Relato de Caso
Carcinoma Renal Cromófilo Hereditário – Relato de Caso
Chromophilic Hereditary Renal Cell Carcinoma
Luis Alberto Batista Peres1, Sérgio Luiz Bader2, Júlio Ricardo Ramos3, Alexandre Galvão Bueno4
1Professor Assistente da Disciplina de Nefrologia da UNIOESTE, 2Professor auxiliar da disciplina de Urologia da
UNIOESTE, 3Acadêmico do Curso de Medicina da UNIOESTE, 4Patologista do ANATON - Laboratório de Patologia
de Cascavel - PR
RESUMO
O carcinoma renal papilar é uma neoplasia maligna que representa 5 a 10% de todos os carcinomas de células renais, ocorrendo bilateralmente em 0,4 a
5% dos casos. Descrevemos o caso de um paciente de 43 anos, do sexo masculino, branco, história familiar positiva para carcinoma renal papilar, com diagnóstico incidental de tumor cístico múltiplo bilateral, que foi, em um primeiro tempo, submetido a nefrectomia unilateral esquerda. O paciente recusou-se a
seguir a orientação de nefrectomia contralateral e terapia renal substitutiva regular e seu acompanhamento foi perdido. (J Bras Nefrol 2005;27(4):226-227)
Descritores: Carcinoma. Renal. Cromófilo. Papilar. Tumor.
ABSTRACT
Papillary renal cell carcinoma is a malignant neoplasia which represents 5-10% of all renal cell carcinomas, occurring bilaterally in 0.4–5%. We describe a
case of a papillary renal cell carcinoma in a 43-year-old man who was admitted after an incidental diagnosis of a bilateral multiple-cystic tumor. He had a
positive familial history for papillary renal cell carcinoma. As a first step, the patient was submitted to unilateral left nephrectomy. He refused to undergo contralateral nephrectomy and maintenance dialysis and his follow-up was lost. (J Bras Nefrol 2005;27(4):226-227)
Keywords: Carcinoma. Kidney. Chromofilic. Papillary. Tumor.
INTRODUÇÃO
O carcinoma renal papilar ou cromófilo corresponde a 10 a 15% dos tumores de células renais, sendo o segundo
tipo mais freqüente e caracterizado por metástases freqüentes. É mais comum em indivíduos idosos e naqueles residentes
em zonas urbanas1. Pode ocorrer esporadicamente ou devido à transmissão genética autossômica dominante, cujo gene
se localiza no cromossomo 7q31.1-34, 17, por perda no cromossomo Y e ganhos nos cromossomos 3q, 8p, 12q, 16q e
20q. A forma familiar frequentemente se apresenta multifocal e bilateral1-3.
Relatamos, a seguir, um caso de carcinoma renal papilar multifocal e bilateral em paciente jovem do sexo masculino com história familiar.
RELATO DO CASO
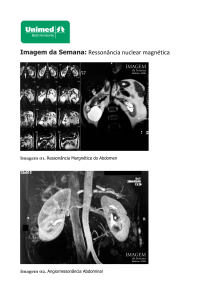
Paciente de 43 anos, branco, sexo masculino, submetido a ecografia de rotina que revelou rim direito com massa
heterogênea em pólo superior medindo 4,2 x 3,3cm e rim esquerdo com contorno lobulado e massa heterogênea em pólo
superior medindo 5,5 x 4,7cm, sem aparente comprometimento extra-renal. História familiar (irmã) positiva para carciRecebido em 18/02/05 / Aprovado em 25/04/05
Endereço para correspondência:
Luis Alberto Batista Peres
R. São Paulo 769, apto. 901
85801-020 Cascavel, PR
Telefone: (45) 3327-2295
Fax: (45) 3327-3413
E-mail: [email protected]
J Bras Nefrol Volume XXVII - nº 4 - Dezembro de 2005
227
Figura 1. Rim esquerdo medindo 13,2 x 10,1cm, com formações
nodulares em meio ao parênquima, medindo entre 0,5 e 1,7cm,
com infiltração de cápsula e tecido adiposo subjacente, com margens cirúrgicas livres.
