DISTROFIA MIOTÔNICA
DE STEINERT
Renan Thomaka
Epidemiologia
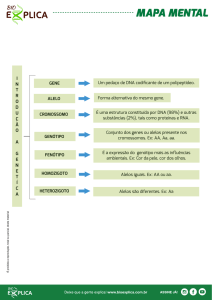
Distrofia Miotônica: DM 1 (Steinert) e DM 2.
É a forma de Distrofia Muscular mais comum
nos adultos.
Prevalência: 1/20.000.
Características Gerais
Doença multisistêmica caracterizada por
uma dificuldade de relaxamento muscular.
Afeta músculo esquelético e liso, sistema
endócrino e sistema nervoso central, bem
como o olho e o coração.
É dividido em 3 fenótipos: leve, clássico e
congênito.
Etiologia
Doença autossômica dominante.
É causada por uma grande quantidade de
repetição CTG no Gene DMPK (19q13.2-q13.3)
- Proteína Quinase da Distrofia Miotônica.
A gravidade da doença é proporcional ao
número de repetições CTG.
Etiologia
Alelos normais: 5 a 34 repetições.
Alelos normais mutáveis (pré-mutação): 35 a
49 repetições.
Alelos de penetrância completa: maior que 50
repetições.
Etiologia
Doença caracterizada pelo fenômeno de
antecipação: com o passar das gerações, a
doença pode se manifestar cada vez mais cedo
e mais grave.
Isso acontece porque alelos com mais de 34
repetições são instáveis e podem gerar mais
repetições durante a meiose.
Manifestações Clínicas
DM 1 Leve: Indivíduos podem ter catarata,
miotonia leve ou Diabetes Mellitus. Vida
normal ou expectativa de vida minimante
reduzida.
DM 1 Clássica: Indivíduos geralmente com
fraqueza muscular e caquexia, miotonia,
catarata, distúrbios de condução e de
freqüência cardíaca.
Manifestações Clínicas
Sintoma predominante é a fraqueza muscular distal,
levando à queda do pé, distúrbio de marcha e
dificuldades em realizar tarefas que exigem destreza
fina das mãos
Fáceis miotônica e calvície masculina precoce
Participação no músculo liso: disfagia, constipação
ou diarréia, oftalmoplegia e disartria
Defeitos de condução cardíaca em diferentes graus.
Manifestações Clínicas
Déficit intelectual em graus variáveis.
Endocrinopatias como hiperinsulinismo e atrofia
testicular.
Mulheres: maior risco de complicações durante a
gravidez.
Redução da expectativa de vida.
As causas mais comuns de morte são pneumonia,
insuficiência respiratória, doenças cardiovasculares,
morte súbita / arritmia, e neoplasias.
Manifestações Clínicas
DM 1 Congênita: As principais características
são grave fraqueza generalizada, hipotonia e
comprometimento respiratório; fácies
miotônica típica: lábio superior em forma de
“V” invertido, bochechas pequenas, músculos
temporais recortados, côncavos; cabeça
estreita, palato em ogiva.
Manifestações Clínicas
Manifestações Clínicas
Geralmente a DM 1 Congênita é herdada por
transmissão materna do alelo DMPK, sendo a relação
de transmissão-distorção na concepção fator favorável
à transmissão de repetições CTG maiores do que
aqueles presentes no pai.
Gestação: polidrâmnio e circulação fetal reduzida.
Retardo mental comum e manifestações iguais ao
adulto.
Alta mortalidade devido à insuficiência respiratória.
Fenótipo mais precoce e mais grave: expectativa de
vida reduzida.
Correlação entre o fenótipo e a quantidade de
repetição CTG na DM 1
A partir de Die-Smulders et al 1998, Mathieu et al 1999, Segundo IDMC 2000
Diagnóstico
Clínico
DM 1 é suspeita em adultos com o seguinte:
-Fraqueza muscular, especialmente distal da perna, mão,
pescoço e face.
