
mesmo ser evitados em pacientes com história de asma. Os
antagonistas beta-1 seletivos podem ser preferíveis em pacientes
com diabete ou com doença vascular periférica, quando há
necessidade de terapia com beta-bloqueador, visto que os
receptores beta-2 são provavelmente importantes no fígado
(recuperação da hipoglicemia) e nos vasos sanguíneos
(vasodilatação).
Vários estudos demonstram um efeito benéfico do
propranolol na redução da freqüência e da intensidade da
enxaqueca.
A cafeína é um antagonista competitivo dos receptores
de adenosina no SNC. Esses receptores, que estão localizados
em neurônios noradrenérgicos pré-sinápticos, quando ativados
pela adenosina, atuam para inibir a liberação da noradrenalina
em sinápses no SNC. O antagonismo desses receptores de
adenosina pela cafeína faz com que a liberação de noradrenalina
não seja inibida, produzindo os efeitos estimulantes
característicos da droga.
O nadolol é notável pela sua duração de ação muito
longa. Seu espectro de ação se assemelha ao do timolol. O
timolol é um fármaco não-seletivo desprovido de atividade
anestésica local. Possui excelentes efeitos hipotensores oculares
quando administrado topicamente no olho. Por outro lado, o
esmolol é um antagonista dos receptores adrenérgicos beta-1
seletivos de ação ultra-curta. O esmolol é potencialmente muito
mais seguro do que os antagonistas de ação mais longa para
pacientes criticamente enfermos que necessitam de antagonista
dos receptores beta-adrenérgicos. O esmolol pode ser útil no
controle das arritmias supraventriculares, das arritmias
associadas à tireotoxicose, da hipertensão peri-operatória e da
isquemia do miocárdio em pacientes agudamente doentes.
Inibidores da síntese de catecolaminas
os inibidores da síntese de catecolaminas possuem
utilidade clínica limitada, visto que esses agentes inibem de
modo inespecífico a formação de todas as catecolaminas. A
alfa-metiltirosina é um análogo estrutural da tirosina que é
transmportado nas terminações nervosas, onde inibe a tiroxina
hidroxilase, a primeira enzima na via de biossíntese das
catecolaminas. Esse agente é utilizado em certas ocasiões no
tratamento da hipertensão associada ao feocromocitoma.
O carvedilol é um antagonista não-seletivo dos
receptores beta que tem alguma capacidade de bloquear os
receptores alfa-1 adrenérgicos. Esse fármaco antagonisa as
ações das catecolaminas com mais potência nos receptores beta
do que nos receptores alfa.
A tiramina é uma amina, presente na dieta, normalmente
metabolizada no trato gastrintestinal e no fígado pela MAO. Em
pacientes que fazem uso de inibidores da MAO (IMAO), a
tiramina é absorvida no intestino, transportada pelo sangue e
captada por neurônios simpáticos, onde é transportada até as
vesículas sinápticas pelo VMAT. Através desse mecanismo, um
estímulo agudo com grandes quantidades de tiramina (como a
ingestão de queijo envelhecido e vinhos)
As drogas antagonistas beta-adrenérgicas são utilizadas
no tratamento da hipertensão, geralmente em associação com
um diurético. Os bloqueadores beta-adrenérgicos reduzem a
freqüência dos episódios de angina e melhoram a tolerância ao
exercício em muitos pacientes com angina. Essas ações estão
relacionadas ao bloqueio dos receptores beta cardíacos,
resultando em diminuição do trabalho cardíaco e da demanda de
oxigênio. A redução da freqüência cardíaca e a sua
regularização podem contribuir para os benefícios clínicos
Pode provocar deslocamento agudo da
noradrenalina vesicular e liberação não-vesicular maciça de
noradrenalina das terminações nervosas, através da reversão do
NET.
6
Marcelo A. Cabral
Tabela 1.1 - Efeitos da ativação dos receptores alfa-1 adrenérgicos: 1) Agonista se liga ao receptor α1, 2) mudança
conformacional proteína Gq– substituição de GDP por GTP e liberação da subunidade αq – 3) ativação da fosfolipase C que catalisa a liberação de IP3 e DAG a
partir do PIP2 – 4) IP3 provoca liberação de Ca+ dos retículos endoplasmáticos – 5) o Ca+ liberado e o DAG ativam a proteína cinase C que fosforila proteínas
– 6) as PTN fosforiladas produzem ações fisiológicas finais.
Local
Efeito fisiológico
Músculo
vascular
liso
Músculo
intestinal
liso
Mecanismo
Estimulação dos receptores alfa-1 aumenta a concentração de Ca+ intracelular
devido a geração de IP3, ativação da calmodulina, fosforilação da cadeia leve de
miosina, interação de actina e miosina é aumentada provocando a contração
muscular.