Figura 2. Neoplasia de origem epitelial com arranjos papilares,
corpos psamomatosos e células contendo núcleos hipercromáticos e pleomórficos.
noma papilar. Submetido à tumorectomia que revelou, ao
exame microscópico, presença de neoplasia de origem epitelial com arranjos papilares, células contendo núcleos hipercromáticos e pleomórficos, além de corpos psamomatosos, confirmando o diagnóstico de carcinoma de células
renais papilífero. Submetido 40 dias depois à nefrectomia
radical esquerda (figura 1), devido a controle ecográfico
que revelou recidiva de múltiplos nódulos tumorais de
diâmetros > 4cm, cujo anátomo-patológico revelou infiltração da cápsula e tecido adiposo subjacente, estroma com
focos de calcificação (figura 2), margens cirúrgicas livres,
sendo classificado como carcinoma de células renais
cromófilo papilar de grau 2 e estadiado como T2N0Mx.
Após indicação pela equipe de nefrectomia contralateral e tratamento dialítico crônico, perdemos o
seguimento do paciente.
A cirurgia conservadora pode ser feita com baixo
risco de recidiva em tumores menores de 4cm, únicos,
com rim contralateral não comprometido; no entanto, em
síndromes tumorais bilaterais, a cirurgia conservadora
apresenta alto risco de recidiva, muitas vezes optando-se
por nefrectomia radical bilateral seguida por tratamento
dialítico definitivo6.
No caso aqui descrito foi realizada nefrectomia
unilateral e houve perda de seguimento do paciente, que
se apresentou resistente à nefrectomia bilateral e ao tratamento dialítico.
DISCUSSÃO
Descrevemos o caso de um carcinoma renal papilar ou cromófilo hereditário. É o tumor mais comum em
indivíduos idosos, ocorrendo geralmente após a quarta
década de vida. Consideramos este caso como a forma
hereditária, pelo fato de o paciente ter história familiar de
tumor renal bilateral e multifocal do tipo papilar.
O carcinoma de células renais bilateral pode apresentar-se de forma sincrônica ou assincrônica, e estima-se
que representa 0,4 a 5% de todos os casos de carcinomas
renais4. Há casos descritos de carcinoma renal bilateral
familiar, com acometimento de vários membros de uma
mesma família. O diagnóstico diferencial se faz com a
enfermidade de Von Hippel-Lindau, carcinoma de células
renais papilar hereditário, síndrome de Birt-Hogg-Dube e
oncocitoma renal familiar5.
REFERÊNCIAS
1. Kuroda N, Toi M, Hiroi M, Enzan H. Review of papillary renal
cell carcinoma with focus on clinical and pathobiological
aspects. Histol Histopathol 2003;18:487-94.
2. Gunawan B, von Heydebreck A, Fritsch T, Huber W, Ringert
R, Jakse G, et al. Cytogenetic and morphologic typing of 58
papillary renal cell carcinomas: evidence for a cytogenetic
evolution of type 2 from type 1 tumors. Cancer Res
2003;63:6200-5.
3. Zbar B, Tory K, Merino M, Schmidt L, Glenn G, Choyke P, et
al. Hereditary papillary renal cell carcinoma. J Urol
1994;151(3):561-6.
4. Petritsch PH, Rauchenwald M, Zechner O, Ludvik W, Pummer K, Urlesberger H, et al. Results after organ-preserving
surgery for renal cell carcinoma an Australian multicenter
study. Eur Urol 1990;18:84.
5. Herring JC, Enquist EG, Chernoff A, Linehan WM, Choyke
PL, Walther MM. Parenchimal sparing surgery in patients with
hereditary renal cell carcinoma: 10-year experience. J Urol
2001;165(3):777-81.
6. Domínguez MD, Rodríguez RQ, Suárez RI, Maestre JM,
González JE. Carcinoma de células renales bilateral sincrónico. Presentación de un caso. Actas Urol Esp
2002;26(10):796-80.