-Miotonia (contração muscular sustentada), que muitas
vezes se manifesta como a incapacidade de lançar
rapidamente um aperto de mão e que pode ser demonstrado
através de toque de um músculo (por exemplo, os músculos
tenar) com um martelo de reflexos.
-Catarata subcapsular posterior detectável como
vermelho e verde fosco iridescente na biomicroscopia.
Diagnóstico
Clínico
DM 1 é suspeita em crianças com o seguinte:
-Hipotonia.
-Fraqueza do músculo facial.
-Fraqueza generalizada.
-Insuficiência respiratória.
Eletromicrografia
Verifica se o padrão elétrico de músculos distais são
sugestivos de miotonia.
Diagnóstico
Biópsia muscular
A miotonia gera um padrão histológico muscular típico .
Pesquisa de Expansão CTG no gene DMPK
Padrão-ouro: o gene DMPK é o único gene conhecido
associado a DM 1. Essencialmente 100% dos indivíduos com
DM 1 possuem expansão CTG no gene.
Diagnóstico Diferencial
DM 1 x DM 2
Ambas possuem manifestações clínicas semelhantes, sendo
muito difícil um diagnóstico apenas clínico. Em teste
moleculares, verifica-se que a DM 1 está associada ao gene
DMPK e a DM 2 ao gene ZNF9.
DM1 x Miopatias Hereditárias
- Miosite com corpo de inclusão hereditária
- Miopatia miofibrilar hereditária
- Distrofias musculares distais
- Miotonia congênita (Doença de Becker)
Diagnóstico Diferencial
Ocasionalmente, DM1 tem sido diagnosticada como
doença do neurônio motor , paralisia cerebral,
retardo mental inespecífico, ou, por causa de 'cara
tapada "e movimentos lentos, parkinsonismo.
Portanto, é importante fazer o diagnóstico diferencial
nesses casos.
O diagnóstico diferencial, excetuando-se a DM 2,
com miopatias hereditárias ou outras doenças pode
ser feito clinicamente associado a concentração
sérica de CK e biópsia muscular.
Manejo Clínico
Avaliações após o diagnóstico inicial
-Exame neurológico , oftalmológico e da função tireoidiana.
-Avaliação da capacidade motora e cognitiva.
-Determinação da glicemia de jejum.
-ECG, Holter e ecocardiograma para avaliar a síncope,
palpitações e outros sintomas potenciais de origem cardíaca.
Tratamento das manifestações
Não existe um tratamento específico para a fraqueza
progressiva . A fisiatria, a terapêutica psíquica e ocupacional
podem ajudar a avaliar a necessidade de órteses tornozelopé, cadeira de rodas ou outros dispositivos de apoio.
Manejo Clínico
Tratamento das manifestações
-Manejo da dor – analgésicos.
-Consultas rotineiras com um cardiologista.
-Remoção de catarata caso comprometa a visão.
-Tratamento da diabetes se houver.
-Tratamento de outros distúrbios (endócrinos,
musculares lisos, etc)
Aconselhamento Genético
Fornecer informações para os indivíduos e as famílias a
respeito da história natural, o tratamento, o modo de
herança, e os riscos genéticos para outros membros da
família, bem como informações sobre recursos disponíveis.
Oferecer o teste genético molecular em parentes de adultos
de risco para permitir o diagnóstico e tratamento das
manifestações precocemente.
O tempo ideal para a determinação do risco genético e
discussão sobre a disponibilidade de testes de pré-natal é
antes da gravidez.
Importância do pré-natal.
Referência Bibliográfica
www.ncbi.nlm.nih.gov/books/genereviews
Acesso em 1 de abril de 2010, às 13:00.
http://www.chc.min-saude.pt/servicos/Genetica/dm.htm
Acesso em 1 de abril de 2010, às 13:00.
http://genoma.ib.usp.br/pesquisas/doencas_distrofiamiotonica-steinert.php
Acesso em 1 de abril de 2010, às 13:00.