Contração
Músculo
liso
genitourinário
Coração
Aumento
inotropoismo
excitabilidade.
do
e
Fígado
Glicogenólise
gliconeogênese.
e
Efeitos da ativação dos receptores alfa-2 adrenérgicos: 1) O agonista se liga ao receptor α2 – 2) mudança conformacional da
proteína Gi– substituição de GDP por GTP e liberação da subunidade αi – 3) a subunidade αi se une e inibe a adenilato ciclase causando uma diminuição dos
níveis de AMPC. Além disso, os rcpts α2 ativam os canais de K+ e inibem os canais de Ca+ controlados pela proteína G (subunidades By da ptn Gi) levando à
hiperpolarização da membrana celular - produção da ação fisiológica final (relaxamento, diminuição das atividades).
Local
Efeito fisiológico
Mecanismo
Terminações
pré-sinápticas
Inibição da transmissão
sináptica.
Atuam como auto-receptores para mediar a inibição da transmissão sináptica
por retroalimentação.
Células beta do
pâncreas
Inibição da liberação
de insulina
Plaquetas
Inibição da agregação
plaquetária
SNC
Diminuição
da
descarga simpática na
periferia
Diminuição da liberação de noradrenalina nas terminações simpáticas, causando
diminuição da contração do músculo liso vascular - diminuição da pressão
arterial.
Efeitos da ativação dos receptores beta-1 adrenérgicos: 1) Agonista se liga ao receptor β1 , 2) mudança conformacional proteína Gs–
substituição de GDP por GTP e liberação da subunidade αs – 3) ativação da adenilato ciclase que catalisa conversão de ATP em AMPC – 4) o AMPC ativa as
proteínas cinases. Estas por sua vez fosforilam proteínas, incluindo canais iônicos, produzindo ações fisiológicas finais.
Local
Efeito fisiológico
Mecanismo
Coração
Aumeto
do
cronotropismo
e
inotropismo = aumento
do débito cardíaco.
O efeito inotrópico é mediado pela fosforilação dos canais de Ca+, incluindo os
canais de cálcio no sarcolema. O aumento do cronotropismo resulta de um
aumento mediado pelos receptores beta-1 na taxa de despolarização da fase 4
das células marca-passo do nodo sinoatrial.
Coração
Aumento da velocidade
de condução AV.
O aumento da entrada de cálcio estimulado pelos receptores beta-1 aumenta a
taxa de despolarização das células do nodo atrioventricular.
Células
justaglomerulares
renais
Secreção de renina.
Efeitos da ativação dos receptores beta-2 adrenérgicos: 1) Agonista se liga ao receptor β1 , 2) mudança conformacional proteína Gs–
substituição de GDP por GTP e liberação da subunidade αs – 3) ativação da adenilato ciclase que catalisa conversão de ATP em AMPC – 4) o AMPC ativa as
proteínas cinases. Estas por sua vez fosforilam proteínas, incluindo canais iônicos, produzindo ações fisiológicas finais.
Local
Músculo liso
Efeito fisiológico
Mecanismo
Relaxamento
A proteinocinase A fosforila diversas proteínas contráteis, especialmente a
cinase da cadeia leve de miosina. Essa fosforilação diminui a afinidade da
cadeia leve de miosina pela cálcio-calmodulina, resultando em relaxamento do
aparelho contrátil.
No músculo liso brônquico, o efluxo aumentado de K+ leva a hiperpolarização
das células musculares lisas e, portanto, opõe-se à despolarização necessária
para produzir contração.
Fígado
Glicogenólise (...)
gliconeogênese (...).
e
Nos hepatócitos, a ativação da cascata de sinalização dá início a uma série de
eventos de fosforilação intracelulares, que resultam em ativação da glicogêniofosforilase e catabolismo do glicogênio. Por conseguinte, o resultado da
estimulação dos hepatócitos pelos receptores beta-2 consiste em aumento dos
níveis plasmáticos de glicose.
Músculo
esquelético
Glicogenólise
captação de K+.
e
A ativação das vias de sinalização intracelular estimula a glicogenólise e
promove a captação de K+.
Tecido adiposo
–
receptores
beta-3.
Lipólise.
A ativação dos receptores beta-3...
Referências Bibliográficas
1. RANG, H. P. et al. Farmacologia. 4 edição. Rio de Janeiro: Guanabara Koogan, 2001;
2. KATZUNG, B. G. Farmacologia: Básica & Clinica. 9 edição. Rio de Janeiro: Guanabara Koogan, 2006;
3. CRAIG, C. R.; STITZEL, R. E. Farmacologia Moderna. 6 edição. Rio de Janeiro: Guanabara Koogan, 2005;
4. GOLAN, D. E. et al. Princípios de Farmacologia: A Base Fisiopatológica da Farmacoterapia. 2 edição. Rio de Janeiro: Guanabara
Koogan, 2009;
5. FUCHS, F. D.; WANNMACHER, L.; FERREIRA, M. B. C. Farmacologia Clínica. 3 edição. Rio de Janeiro: Guanabara Koogan,
2004.
6. GILMAN, A. G. As Bases farmacológicas da Terapêutica. 10 edição. Rio de Janeiro: Mc-Graw Hill, 2005.
7. CONSTANZO, L. S. Fisiologia. 2 edição. Rio de Janeiro: Elsevier, 2004.
ЖЖЖЖЖЖ